
 |
 |
 |
 |
 |
Educação
|
|
| Uma forma simplificada de deduzir as equações de Hartree e Hartree-Fock A simplified route to obtain the Hartree and Hartree-Fock equations |
|
Rogério Custodio*
Instituto de Química, Universidade Estadual de Campinas, Barão Geraldo, 13083-970 Campinas - SP, Brasil Recebido em 13/01/2015 *e-mail: roger@iqm.unicamp.br Alternative considerably simpler ways of obtaining the Hartree and Hartree-Fock equations are presented. These alternatives do not replace the formal demonstrations, which should be introduced in undergraduate or graduate courses according to the required level of student training. However, the use of the present approaches allows a student-friendlier introduction of the basic principles of electronic structure calculations as a prior teaching resource to the formal demonstrations. General implications and comparisons between the Hartree and Hartree-Fock energies are discussed. INTRODUÇÃO Em artigo recente do Monte e Ventura1 apresentaram uma abordagem do desenvolvimento teórico do método de Hartree2,3 indicando a importância em se introduzir conceitos de estrutura eletrônica empregando este método pioneiro baseado em equações autoconsistentes. Em termos históricos, um dos aspectos que mais chama a atenção no trabalho de Hartree é a proposta de determinar a distribuição eletrônica de átomos empregando a formulação de partículas independentes e resolvendo as equações diferenciais por meio de métodos numéricos. Enquanto este tipo de procedimento pode ser aplicado atualmente sem maiores dificuldades para diversas aplicações utilizando-se computadores, na época de Hartree estes equipamentos não passavam de sonhos de tecnologia que poderiam surgir no futuro. Neste sentido, pode-se sugerir que Hartree foi um visionário e anteviu o avanço tecnológico de equipamentos que tornassem o seu procedimento factível e permitissem a aplicação rotineira de métodos numéricos em problemas de estrutura eletrônica. Ainda hoje, em termos didáticos, introduzir o método de Hartree em disciplinas de graduação ou mesmo pós-graduação é um desafio e exige a apresentação de uma série de conceitos e técnicas matemáticas, tais como cálculo variacional, cálculo funcional, transformação unitária, multiplicadores de Lagrange, o conceito de potencial médio, dentre outras, para finalmente se chegar às equações autoconsistentes, como indicado no artigo de do Monte e Ventura.1 O problema é ainda dificultado pela inclusão de efeitos de antissimetria eletrônica no desenvolvimento do conjunto de equações também autoconsistentes de Hartree-Fock.4 Muito provavelmente em função da bagagem matemática necessária para a dedução rigorosa destas equações, grande parte dos livros-textos didáticos trazem apenas uma descrição semiqualitativa do método e a associação das energias orbitais com tendências periódicas dos elementos.5-7 As equações de Hartree-Fock formam o ponto de partida para a grande maioria dos métodos de estrutura eletrônica utilizados atualmente. Para que estas equações sejam aplicáveis a sistemas moleculares grandes, a utilização de expansão de funções de onda de 1-elétron (orbital se considerar apenas coordenadas espaciais ou spin-orbital se incluir também coordenadas de spin) em termos de funções de base ou também popularmente conhecido como método de combinação linear de orbitais atômicos é uma técnica muito utilizada e, para a sua compreensão e de suas consequências, a demonstração da obtenção das equações de Hartree-Fock é extremamente importante. Além disso, os orbitais moleculares canônicos e suas energias são muito utilizados em métodos correlacionados pós-Hartree-Fock. Logo, algumas das propriedades relevantes desses cálculos mais elaborados ficam evidentes durante a demonstração da obtenção das equações de Hartree-Fock. Neste sentido, fica estabelecido como objetivo deste artigo a apresentação de uma forma simplificada de desenvolvimento das equações de Hartree e Hartree-Fock para problemas de muitos elétrons.
DESENVOLVIMENTO TEÓRICO Informações gerais do modelo de partículas independentes O operador Hamiltoniano em unidades atômicas para um sistema de n elétrons pode ser escrito como:  sendo ĥi o operador de um elétron constituído pela energia cinética e atração nuclear do i-ésimo elétron. O último termo à direita da Eq. 1 corresponde ao operador de dois elétrons de repulsão eletrônica, sendo rij a distância entre os elétrons i e j. Excluindo-se o termo de repulsão eletrônica e escrevendo-se a equação de Schrödinger independente do tempo com uma função de onda de n-elétrons tem-se: 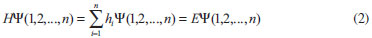 Para resolver-se esta equação analiticamente, basta considerar que a função de onda possa ser descrita a partir de um produto de funções spin-orbitais, φi. Esta aproximação é denominada de modelo de partículas independentes ou produto de Hartree, e é representada matematicamente por:  sendo cada spin-orbital definido como o produto de uma função orbital (Φi) por uma função de spin (α ou β). Na representação dada pela Eq. 3, as coordenadas espaciais e de spin correspondem aos números entre parênteses. Substituindo-se a função de onda dada pela Eq. 3 na Eq. 2 verifica-se que a equação de n-elétrons (Eq. 2) é separável em n-equações de 1-elétron do tipo:  e com energia eletrônica total igual a:  A inclusão das repulsões intereletrônicas impossibilita a solução analítica da equação de Schrödinger. Porém, pode-se considerar a utilização da função de onda definida pela Eq. 3 como uma aproximação e determinar-se a energia eletrônica do sistema incluindo as repulsões intereletrônicas. Neste caso, a energia eletrônica total pode ser estimada utilizando-se o teorema do valor médio, que em notação de Dirac é dado por: 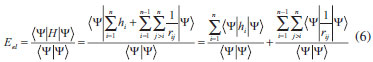 As integrais nos numeradores correspondem às energias do sistema e a integral nos denominadores corresponde à integral de normalização da função de onda. Para simplificar, assumindo-se que a função de onda é normalizada, 〈Ψ⎢Ψ〉 = 1, e substituindo-se a Eq. 3 na Eq. 6 têm-se: 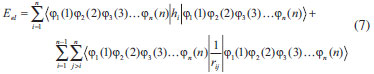 Uma vez que o operador ĥi só atua sobre as coordenadas do i-ésimo elétron e o operador 1/rij só atua sobre as coordenadas dos elétrons i e j, a Eq. 7 será então escrita como: 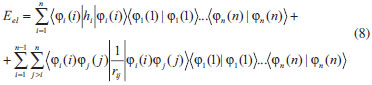 Para que a função de onda total definida pela Eq. 3 seja considerada normalizada, as funções spin-orbitais devem também ser normalizadas, ou seja: 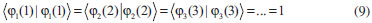 e para que as funções spin-orbitais sejam normalizadas, as funções orbitais (Φi) e de spin (α ou β) também devem sê-lo, ou seja: 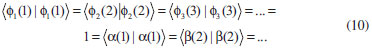 Assim, a Eq. 7 pode ser simplificada para: 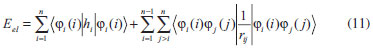 Nesta equação o primeiro somatório à direita da igualdade corresponde às integrais de energia cinética e atração nuclear e o segundo somatório corresponde às integrais de repulsão eletrônica ou integrais de Coulomb. No método de Hartree os elétrons são distinguíveis, ou seja, cada elétron é representado por uma determinada função spin-orbital. Caso as integrais na Eq. 11 sejam integradas nas coordenadas de spin, tem-se que a energia eletrônica será: 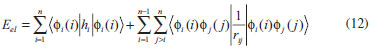 Algo que não está explicitado na Eq. 12 é como se estabelece a distribuição eletrônica. Não há qualquer indicação de como se obtém os orbitais e quantos elétrons podem ser colocados em cada um deles ou se os orbitais devem ser ortogonais ou não. Ao se resolver as equações de Hartree numericamente pelo procedimento convencional a condição de ortogonalidade dos orbitais atômicos ou moleculares é necessária. Sem ela todos os elétrons ocuparão o orbital de mais baixa energia, algo que produzirá resultados de propriedades eletrônicas que não se assemelham a nenhuma informação experimental existente. As funções de spin α e β automaticamente tornam duas funções spin-orbitais, construídas a partir de uma mesma função orbital, ortogonais entre si, mas funções spin-orbitais com a mesma função de spin serão distintas com a imposição da condição de ortogonalidade entre as funções orbitais. É possível trabalhar-se com funções orbitais não ortogonais, mas este tema não será abordado neste trabalho. Em uma dedução rigorosa, a determinação das funções orbitais a partir da Eq. 12 pode ser feita a partir da minimização da energia em relação a variações em cada função orbital, impondo-se o vínculo de ortogonalização e empregando-se o método de multiplicadores de Lagrange.8-10 Este é o caminho formal usualmente seguido não só pelo método de Hartree, mas também pelo método de Hartree-Fock e outras técnicas de estrutura eletrônica. Após desenvolvimento apropriado se chega às equações de Hartree ou Hartree-Fock. Uma dedução alternativa da equação de Hartree Uma rota alternativa para se chegar às equações de Hartree e Hartree-Fock será apresentada como mecanismo didático preservando grande parte dos conceitos fundamentais e possibilitando aos alunos a obtenção destas equações extremamente importantes que permeiam grande parte dos métodos de estrutura eletrônica. A primeira demonstração será feita considerando-se apenas o método de Hartree e ao invés de uma demonstração genérica, optou-se pelo caso particular de um sistema arbitrário contendo três elétrons representados por três spin-orbitais. Para este sistema a Eq. 11 será escrita como: 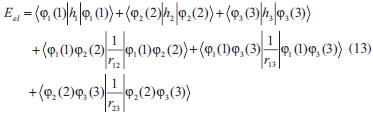 sendo as três primeiras integrais à direita da igualdade as energias cinéticas e de atração nuclear e as três últimas as repulsões eletrônicas entre os três elétrons em três diferentes spin-orbitais. Por conveniência, pode-se simplificar a notação da Eq. 13 definindo-se o que se conhece como operador de Coulomb nas integrais de repulsão eletrônica. Para o caso, por exemplo, da integral envolvendo os elétrons 1 e 2, pode-se escrever a integral de Coulomb como: 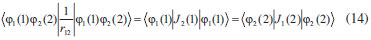 Nesta equação verifica-se que a integral pode ser escrita em termos de um operador de Coulomb, Ĵ2(1) ou Ĵ1(2), ou seja: 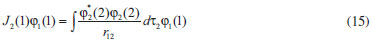 ou  Nas equações de Hartree o índice que identifica o elétron é o mesmo que identifica spin-orbital, mas em outros modelos isto usualmente não ocorre. A integração na Eq. 15 é realizada nas coordenadas do elétron 2 e como o integrando depende também das coordenadas do elétron 1 em r12, o resultado será dependente das coordenadas do elétron 1. O resultado, portanto, será representado por Ĵ1(2). A notação usualmente empregada considera o subíndice de J como identificação do orbital ou spin-orbital usado na integração e o índice entre parênteses a identificação das coordenadas do elétron que estão sendo integradas. Da mesma forma, na Eq. 16 tem-se a integração sendo realizada nas coordenadas do elétron 1 e o resultado da integral será dependente das coordenadas do elétron 2, representado por J1(2). Independente de qual integral se resolva primeiro na Eq. 14, o resultado final será o mesmo e a integral de Coulomb entre os elétrons 1 e 2 podem ser escritas de diferentes maneiras (ver Eq. 14). Além do uso dos operadores de Coulomb, a Eq. 13 pode ser escrita com uma notação ainda mais compacta. Para as integrais de um e dois elétrons serão considerados: 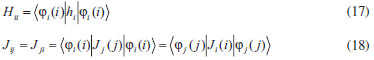 Assim, a Eq. 13 passa a ser escrita apenas como: 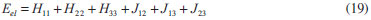 O objetivo no momento é possibilitar que a Eq. 19 seja escrita em termos de uma soma de energias orbitais, como na Eq. 5. Isto é possível, como será discutido posteriormente no texto, mas com o intuito de preservar a estrutura das equações de Hartree será considerada apenas a alternativa de procurar definir energias orbitais. Uma alteração que não modifica a energia eletrônica total do sistema pode ser obtida se somarmos na Eq. 19 integrais de Coulomb (Jij - Jij), ou seja: 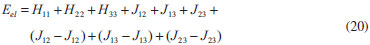 Estas integrais podem ser reorganizadas como: 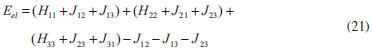 Pode-se verificar que os termos entre parênteses correspondem às energias envolvendo o spin-orbital i e todos os outros elétrons do sistema, assim define-se energia orbital como:  ou genericamente:  Voltando a escrever as expressões para as energias orbitais de forma explícita, tem-se: 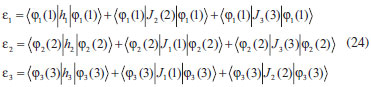 Repare que a forma com que se escolhe a definição do operador de Coulomb é arbitrária e é feita por conveniência para manter a estrutura das integrais definidas em termos da função spin-orbital de interesse em cada expressão. Com exceção da própria energia orbital, todos os outros termos são apresentados na forma integral. Uma vez que as funções orbitais são consideradas normalizadas, pode-se multiplicar cada uma das energias orbitais dadas pelas Eqs.24 pelas integrais de normalização, ou seja: 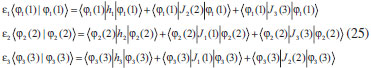 Uma vez que as energias orbitais são constantes, elas podem ser colocadas dentro das integrais de normalização, ficando: 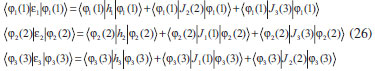 Estas integrais podem ser rearranjadas colocando-se as funções spin-orbitais em evidência e escritas como: 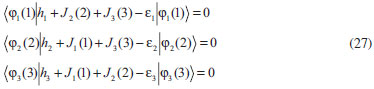 Para que estas igualdades sejam verdadeiras, basta que: 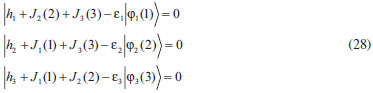 ou rearrajando novamente: 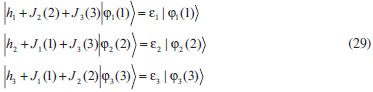 Estas equações podem ser escritas uma para cada elétron e definidas genericamente como:  e são conhecidas como equações de Hartree ou simplesmente:  sendo f (i) o operador de Hartree e sua representação está definida entre colchetes na Eq. 30, ou seja:  Uma dedução alternativa da equação de Hartree-Fock A função de onda definida pela Eq. 3 possui dois erros conceituais. O primeiro deles é que o produto de Hartree não considera a indistinguibilidade eletrônica. Elétrons são partículas indistinguíveis e não é aceitável considerar, por exemplo, que o elétron 1 seja representado apenas pelo spin-orbital 1, o elétron 2 seja representado apenas pelo spin-orbital 2, etc. O segundo problema com a função de onda de Hartree diz respeito à simetria da mesma. Ao efetuar-se uma distribuição eletrônica, deve-se considerar que está sendo feita uma distribuição de férmions e toda distribuição de férmions deve ser representada por funções de onda antissimétricas. Funções de onda antissimétricas são caracterizadas pela troca de sinal ao se trocar as coordenadas de dois elétrons quaisquer. Uma maneira prática e muito simples de se construir uma função de onda antissimétrica é por meio de um determinante de Slater. Para um sistema de n-elétrons representado por n spin-orbitais a função de onda poderá ser escrita como: 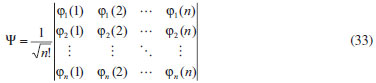 O termo imediatamente à direita do sinal de igualdade representa um fator de normalização da função de onda. Da mesma forma como feito para a função de onda definida pela Eq. 3, determina-se a expressão para o valor médio da energia eletrônica total utilizando a função de onda antissimétrica apresentada na Eq. 33 na Eq. 6. O uso da Eq. 6 empregando determinantes de Slater normalizado para a obtenção da energia média de um sistema qualquer está muito bem descrita na literatura9,10 e, uma vez que as funções spin-orbitais são consideradas também normalizadas e ortogonais, para um sistema de n-elétrons, têm-se que: 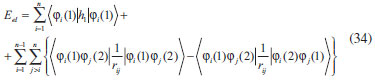 Comparando-se a Eq. 34 com a Eq. 11 verifica-se algumas diferenças importantes. A mais evidente corresponde ao surgimento de uma nova integral de repulsão eletrônica. O último termo à direita da Eq. 34 é denominado de integral de troca e surge naturalmente como consequência da antissimetria da função de onda. Outro aspecto que chama a atenção é o fato de que não há mais uma correspondência direta entre a função spin-orbital e as coordenadas eletrônicas. No método de Hartree o índice característico das coordenadas eletrônicas era o mesmo da função spin-orbital. Agora, como os elétrons são indistinguíveis, qualquer índice para caracterizar as coordenadas eletrônicas pode ser utilizado. Normalmente utilizam-se as coordenadas dos elétrons 1 e 2, mas poderiam ser utilizadas as coordenadas de qualquer par eletrônico. De forma semelhante ao realizado para integrais de Coulomb, pode-se definir operadores de troca. Para o caso dos elétrons 1 e 2 estes operadores de troca serão escritos como: 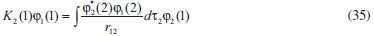 ou 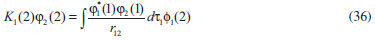 De maneira semelhante ao realizado para as integrais de Coulomb, as integrais de troca também poderão ser escritas como: 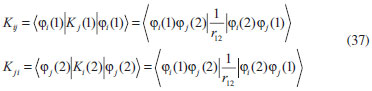 A energia eletrônica para um sistema de n-elétrons será então escrita em notação compacta como:  Quando i=j as integrais de Coulomb e de troca são idênticas, ou seja, Jii = Kii e se cancelam entre si. Portanto, a sua inclusão não afeta a energia eletrônica total que pode ser escrita como: 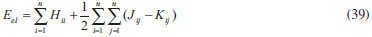 Assim, para um sistema contendo três elétrons e utilizando-se a Eq. 39 tem-se: 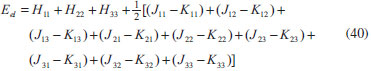 Novamente, o objetivo será procurar definir uma energia orbital como feito para a energia de Hartree. Uma alternativa consiste em somar e subtrair o termo de repulsão eletrônica de Coulomb e de troca, (Jij - Kij) - (Jij - Kij), levando a Eq. 40 a ser reescrita como: 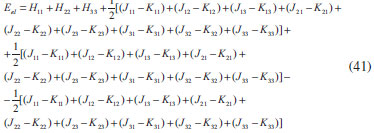 Reorganizando a Eq. 41, obtém-se: 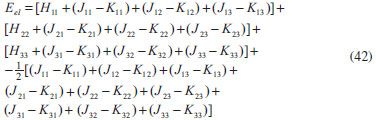 Desta forma, também se pode definir uma energia orbital e se escolher a notação mais conveniente para os operadores de coulomb e troca (ver Eq. 18 e 37) como: 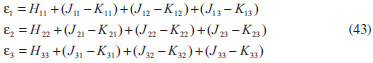 ou genericamente:  Com a definição de energia orbital, verifica-se que a energia eletrônica total (Eq. 42) também pode ser determinada por:  que é a expressão usualmente encontrada em todos os livros-textos que tratam do tema. Em sua forma mais explícita as energias orbitais são: 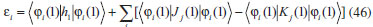 Multiplicando-se a energia orbital por 1 não se altera a equação e, uma vez que estamos considerando que as funções spin-orbitais são normalizadas, tem-se que 1 = 〈φi(1)|φi(1)〉. Assim, a Eq. 46 pode ser escrita como: 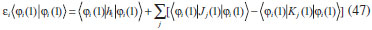 ou 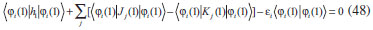 A soma das integrais pode ser escrita como a integral da soma colocando-se a função spin-orbital em evidência, que leva à: 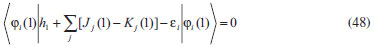 Para que esta igualdade seja verdadeira, basta que: 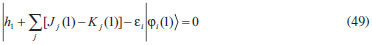 ou 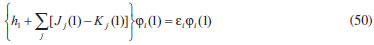 que correspondem à equação canônica de Hartree-Fock ou simplesmente:  cujo operador de Fock corresponde a:  É importante mencionar que, por definição, os operadores de Coulomb (Eq. 15-16) e troca (Eq. 35-36) dependem de funções spin-orbitais, mas estas funções devem ser obtidas como solução das próprias equações canônicas de Hartree e Hartree-Fock. Assim, em um primeiro momento, um iniciante pode ter a impressão de que é um problema sem solução. Porém, Hartree utilizou-se engenhosamente de uma alternativa conhecida como método autoconsistente. Uma vez que não se conhece os spin-orbitais, uma alternativa corresponde a ignorar os operadores de Coulomb e troca em uma primeira tentativa e resolver-se as equações de Hartree ou Hartree-Fock sem o termo de repulsão eletrônica. Com isso, tem-se uma representação muito grosseira dos spin-orbitais. Com essa forma bruta dos spin-orbitais pode-se obter os operadores de Coulomb e troca e resolver-se novamente as equações, corrigindo os spin-orbitais anteriores. O processo deve ser repetido até que o sistema atinja um nível de precisão desejado em termos de energia total ou de densidade eletrônica. Também é possível realizar o primeiro cálculo com funções aproximadas. É por isso que tanto as equações de Hartree quanto de Hartree-Fock são conhecidas como possuindo a propriedade de autoconsistência.
DISCUSSÃO Aspectos gerais das equações autoconsistentes Para átomos ou moléculas diatômicas e triatômicas, as equações de Hartree e Hartree-Fock podem ser resolvidas numericamente. A referência 1 apresenta o caso particular do método de Hartree, em que se conhecendo a simetria dos orbitais atômicos, reduz-se o problema de resolução das Eqs.31 à determinação das funções radiais em átomos. A Eq. 51 também pode ser resolvida da mesma forma. Algoritmos eficientes como o método de Noumerov podem ser empregados para a obtenção das raízes dessas equações para o estado atômico fundamental.7 Para moléculas é conveniente considerar-se o uso de combinação linear de funções de base, ou como se denominava originalmente, combinação linear de orbitais atômicos para resolver as equações de Hartree ou Hartree-Fock, o que nos leva ao método Hartree-Fock-Roothaan.10-12 Porém, considerando-se os métodos de Hartree e Hartree-Fock e os desenvolvimentos simplificados apresentados acima, algumas informações são simples de serem observadas e outras não. Uma das primeiras constatações em relação aos procedimentos acima é que não aparece em momento algum qualquer indicação de que a energia eletrônica do sistema está sendo minimizada. O caminho da demonstração formal em que a energia é minimizada com relação às funções orbitais leva exatamente às Eqs.31 e 51. Portanto, embora não tenha sido explicitamente utilizada, a energia eletrônica resultante destas equações corresponde a um mínimo. Outra característica importante da demonstração formal está no fato de que os operadores de Hartree (Eq. 32) e de Fock (Eq. 52) são invariantes com relação a transformações unitárias, o que indica que os spin-orbitais obtidos pela solução das equações de Hartree e Hartree-Fock não são únicos. Os spin-orbitais obtidos no formato apresentado neste trabalho e em praticamente todos os textos na literatura são denominados de spin-orbitais canônicos. Substituindo-se, por exemplo, a Eq. 52 na Eq. 48 e rearranjando-se, verifica-se que as energias canônicas também podem ser definidas pela equação: εi = 〈φi (1)| ƒ1| φi (1)〉, em que o índice i caracteriza o spin-orbital de interesse. Os orbitais canônicos satisfazem ainda a condição: εij = 〈φi (1)| ƒ1| φi (1)〉 = 0 para spin-orbitais diferentes, φi ≠ φj. Desta forma, diz-se que a matriz de energias orbitais é diagonal, ou seja, somente as energias diagonais são diferentes de zero. Está é uma solução particular das equações Hartree-Fock ou de Hartree. Caso seja possível produzir um conjunto de spin-orbitais que satisfaça o princípio variacional e apresente uma distribuição eletrônica apropriada, muito provavelmente a matriz de energias orbitais não será diagonal. A obtenção desses orbitais não-canônicos é possível e não será tratada neste texto. No final deste artigo será comentado um caso importante de transformação unitária dos orbitais. Também não está explicitado durante o procedimento desenvolvido no presente artigo se os spin-orbitais são ortonormais ou não. Em nenhum momento das demonstrações isto está sendo explicitamente considerado. No caso do método de Hartree a exigência de ortonormalidade não é explicitamente necessária em nenhum momento da demonstração, embora ela seja implicitamente necessária para manter a consistência entre a Eq. 30 e a Eq. 23. No caso do método Hartree-Fock a condição de ortonormalidade surge no desenvolvimento da Eq. 34 a partir da função de onda antissimétrica (Eq. 33). Sem a condição de ortonormalização, diversas integrais seriam diferentes de zero e a expressão da energia eletrônica (Eq. 34) seria consideravelmente mais complicada. Quando se resolve numericamente as equações de Hartree ou Hartree-Fock sem a condição de ortonormalização dos orbitais, todas as equações dos respectivos métodos produziriam a mesma função orbital, ou seja, todos os elétrons ocupariam o orbital de menor energia, o que não é compatível com informações experimentais. A inclusão de antissimetria por Fock se tornou tão popular que não se questiona se no método de Hartree ela é importante ou não. A função de onda que descreve o modelo de Hartree corresponde à representação de um evento em que a probabilidade que envolve, por exemplo, o elétron i é completamente independente da probabilidade que envolve o elétron j. Matematicamente, esta afirmação pode ser verificada pela equação: 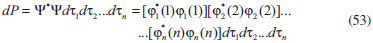 Nota-se que a densidade de probabilidade de cada elétron em cada spin-orbital não envolve as coordenadas de qualquer outro elétron. Eventos deste tipo são considerados como estatisticamente não correlacionados e, portanto, o método de Hartree apresenta deficiências de ausência completa de correlação eletrônica. No caso do método de Hartree-Fock, o uso da função de onda antissimétrica produz densidades de probabilidades que combinam spin-orbitais com coordenadas eletrônicas de diferentes elétrons. Portanto, o método pode ser considerado como incluindo algum efeito de correlação eletrônica. Uma análise mais profunda pode ser realizada na obtenção da energia eletrônica total indicando que a antissimetria possibilita a correção de efeitos de correlação eletrônica entre elétrons com mesmo spin.13 Variacionalmente pode-se considerar que a energia eletrônica obtida pelo método Hartree-Fock está mais próxima do valor exato do que a energia obtida pelo método de Hartree e, portanto, EHartree-Fock < EHartree. Comparando-se qualitativamente as Eqs. 11 e 34 verificam-se ainda duas observações importantes. A primeira é bem conhecida e sugere que se a definição das energias orbitais (Eqs. 23 e 44) forem substituídas nas respectivas energias eletrônicas totais (Eqs.11 e 34), as respectivas energias eletrônicas totais não correspondem à soma das energias orbitais. É possível definir arbitrariamente um conjunto de energias orbitais que somadas proporcionem a energia eletrônica total. Na demonstração apresentada acima bastaria, por exemplo, na Eq. 19, considerar que as integrais de Coulomb fossem divididas por dois, ou seja, ao invés de realizar a soma de (Jij - Jij) na Eq. 20, o termo de repulsão Coulombico poderia ser dividido por dois, ou seja, Jij = 0.5Jij + 0.5Jij. A diferença na equação de Hartree ou de Hartree-Fock (usando procedimento semelhante nas equações de Hartree-Fock) seria o aparecimento de um fator de 0.5 nas energias de repulsão eletrônica nas respectivas equações finais (Eqs. 30 e 50). Energias orbitais permitem manipulação apropriada para tornarem-se aditivamente equivalentes à energia eletrônica total de cálculos Hartree-Fock,14,15 porém, a associação da definição original das energias orbitais com o negativo das energias de ionização, demonstrada pelo teorema de Koopmans, favorece a manutenção da definição de energias orbitais como indicado acima e na literatura. Outro aspecto que deve ser ressaltado na escolha dos orbitais canônicos está na possibilidade de se resolver as equações de Hartree e Hartree-Fock por meio de métodos de solução de equações de autovalores, que são extremamente práticos e eficientes. Finalmente, a diferença óbvia entre as equações de Hartree e Hartree-Fock está na presença do termo de troca nas energias obtidas pelo método de Hartree-Fock. Como mencionado acima, este termo está associado com correções nos efeitos de correlação entre elétrons de mesmo spin. Quando se pensa em representar a estrutura de uma molécula qualquer, intuitivamente químicos tendem a utilizar representações de Lewis. Em cálculos Hartree-Fock para moléculas, os orbitais moleculares gerados apresentam natureza completamente deslocalizada, ou seja, não se concentram em regiões específicas da molécula de acordo com as representações de Lewis. Ao contrário, as distribuições eletrônicas orbitais geralmente se estendem por mais do que dois átomos. Existem diferentes formas de se localizar orbitais com critérios completamente distintos.16 Porém, uma das primeiras tentativas de procurar resgatar um caráter localizado da distribuição eletrônica orbital a partir de orbitais deslocalizados foi realizada por Edmiston e Ruedenberg.17 Em trabalho clássico esses autores demonstraram que se as integrais de troca entre os orbitais fossem minimizadas e as integrais de Coulomb fossem maximizadas, os orbitais deslocalizados do método Hartree-Fock seriam convertidos em orbitais localizados. Numericamente o valor da energia eletrônica total e de outras propriedades globais seriam invariantes com relação a esta transformação, uma vez que se trata de uma transformação unitária. Esta perspectiva levou Levy, Nee e Parr18 a sugerirem que a utilização do método de Hartree poderia ser considerada como uma forma direta de obtenção de localização de orbitais. Portanto, embora o método de Hartree possa produzir energias variacionalmente maiores do que o método de Hartree-Fock, os orbitais resultantes apresentam a conveniência de apresentarem um caráter localizado compatível com as estruturas de Lewis. Na prática, entretanto, opta-se pelo uso do método Hartree-Fock e posterior localização dos orbitais para que se tenha uma estrutura eletrônica mais próxima da função de onda exata.
CONCLUSÃO Este trabalho procurou explorar formas alternativas e consideravelmente simples de se obter as equações de Hartree e Hartree-Fock de forma genérica. Usualmente, a abordagem desses métodos tanto em nível de graduação quanto em nível de pós-graduação é feita de forma elaborada e exige a introdução de métodos matemáticos sofisticados que levam os alunos a terem dificuldade de assimilar as ideias principais associadas aos respectivos métodos. A apresentação das alternativas acima não substitui as demonstrações formais, que devem ser introduzidas de acordo com o nível de formação esperado, mas a experiência que o autor tem tido ao introduzir estes caminhos como recurso didático, preliminarmente às demonstrações formais quando exigidos, mostra que há melhor assimilação do modelo e sugere que os alunos superam com menores dificuldades os desenvolvimentos formais.
AGRADECIMENTOS Gostaríamos inicialmente de agradecer os comentários feitos pelos revisores deste trabalho. Agradecemos também o apoio financeiro concedido pelas seguintes agências de fomento: FAPESP (Fundação de Amparo à Pesquisa do Estado de São Paulo - Center for Computational Engineering and Sciences - Grant 2013/08293-7), CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico), e FAEPEX-UNICAMP (Fundo de Apoio ao Ensino, à Pesquisa e à Extensão da UNICAMP).
REFERÊNCIAS 1. Do Monte, S. A.; Ventura, E.; Quim. Nova 2011, 34, 527. DOI: http://dx.doi.org/10.1590/S0100-40422011000300028 2. Hartree, D. R.; Proc. Cambridge Philos. Soc. 1928, 24, 111. DOI: http://dx.doi.org/10.1017/S0305004100011920 3. Hartree, D. R.; Proc. Cambridge Philos. Soc. 1928, 24, 89. DOI: http://dx.doi.org/10.1017/S0305004100011920 4. Fock, V.; Z. Phys. 1930, 61, 126. DOI: http://dx.doi.org/10.1007/BF01340294 5. Levine, I.; Quantum Chemistry; 6th ed.; Prentice Hall: New Jersey, 2008. 6. Pilar, F. L.; Elementary Quantum Chemistry; 2nd ed.; Dover Publications: New York, 2001. 7. McQuarrie, D. A.; Simon, J. D.; Physical Chemistry: A Molecular Approach; University Science Books: Sausalito, 1997. 8. Arfken, G. B.; Weber, H. J.; Harris, F. E.; Mathematical Methods for Physicists: A Comprehensive Guide; 6th ed.; Academic Press: New York, 2005. 9. Métodos de Química Teórica e Modelagem Molecular; Morgon, N. H.; Coutinho, K., Eds.; Editora Livraria da Física: Sao Paulo, 2007. 10. Pople, J. A.; Beveridge, D. L.; Approximate Molecular Orbital Theory; McGraw-Hill: New York, 1970. 11. Custodio, R.; Politi, J. R. dos S.; Segala, M.; Haiduke, R. L. A.; Cyrillo, M.; Quim. Nova 2002, 25, 159. DOI: http://dx.doi.org/10.1590/S0100-40422002000100025 12. Roothaan, C. C. J.; Rev. Mod. Phys. 1960, 32, 179. DOI: http://dx.doi.org/10.1103/RevModPhys.32.178 13. Szabo, A.; Ostlund, N. S.; Modern Quantum Chemistry: Introduction to Advanced Electronic Structure Theory; Dover Publications: New York, 1996. 14. Coulson, C. A.; Neilson, A. H.; Discuss. Faraday Soc. 1963, 35, 71. DOI: http://dx.doi.org/10.1039/df9633500071 15. Davidson, E. R.; J. Chem. Phys. 1972, 57, 1999. DOI: http://dx.doi.org/10.1063/1.1678521 16. Weinstein, H.; Pauncz, R.; Cohen, M.; Adv. At. Mol. Opt. Phys. 1971, 7, 97. 17. Edmiston, C.; Ruedenberg, K.; Rev. Mod. Phys. 1963, 35, 457. DOI: http://dx.doi.org/10.1103/RevModPhys.35.457 18. Levy, M.; Nee, T.; Parr, R. G.; J. Chem. Phys. 1975, 63, 316. DOI: http://dx.doi.org/10.1063/1.431100 |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access