
 |
 |
 |
 |
 |
Artigo
|
|
| Dados espectroscópicos de diterpenos labdânicos: uma análise teórica via RMN e DFT Spectroscopic data of labdane diterpenes: a theoretical analysis via NMR and DFT |
|
Fabrine S. de SouzaI; Silvana de O. SilvaI; Lênio J. G. de FariaII; Cláudio N. AlvesI; Adolfo H. MullerIII; Giselle M. S. P. GuilhonI; Davi do S. B. BrasilII,*
IInstituto de Ciências Exatas e Naturais, Universidade Federal do Pará, 66075-110 Belém - PA, Brasil Recebido em 26/09/2014 *e-mail: davibb@ufpa.br Labdane diterpenes exhibit important bioactivities such as cardiovascular effects in rats as well as effects in the treatment of autoimmune diseases and Alzheimer syndrome. Recently, the labdane diterpenes ent-13-epi-manoil oxide, ribenone and ribenol were isolated from Croton palanostigma. The computational method DFT/B3LYP/cc-pVDZ was used to optimize the structures of these diterpenes and to calculate infrared data. Chemical shifts (δH and δC) of the minimum energy structures (local minimum) were calculated and compared with the experimental data. Comparison of the NMR data by simple linear regression (SLR) showed satisfactory statistical results with a correlation coefficient (R2) and predictive ability (Q2) of over 98%. The predicted NMR data were used to confirm the δH values that have not been published. INTRODUÇÃO Os diterpenóides possuem 20 átomos de carbono e apresentam formação de quatro unidades isopropênicas.1 Pertencem à classe dos terpenos, que tem como destaque os diterpenos de esqueleto clerodano (metabólitos secundários predominantes no gênero Croton), detentores de um grupo de substâncias extremamente diversificadas que compreendem mais de 800 compostos já conhecidos e, em menores proporções, os esqueletos traquilobanos, cauranos e labdanos2-3, sendo de origem fúngica4 ou vegetal,3 além de serem conhecidos como um grupo com elevado número de substâncias biologicamente ativas.5 Dentre as importantes atividades biológicas relacionadas aos diterpenos labdânicos, pode ser mencionado o efeito cardiovascular de um labdano isolado de Xylopia langsdorffiana St.Hil & Tul. (Annonaceae) testado em ratos,6 além dos relevantes efeitos de compostos labdânicos derivados de Adrographis paliculata contra doenças autoimunes e mal de Azheimer, devidamente patenteados em 2007.7 De acordo com Costas e Konstantinos (2001),5 a falta ou insuficiência de dados cromatográficos de compostos de referência para a maioria dos labdanos é a principal razão para a ausência de registros químicos de extratos vegetais dessas substâncias. Nos últimos anos, as técnicas de modelagem molecular têm sido amplamente utilizadas em estudos de biomoléculas e, em especial, têm demonstrado ser uma ferramenta útil para interpretação e elucidação de dados espectroscópicos de compostos orgânicos. A utilização de técnicas de RMN (Ressonância Magnética Nuclear) na elucidação estrutural de produtos naturais é frequente em laboratórios de química. Entretanto, indefinições de deslocamentos químicos e de estereoquímica são problemas recorrentes durante a análise experimental de substâncias químicas via RMN. Diversos pesquisadores8 têm aplicado ferramentas de modelagem molecular em seus trabalhos como uma alternativa promissora em auxílio às técnicas experimentais tradicionais de RMN, alcançando resultados precisos em um tempo de obtenção de dados significativamente pequeno. Como ressalta Farias (2013), o elo entre as previsões teóricas e os resultados experimentais evidencia a importância das simulações computacionais na ciência contemporânea. Pode-se dizer então que, atualmente, a simulação computacional é um ramo da ciência tão importante quanto as técnicas experimentais9 e o desenvolvimento de computadores mais modernos atrelado à melhoria dos algoritmos utilizados em softwares de cálculos quânticos fez com que cálculos ab initio, com metodologia DFT, de moléculas orgânicas de tamanho médio, se tornassem uma ferramenta regular no estudo de parâmetros geométricos, de dados espectroscópicos na região do infravermelho e de dados de RMN de produtos naturais.10 Do pericarpo de Solenostemma argel (Apocynaceae) foram isolados dois novos esteroides denominados como estemosideos. Durante a elucidação estrutural, as configurações relativas dessas moléculas foram determinadas utilizando-se uma estratégia baseada na simulação de parâmetros de RMN de todos os possíveis estereoisômeros.11 Burgueño-Tapia e Joseph-Nathan (2008) determinaram inequivocamente as configurações absolutas do produto natural 6-hidroxieuriopisina, de dois de seus derivados acetil e da eremofilanolida através de cálculos teóricos a nível DFT.12 Cerda-Garcia-Rojas et al., em trabalho semelhante, também conseguiram definir a configuração absoluta de moléculas isoladas de Lippia integrifolia através de espectros de dicroísmo circular vibracional (VCD).13 Essa técnica consiste na comparação entre o espectro VCD experimental e a curva teórica correspondente para o enantiômero adequado, obtida via DFT. A atribuição dos dados de RMN da substância nimbosodiona,14 inicialmente feita de forma equivocada,14-16 somente foi corrigida após a utilização de ferramentas de modelagem molecular, comprovando que a utilização de cálculos teóricos tornou-se uma ferramenta importante na elucidação estrutural de moléculas estruturalmente complexas.14 Neste sentido, o presente trabalho vem contribuir para o estado da arte referente ao uso de técnicas de modelagem molecular na identificação espectral via RMN de produtos naturais. Três diterpenos labdanos: óxido ent-13-epi-manoil (1), óxido ent-3-oxo-13-epi-manoil (ribenona) (2) e o ribenol (3), Figura 1, derivados da espécie Croton palanostigma, isolados em trabalhos anteriores por Brasil (2008),17 e identificados através de espectro de Ressonância Magnética Nuclear, modelo Mercury (300 MHz), foram submetidos a cálculos quânticos a nível DFT para obtenção de dados de infravermelho (IV) e deslocamentos químicos (δH e δC). Os valores teóricos dos δH e δC obtidos foram comparados aos experimentais via regressão linear simples, a fim de verificar o grau de correlação, significância e previsibilidade dos modelos obtidos.
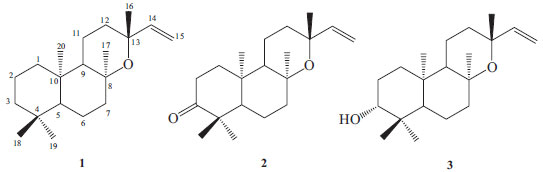 Figura 1. Estruturas das substâncias 1, 2 e 3
MÉTODOS Método computacional Foram realizados estudos de modelagem molecular utilizando inicialmente o programa HyperChem Professional 7.518 na manipulação gráfica das estruturas 1, 2 e 3 e para realizar uma pré-otimização com o método semi-empírico PM3 (Parametric Method 3). Adicionalmente, o método DFT a nível de B3LYP/cc-pVDZ foi empregado para a otimização estrutural das três substâncias estudadas. As análises vibracionais foram feitas pelo método DFT/B3LYP/cc-pVDZ para confirmação dos pontos estacionários. As conformações representativas do mínimo local de energia (Figura 2) foram utilizadas como entrada nos cálculos dos deslocamentos químicos de 1H e 13C, aplicando o método DFT/B3LYP/cc-pVDZ em fase gasosa, no programa Gaussian® 03.19 O tetrametilsilano (TMS), utilizado como padrão interno, foi calculado em fase gasosa pelo método DFT/B3LYP/cc-pVDZ. Os valores obtidos para as constantes de blindagem teóricas, referentes aos núcleos de 13C e 1H, foram de 192,2336 e 31,3046 ppm, respectivamente. Cálculos realizados em fase gasosa têm demonstrado eficiência na simulação de dados de RMN quando o solvente usado na aquisição do espectro experimental é o CDCl3, alcançando resultados de δH e δC teóricos condizentes com os obtidos experimentalmente.8
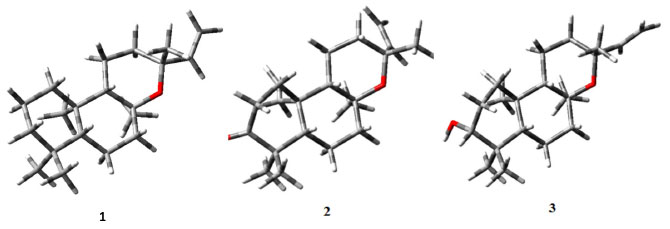 Figura 2. Estruturas das substâncias 1, 2 e 3 otimizadas em B3LYP/cc-pVDZ (fase gasosa)
Análise estatística Os dados teóricos de RMN foram comparados aos experimentais via regressão linear e parâmetros estatísticos importantes como grau de ajuste (R2), desvio-padrão (s), grau de significância (F) e previsibilidade (Q2 e sPRESS ) foram obtidos com o auxílio do software MINITAB 14.20 As equações lineares foram obtidas para a predição dos valores de deslocamento químico de RMN. Foram determinados outros parâmetros estatísticos para o aprimoramento da análise dos dados de RMN teóricos obtidos, tais como coeficientes a e b da regressão linear (δcalc = a + bδexp); erro absoluto médio (EAM), definido como EAM = Σn |δcalc - δexp|/n, e erro absoluto médio corrigido (EAMC), definido por EAMC = Σn |δcorr - δexp| /n, onde δcorr = (δcalc - a)/b.21 As equações lineares obtidas foram testadas para seus poderes de ajuste, significância e previsibilidade usando um procedimento de validação cruzada. Após a obtenção de cada modelo linear, os dados omitidos são preditos e as diferenças entre valores verdadeiros (deslocamentos químicos experimentais) e valores preditos (ŷ) (deslocamentos químicos obtidos com o modelo linear) são calculados. A soma dos quadrados dessas diferenças é determinada e, finalmente, o desempenho do modelo (sua habilidade preditiva) pode ser dada por PRESS (Predictive Sumo of Squares) e sPRESS (Standart Deviation of Cross-validation)22 Eq. (1), 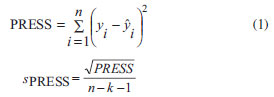 em que y é o valor experimental, ŷ é o valor predito, n é o número de amostras usadas para obter o modelo e k é o número de parâmetros de RMN.22 A habilidade preditiva dos modelos foi também quantificada em termos de Q2, o qual é definido como:22 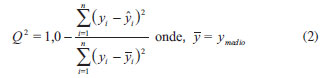
RESULTADOS E DISCUSSÃO Os resultados estatísticos revelam que todos os modelos lineares de RMN de 1H e 13C dos labdanos 1, 2 e 3 calculados em fase gasosa apresentaram boa correlação linear (R2 > 98%) e resultados satisfatórios após o procedimento de validação cruzada. Entretanto, o melhor conjunto de resultados para RMN de 1H foi obtido para a substância 1: (R2 = 99,60; F = 1894,35; s = 0,1392; sPRESS = 0,0702 e Q2 = 99,14%), enquanto que, para RMN de 13C, foi obtido para a substância 2: (R2 = 99,90; F = 13903,46; s = 1,8561; sPRESS = 0,4973 e Q2 = 99,83%), como disposto na Tabela 1. Da mesma forma que o grau de ajuste (R2), a previsibilidade (Q2) para RMN de 1H e 13C das três substâncias apresentaram valores superiores a 98%. Percebe-se, após avaliação dos dados estatísticos, que, em geral, o modelo de regressão dos deslocamentos de RMN de 13C e 1H das estruturas 1, 2 e 3 é significativo e está bem ajustado. Essa análise indica que há uma concordância linear entre os dados de deslocamentos químicos teóricos de RMN de 1H e 13C obtidos em relação aos valores experimentais para as substâncias em estudo, pois eles são quantitativamente semelhantes aos deslocamentos químicos citados na literatura, mostrando que o método teórico aplicado pode auxiliar de forma eficaz o uso das técnicas de Ressonancia Magnética Nuclear.
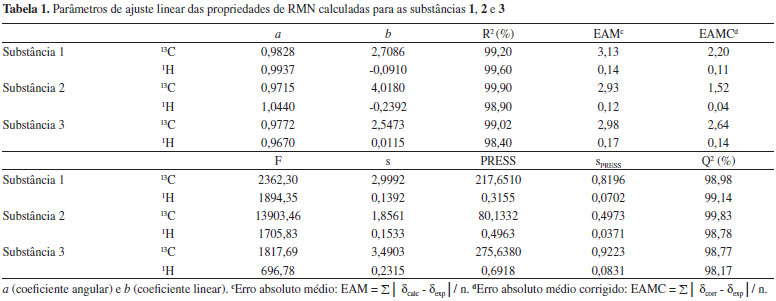
O tratamento estatístico baseado em regressão linear simples e procedimentos de validação cruzada reforça a proximidade existente entre os dados teóricos e calculados para os três labdados estudados, alcançando graus de correlação e previsibilidade superiores a 98%, além de alto grau de significância, o que gerou modelos lineares com boa representatividade estatística, indicando que o método teórico utilizado pode ser aplicado a produtos naturais similares aos labdanos citados. Sendo assim, os valores calculados de δ a nível de B3LYP/cc-pVDZ foram próximos aos experimentais como pode ser observado nas Tabelas 2, 3 e 4, apresentando baixos valores residuais (diferença entre deslocamentos químicos experimentais e teóricos) e δ similares aos seus respectivos valores preditos (obtidos com os modelos lineares). Alguns pontos observados para os δH de 2 demonstraram valores de δ relativamente distantes aos experimentais, quando comparados aos pontos de 1 e de 3.
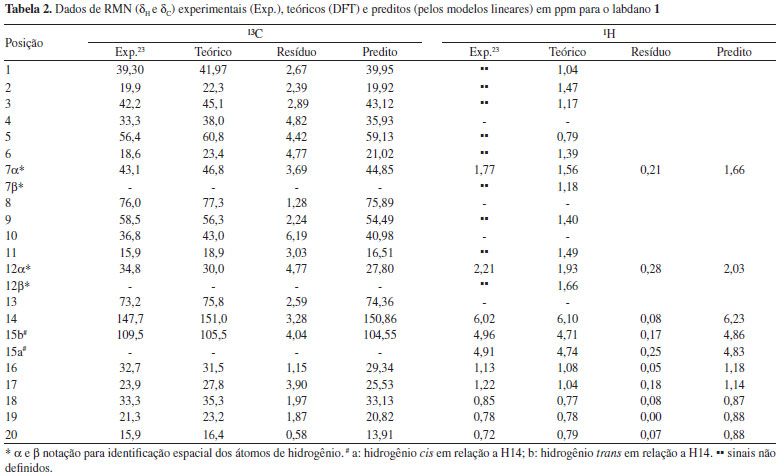
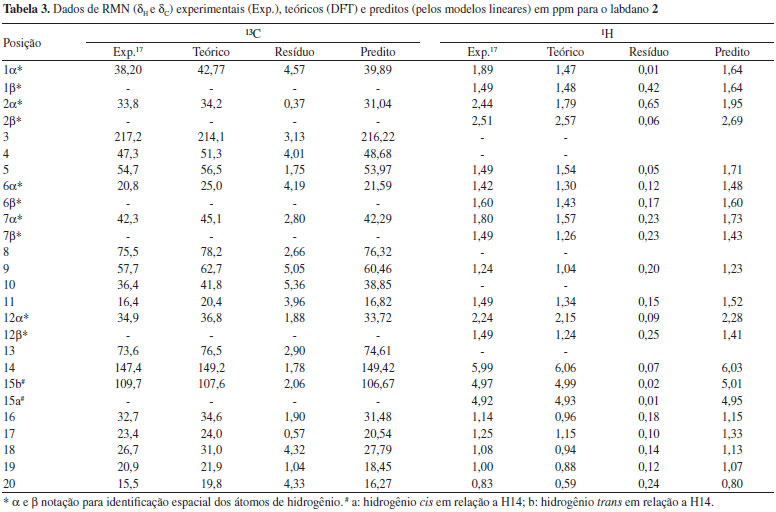
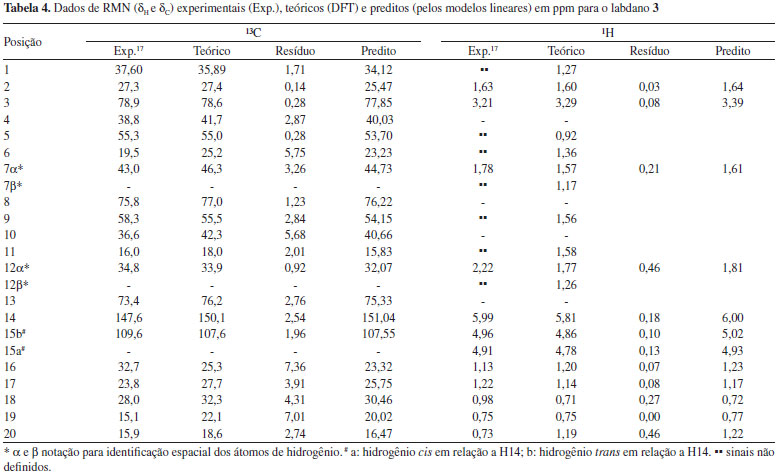
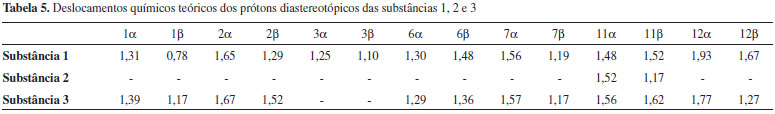
Entretanto, a análise estatística apresentada na Tabela 1 reforça a tendência linear existente entre os deslocamentos químicos teóricos e experimentais para os núcleos de hidrogênio desta substância. O ponto mais afastado foi observado para a posição 2α (δH) de 2 com δ teórico e experimental iguais a 1,79 ppm e 2,51 ppm, respectivamente, o que poderia sugerir uma inversão de sinais. Todos os possíveis centros estereogênicos das moléculas estudadas foram anteriormente definidos no trabalho de Konishi e co-autores em 1996.23 Os três labdanos estudados apresentam alguns núcleos de 1H que não tiveram seus deslocamentos químicos elucidados experimentalmente,17,23 como pode ser observado nas Tabelas 2, 3 e 4. Entretanto, deve-se considerar a relevância dos dados teóricos obtidos, uma vez que, na maioria dos casos, os valores de deslocamentos químicos mostraram-se próximos aos experimentais, reforçando a ideia de que métodos computacionais podem ser empregados em auxílio à interpretação de dados experimentais, fazendo com que tais métodos ganhem cada vez mais espaço no campo científico. Os valores preditos, determinados para δ através do modelo linear obtido, foram comparados aos valores experimentais, como apresentado nas Tabelas 2, 3 e 4. Nas últimas décadas, o avanço das tecnologias computacionais de hardware e software possibilitou uma considerável melhoria na precisão de informações obtidas para as diversas áreas da química. Cálculos computacionais têm sido utilizados amplamente em auxílio às análises espectroscópicas, contribuindo, desta forma, na elucidação estrutural de substâncias orgânicas. Os dados espectrocópicos experimentais não identificados dificultam a aquisição de informações sobre várias propriedades químicas e estruturais, sendo a modelagem molecular uma ferramenta promissora para o entendimento dessas características. Para as três substâncias, os cálculos teóricos permitiram sugerir os deslocamentos químicos de vários átomos de hidrogênios: substância 1: posições 1, 2, 3, 5, 6, 7β, 9, 11, 12α e 12β; substância 2: posições 2α, 2β, 6α, 6β, 12α e 12β; substância 3: posições 1, 5, 6, 7β, 9, 11 e 12β. No caso específico dos hidrogênios da posição 2 da substância 2 os resultados teóricos permitiram propor uma inversão nos valores descritos na literatura para as posições α e β. Inversões de deslocamentos químicos de prótons diastereotópicos são comuns, já que possuem valores tão próximos que a não equivalência muitas vezes não é identificada em espetros de RMN de 1H. Isso ocorre quando hidrogênios diastereotópicos estão muito afastados para que a assimetria do centro quiral tenha um efeito significativo. Todos os grupos metilênicos das moléculas 1, 2 e 3 possuem prótons diastereotópicos. Entretanto, somente a substância 2 obteve seus hidrogênios diastereotópicos observados experimentalmente (com exceção de 11α e 11β), porém inversamente atribuídos na posição 2, como apresentado na Tabela 3. Para as substâncias 1 e 3, apenas os hidrogênios da posição 15 foram elucidados a nível experimental (Tabelas 2 e 4). A simulação computacional de RMN de 1H permite identificar hidrogênios não equivalentes, sendo uma ferramenta auxiliar útil à elucidação de moléculas orgânicas. Com o método teórico testado, os prótons Hα e Hβ foram satisfatoriamente atribuídos para as substâncias estudadas, apresentando valores relativamente próximos, como disposto na Tabela 5.
CONCLUSÕES Os deslocamentos químicos obtidos para os núcleos de 1H e 13C utilizando o método DFT B3LYP/cc-pVDZ em fase gasosa são condizentes aos experimentais, apresentando baixos resíduos. Desta forma, corrobora-se a utilização do método empregado em labdados similares aos estudados que não tenham sido completamente elucidados por técnicas experimentais tradicionais. A contribuição principal deste trabalho foi a proposição de deslocamentos químicos de 1H que não foram identificados a nível experimental, especialmente os prótons diastereotópicos que por possuírem valores muito próximos, a observação experimental muitas vezes é prejudicada, o que evidencia a importância da utilização de métodos computacionais em auxílio à ciência moderna. A análise de espectros de RMN nem sempre é uma tarefa fácil, contudo, técnicas de simulação computacionais tem ganhado espaço significativo no meio científico, oferencendo boas contribuições à Química Orgânica. No caso dos labdanos estudados, todos os carbonos e prótons foram satisfatoriamente simulados, gerando modelos lineares (experimental x teórico) com graus de correlação estatística superiores a 98%.
AGRADECIMENTOS Os autores agradecem ao CNPq, FAPESPA e PIBIC/UFPA pelo auxílio financeiro concedidos para a realização deste trabalho.
REFERÊNCIAS 1. Ootani, M. A.; Aguiar, R. W.; Ramos, C. C.; Brito, D. R.; Silva, J. B.; Cajazeira, J. P.; J. Biotechnol. Biodiversity 2013, 4, 162. 2. Salatino, A.; Salatino, M. L.; Negri, G.; J. Braz. Chem. Soc. 2007, 18, 11. DOI: http://dx.doi.org/10.1590/S0103-50532007000100002 3. Lopes, E. L.; Andrade Neto, M.; Silveira, E. R.; Pessoa, O. D. L.; Braz-Filho, R.; Quim. Nova, 2012, 35, 2169. DOI: http://dx.doi.org/10.1590/S0100-40422012001100012 4. Fraga, B. M.; Hernández, M. G.; González, P.; López, M.; Suárez, S.; Tetrahedron 2001, 57, 761. DOI: http://dx.doi.org/10.1016/S0040-4020(00)01052-8 5. Costas, D.; Konstantinos, D. S.; Stud. Nat. Prod. Chem. 2001, 25, 235. 6. Ribeiro, L. A. A.; Tavares, J. F.; Andrade, N. C.; Silva, M. S.; Silva, B. A.; Rev. Bra. Farmacogn. 2007, 17, 197. DOI: http://dx.doi.org/10.1590/S0102-695X2007000200011 7. Orozco, J. L. H.; Aguilera, R. A.; Patentes Online, PI 0418053-4 A, 2007. 8. Amador, D. H. T.; Silva, S. de O.; Deus, R. J. A.; Castro, K. C. F.; Docelli, M. D.; Martins, F. T.; Ellena, J.; Polikarpov, I.; Da Silva, M. N.; Arruda, M. S. P.; Macedo, L. G.; Alves, C. N.; Brasil, D. do S. B.; Adv. Sci. Lett. 2012, 18, 150; Brasil, D. S. B.; Muller, A. H.; Guilhon, G. M. S. P.; Alves, C. N.; Peris, G.; Llusard, R.; Molinerd, V.; J. Braz. Chem. Soc. 2010, 21, 731; Burgueño-Tapia, E.; Joseph-Nathan, P.; Phytochemistry 2008, 69, 2251; Silva, S. O.; Peixoto, R. N. S.; Silva, J. R. A.; Alves, C. N.; Guilhon, G. M. S. P.; Santos, L. S.; Brasil, D. S. B.; Int. J. Mol. Sci. 2011, 12, 9389. DOI: http://dx.doi.org/10.1166/asl.2012.4398 9. Farias, R. R.; Cardoso, L. A. M.; Oliveira-Neto, N. M.; Junior, B. B. N.; Quim. Nova, 2013, 36, 729. DOI: http://dx.doi.org/10.1590/S0100-40422013000500020 10. Steinbeck, C.; Nat. Prod. Rep. 2004, 21, 512. DOI: http://dx.doi.org/10.1039/b400678j PMID: 15282633 11. Plaza, A.; Piacente, S.; Perrone, A.; Hamed, A.; Pizza, C.; Bifulco, G.; Tetrahedron 2004, 60, 12201. DOI: http://dx.doi.org/10.1016/j.tet.2004.10.021 12. Burgueño-Tapia, E.; Joseph-Nathan, P.; Phytochemistry 2008, 69, 2251. DOI: http://dx.doi.org/10.1016/j.phytochem.2008.05.011 PMID: 18599095 13. Cerda-Garcia-Rojas, C. M.; Catalán, C. A. N.; Muro, A. C.; Joseph-Nathan, P.; J. Nat. Prod. 2008, 71, 967. DOI: http://dx.doi.org/10.1021/np8000927 PMID: 18500843 14. Bagno, A.; Rastrelli, F.; Saielli, G.; J. Org. Chem. 2007, 72, 7373. DOI: http://dx.doi.org/10.1021/jo071129v PMID: 17718506 15. Ghosh, S.; Ghatak, U.R.; J. Chem. Res., Synop. 1992, 10, 352. 16. Li, A. P.; Bie, P. Y.; Peng, X. S.; Wu, T. X.; Pan, X. F.; Chan, A. S. C.; Yang, T. K.; Synth. Commun. 2002, 32, 605. DOI: http://dx.doi.org/10.1081/SCC-120001519 17. Brasil, D. S. B.; Dissertaçao de Mestrado, Universidade Federal do Pará, Brasil, 2008. 18. HyperChemTM Release 7.5 for Windows Molecular Modeling System. Hypercube: Gainesville, Estados Unidos, 2002. 19. Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Montgomery Jr., J. A.; Vreven, T.; Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G. A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ocheterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaroni, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Gonzalez, C.; Pople, J. A.;Gaussian 03, Revision B.04; Gaussian: Pittsburgh, PA, United States of America, 2003. 20. Minitab Release 14 for Window. Minitab Inc., State College, Pennsylvania, United States of America, 2003. 21. Cimino, P.; Gomez-Paloma, L.; Duca, D.; Riccio, R.; Bifulco, G.; Magn. Reson. Chem. 2004, 42, 526. 22. Gaudio, A. C.; Zandonade, E.; Quim. Nova 2001, 24, 658. DOI: http://dx.doi.org/10.1590/S0100-40422001000500013 23. Konishi, T.; Azuma, M.; Itoga, R.; Kiyosawa, S.; Fujiwara, Y.; Shimada, Y.; Chem. Pharm. Bull. 1996, 44, 229. DOI: http://dx.doi.org/10.1248/cpb.44.145 |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access