
 |
 |
 |
 |
 |
Educação
|
|
| Síntese de ácidos cumarino-3-carboxílicos e sua aplicação na síntese total da aiapina, cumarina e umbeliferona# Synthesis of 3-coumarin-carboxylic acids and their application in the total synthesis of aiapin, coumarin, and umbeliferone |
|
Silvio CunhaI,II,*; Carlos Eduardo Martins IunesI,II; Caio Costa OliveiraI,II; Lourenço Luis Botelho de SantanaI,II
IInstituto de Química, Universidade Federal da Bahia, Campus de Ondina, 40170-290 Salvador - BA, Brasil Recebido em 22/01/2015 *e-mail: silviodc@ufba.br The synthesis of 3-coumarin-carboxylic acids and their application to the total synthesis of the natural products ayapin, coumarin, and umbeliferone in undergraduate organic chemistry experiments is described herein. The synthetic approach consists of a one-pot cyclization between salyciladehydes and Meldrum's acid in water to produce the above mentioned acids, followed by decarboxylation under basic or radical conditions. INTRODUÇÃO As substâncias heterocíclicas têm uma grande importância na vida em geral e predominam entre as substâncias que têm ação terapêutica.1,2 Apesar destes aspectos, o ensino da química dos heterociclos na graduação ainda não recebe ênfase correspondente à importância desta classe de substâncias.3 Salvo melhor juízo, o primeiro livro genuinamente brasileiro devotado aos aspectos teóricos do tema, de autoria de Stefani, foi publicado apenas em 2009.4 A inserção da química dos compostos heterocíclicos é ainda mais incipiente nos cursos experimentais, e quando a síntese de heterociclos é encontrada em manuais práticos, o procedimento empregado é aquele clássico.5-9 Em alguns casos sequer consta a preparação de heterociclo.10 Afortunadamente, é possível lançar mão de publicações recentes em Química Nova que têm contribuído para sanar estas lacunas.11-15 Em função dos aspectos supracitados e do nosso envolvimento com o desenvolvimento de aulas experimentais para o ensino de Química Orgânica,15-19 descrevemos uma abordagem de síntese de cumarinas que permite relacionar a química de heterociclos, tanto como experimento isolado para um curso experimental básico quanto em disciplina que contemple a execução de miniprojetos de síntese orgânica, com a síntese total de produtos naturais contendo o núcleo cumarínico. A seleção de cumarinas como tema de aula foi motivada em função da ocorrência natural dessas substâncias e da sua relevância biológica, Figura 1,20 que pode ser empregado como argumento motivacional para os estudantes e permite explorar aspectos interdisciplinares.
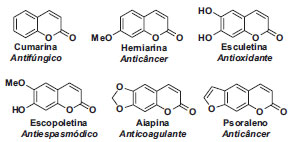 Figura 1. Exemplos de cumarinas boativas
RESULTADOS E DISCUSSÃO São escassas as propostas de síntese de cumarinas descritas em manuais experimentais de química na graduação da segunda metade do século XX. Nos raros casos onde se encontra a preparação de cumarinas, os procedimentos e os exemplos são sempre os mesmos, inclusive nos livros lançados no início do século XXI.9,10 Os métodos clássicos de síntese para preparação de cumarimas estão indicados na Figura 2 e empregam como reagentes fenóis ativados e beta-cetoesteres na reação de Pechmann, 2-carboxifenóis com anidrido acético ou com compostos contendo o grupo metileno ativado, como nas condensações de Perkin e Knoevenagel, respectivamente.4
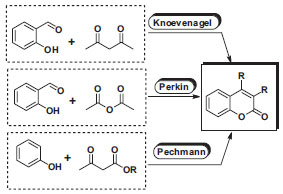 Figura 2. Métodos clássicos de síntese de cumarinas
Síntese dos ácidos cumarino-3-carboxílicos Nos raros exemplos das preparações de cumarinas encontradas em manuais experimentais de química orgânica, representadas no Esquema 1, os procedimentos empregados envolvem longos tempos reacionais e/ou solventes que requerem cuidados na sua manipulação por estudantes ainda inexperientes, tanto na etapa de síntese quanto na purificação.6,9 Por exemplo, na síntese da 4-metilcumarina via reação de Pechmann, apesar do emprego de uma resina ácida reutilizável, a reação usa tolueno como solvente e, na etapa de purificação, é empregado metanol.21,22
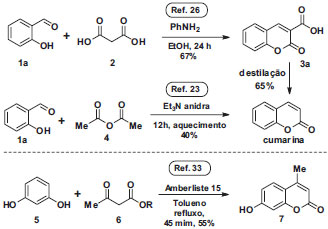 Esquema 1. Sínteses de cumarinas descritas em manuais de laboratório com rendimentos típicos obtidos por estudantes
Para as sínteses das cumarinas aqui apresentadas, selecionamos o procedimento que emprega água como solvente e tem como reagentes o ácido de Meldrum 8 e o saliciladeído 1a, que leva à formação do ácido cumarino-3-carboxílico 9a por meio de uma reação de condensação de Knoevenagel, Esquema 2.23,24 Para tentar diminuir o tempo de reação e inserir o uso de reação orgânica assistida por micro-ondas, a síntese de 9a foi avaliada empregando um reator dedicado. Quantidades equimolares do aldeído 1a e do ácido de Meldrum 8 foram aquecidos em água pelo tempo indicado a 80 ºC (2 min), 100 ºC (2 min) e 140 ºC (10 min) até consumo total de 8, resultando em rendimentos de 10%, 30% e 64% de 9a, respectivamente. Apesar de diminuir o tempo para o consumo de 8, o rendimento não foi maior que o obtido pelo método que emprega aquecimento convencional (97%).
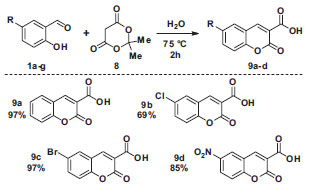 Esquema 2. Síntese de ácidos cumarino-3-carboxílicos
Podem ser empregados vários 2-hidroxi-benzaldeídos substituídos com grupos retiradores de elétrons, fornecendo os correspondentes ácidos cumarino-3-carboxílicos 9b-d em ótimos rendimentos (a reação funciona também com aldeídos com substituintes doadores de elétrons, como será visto adiante), superiores ao que são tipicamente obtidos pelos métodos tradicionais (comparar os Esquemas 1 e 2). A versatilidade do método permite empregar o aldeído mais disponível no laboratório de graduação, ou mesmo inserir a síntese do 2-hidroxi-benzaldeído 1b-c como uma das etapas da aula, uma vez que alguns destes aldeídos são também propostas de aulas experimentais,25-27 assim como o ácido de Meldrum 8 (mas todos reagentes são disponíveis comercialmente).8 A reatividade especial do ácido de Meldrum propicia condições reacionais brandas e o emprego de água como solvente, e não necessita catalisador, solvente orgânico ou outros aditivos. O isolamento do produto ocorre de forma muito prática, pois o heterociclo precipita do meio reacional à medida que é formado, sendo isolado por uma filtração, com pureza adequada para a caracterização posterior, Esquema 2. A síntese dos ácidos cumarino-3-carboxílicos pode ser aplicada numa aula semanal de quatro horas de duração, pois o tempo reacional é relativamente curto e todas as operações unitárias podem ser realizadas nesta primeira aula e a caracterização na aula seguinte. Assim, o instrutor pode aplicar a proposta de aula aqui apresentada em cursos básicos da graduação ou, se o projeto pedagógico do curso permitir, empregar os produtos obtidos nesta aula em miniprojetos de síntese orgânica, como descrito a seguir. Síntese total da aiapina, cumarina e umbeliferona Entre os graduandos dos cursos de química, mesmo os mais experientes, a percepção é de que a síntese total de um produto natural é tarefa muito complexa e que não pode ser executada em aulas experimentais. Alguns cientistas brasileiros têm apresentado alternativas a este cenário.28 Pinto e colaboradores têm contribuído com uma série de sínteses de produtos naturais em aulas experimentais de graduação como, por exemplo, a síntese em uma etapa de aromas de frutas29 e a conversão de borneol à cânfora.30 A síntese total de um produto natural envolvendo a formação de ligações carbono-carbono e carbono-heteroátomo raramente é realizada em cursos de graduação, pois geralmente envolvem várias etapas, reagentes complexos ou condições de reações sofisticadas. A síntese do alcaloide convolutamidina A é um dos poucos experimentos de síntese total para a graduação descrito na primeira década do século XXI por Pinto e colaboradores em Química Nova.31 Para contribuir com a formação em síntese total em cursos experimentais, elaboramos um plano sintético para a síntese de três produtos naturais que contém o núcleo cumarínico: aiapina, cumarina e umbeliferona. Como estratégia, desenvolvemos uma rota alternativa de síntese de cumarinas funcionalizadas, empregando os ácidos cumarino-3-carboxílicos como intermediários, passível de execução por estudantes inseridos em uma disciplina de projeto em química. Todavia, a aplicação da estratégia aqui apresentada foi mais adequada para a síntese da aipina e da cumarina. A aiapina é uma cumarina de origem natural produzida por diversas espécies de plantas, isolada pela primeira vez em 1936 de uma planta da região amazônica conhecida como aiapana (Eupatorium ayapana), que é utilizada por povos indígenas para o tratamento de diversas mazelas.32 Recentemente, foi isolada da imburana-de-cheiro, Amburana cearensis A. C. Smith (Fabaceae), outra planta típica do Brasil que ocorre no sertão do Nordeste.33 A substância natural cumarina é o membro mais simples desta classe de heterociclos e seu nome coincide com o nome desta família de compostos; a umbeliferona é o derivado hidroxilado na posição 7.21 Diversas rotas para a síntese da aiapina estão descritas na literatura.34-36 Destacamos aqui três sínteses recentes que empregam como reagentes substâncias estruturalmente semelhantes àqueles selecionadas na nossa rota de síntese, o aldeído 1g e um derivado do ácido de Meldrum, Esquema 4. Dessa forma, Maes e colaboradores sintetizaram a aiapina por meio da reação entre 4,5-metilenodióxi-2-hidróxibenzaldeído e um ilídeo de fósforo (reação de Wittig).34 Já Fillion e colaboradores sintetizaram a aiapina, entre outras cumarinas, por meio de reações de fenóis (no caso da aiapina, o sesamol) e alquilidenos do ácido de Meldrum em nitrometano, catalisadas por triflato de itérbio.35 Adicionalmente, Trost reportou uma ciclização mediada por paládio baseada na hidroarilação de butinoatos com fenóis ricos em elétrons. Apesar da abordagem elegante e da alta economia atômica, o experimental laborioso, aliado aos longos tempos reacionais, inviabilizam o uso desta metodologia como um experimento de aula prática.36
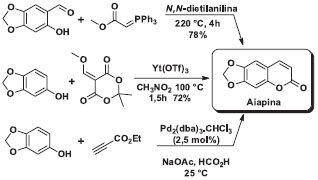 Esquema 3. Sínteses recentes da aiapina
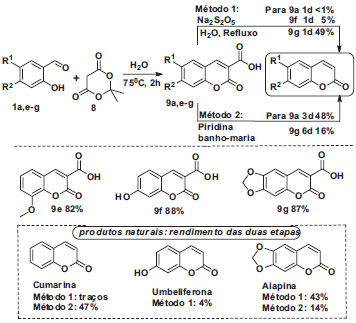 Esquema 4. Síntese total da aiapina, cumarina e umbeliferona
Duas sínteses da cumarina em aulas de graduação estão apresentadas no Esquema 1, em duas etapas com 44% ou em etapa única e 40% de rendimento global. Para a umbeliferona não há descrição de síntese destinada a experimentos de gradução. Os ácidos cumarino-3-carboxílicos são intermediários versáteis para a síntese de outras substâncias com atividade biológica relevante e de cumarinas naturais.37,38 Dessa forma, em cursos nos quais projetos de síntese podem ser incorporados, desenvolvemos uma rota que emprega estes intermediários na síntese total de cumarinas. Os alvos selecionados foram o heterociclo mais simples da família, que recebe o próprio nome de cumarina, e os derivados oxigenados umbeliferona e aipina. Para este objetivo, os ácidos cumarino-3-carboxílicos 9a (Esquema 2) e 9e-g, Esquema 4, foram preparados a partir do orto-hidroxi-benzaldeído correspondente em rendimentos superiores a 80%. Na síntese dos ácidos 9a, 9e e 9f foram empregados aldeídos disponíveis comercialmente. Para completar a síntese dos produtos naturais, estes ácidos foram submetidos à reação de descarboxilação. São diversos os métodos de descarboxilação disponíveis. Para a aula, foi selecionado o método da descarboxilação que emprega água como solvente, Esquema 4.39 O método de descarboxilação que utiliza o metabissulfito de sódio em água mostrou-se limitado pois, quando aplicado aos ácidos 9a-d, forneceu baixíssimo rendimento (9a) ou misturas complexas com os ácidos 9b-d contendo grupos retiradores de elétrons. Mesmo com o ácido 9e, contendo o grupo metoxila, não foi efetivo. Para contornar esta limitação foram testados outros métodos clássicos de descarboxilação, empregando-se o mais simples dos ácidos 9a para otimizar a condição experimental. Todavia, apenas o método que utiliza piridina38 como solvente e aquecimento a 90-95 ºC promoveu a descarboxilação em rendimento ainda adequado para uma aula experimental (48%). Assim, a síntese da cumarina natural mais simples foi alcançada em 47% de rendimento global, Esquema 4. A síntese total da aiapina foi realizada em duas etapas, sendo a primeira uma condensação de Knoevenagel entre o 6-hidróxi-piperonal 1g e o ácido de Meldrum 8, seguida da descarboxilação do ácido 9g utilizando o metabissulfito de sódio (Na2S2O5). O rendimento total da síntese foi de 43%, Esquema 4. O método de descarboxilação com metabissulfito de sódio em água parece ser limitado aos ácidos cumarínicos muito ricos em elétrons, pois quando foi empregado na descarboxilação do ácido 9f forneceu a umbeliferona em apenas 5% de rendimento, o que foi responsável pelo rendimento global de 4% deste produto natural, Esquema 4 Para comparar o método que emprega água como solvente com o que utiliza piridina, esta última condição reacional foi estendida para a síntese da aiapina. Entretanto, de forma contrária ao ácido 9a, o método de descarboxilação que utiliza piridina, quando aplicado ao ácido 9g, promoveu a descarboxilação em 16% de rendimento, resultando em rendimento total da aiapina de 14%, muito menor do que com o metabissulfito de sódio, o que ratifica a importância e utilidade do método em água. Síntese do 6-hidróxi-piperonal Enquanto os aldeídos 1a e 1f empregados na síntese da cumarina e da umbeliferona são disponíveis comercialmente, o 6-hidróxi-piperonal 1g precisa ser preparado. No contexto de um projeto de química orgânica na graduação esta etapa da síntese total pode ser parte integrante da tarefa do graduando, como pode também ser delegado como tarefa aos técnicos de laboratório que auxiliam as aulas práticas, supervisionada pelo instrutor do curso. A decisão é função, portanto, da infraestrutura disponível e da natureza do curso experimental. Para a síntese de 1g, duas rotas tradicionais são descritas na literatura partindo do sesamol 10,34 uma que emprega a formilação de Vilsmeier-Haack e outra que envolve a reação de Gatterman. Em função da disponibilidade dos reagentes, executamos esta última, Esquema 5.
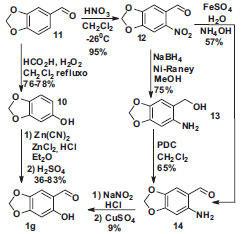 Esquema 5. Síntese do 6-hidróxi-piperonal
O sesamol 10, apesar de ser comercialmente disponível, foi obtido em escala multigrama pela reação de Dakin do piperonal 11 com H2O2 e ácido fórmico, Esquema 5.40 A formilação de Gatterman do sesamol para preparar o 6-hidróxi-piperonal 1g requer assistência direta do professor, pois esta reação é perigosa, uma vez que gera HCN in situ durante 2 horas. Entretanto, esta reação de formilação foi realizada várias vezes por estudante experiente, sempre em capela típica de laboratório de ensino com boa ventilação, e várias medidas práticas foram adotadas para a obtenção de 1g em rendimento satisfatório e com segurança (ver experimental). Sem estes cuidados houve várias tentativas sem sucesso. Para contornar o emprego da reação de Gatterman elaboramos a rota descrita no Esquema 5, que emprega piperonal como reagente de partida e reações comuns em cursos experimentais de química orgânica. Assim, o 6-nitro-piperonal 12 foi obtido a partir da nitração do aldeído 11.41 Redução com NaBH4 na presença de níquel de Raney forneceu o amino-álcool 13, cuja oxidação seletiva do álcool forneceu o amino-aldeído 14 em 46% global a partir do piperonal.42 Alternativamente, e de forma preferencial, foi possível preparar 14 em 54% de rendimento global a partir de 11, com uma etapa reacional a menos e sem o emprego do reagente de cromo (PDC) e do níquel de Raney, por meio da redução seletiva do grupo nitro de 12 empregando-se uma mistura de NH4OH e FeSO4.43 Apesar disso, a purificação do produto foi mais trabalhosa. Para a síntese do fenol 1g via amina 14 foram avaliadas diversas tentativas de diazotação, sem sucesso. A única condição que forneceu o 6-hidróxi-piperonal 1g foi a decomposição do sal de diazônio realizada em solução saturada de CuSO4,44 porém o baixo rendimento desta reação reduziu o rendimento global a 4%, muito inferior à rota via reação de Gatterman, que fornece 1g entre 27-64% de rendimento total, Esquema 5.
CONCLUSÃO As aulas propostas apresentam características multifacetadas, uma vez que tanto a síntese do núcleo heterocíclico das cumarinas foi obtida empregando procedimento rápido, simples e de fácil execução, vidrarias e equipamentos rotineiros, possibilitando a síntese de vários ácidos cumarino-3-carboxílicos, contextualizando o ensino da química de heterocíclicos em aulas experimentais de química orgânica logo nos estágios iniciais da formação dos futuros profissionais, pois a reação de síntese do núcleo heterocíclico emprega água como solvente. Dessa forma, a etapa de síntese dos ácidos cumarino-3-carboxílicos pode constituir uma aula isolada de curso experimental básico de Química Orgânica, pois uma série de salicilaldeídos pode ser empregada, podendo o instrutor lançar mão do reagente que estiver disponível ou inserir a síntese do saliciladeído substituído como parte integrante da aula. Adicionalmente, foi desenvolvida uma rota inédita de síntese total dos produtos naturais aipina, cumarina e umbeliferona, mais eficaz para as duas primeiras cumarinas quanto ao rendimento global, adequada para disciplinas avançadas de Química Orgânica experimental. Mesmo com as limitações impostas por algumas etapas executadas, como observado para a síntese do 6-hidróxi-piperonal (precursor da aiapina) e nas reações de descarboxilação, o estudante trava conhecimento com vários aspectos comuns à execução de projetos de síntese total de produtos naturais, como a limitação de uma reação em função dos substituintes do reagente de partida, rendimento limitado em uma etapa avançada da síntese, interconversão de grupo funcional, formação de ligação carbono-carbono e carbono-heteroátomo. Dessa forma, a execução do projeto de síntese aqui proposto não mascara a realidade da síntese orgânica, ao contrário, a proposta de síntese de cumarinas naturais, empregando uma rota exequível para disciplinas de projetos de química, proporciona a desmitificação da síntese orgânica na graduação, o que pode despertar vocações.
PARTE EXPERIMENTAL Os aldeídos sólidos foram empregados nas reações sem tratamento prévio e os líquidos foram destilados antes do uso. Os pontos de fusão foram determinados em um aparelho de placa aquecida Microquímica MQAPF 301 e não foram corrigidos. Os espectros na região do infravermelho foram obtidos na forma de discos de KBr em um aparelho SHIMADZU IR Affinity-1. Os espectros de RMN foram obtidos nos aparelhos Varian Gemini 300 ou Varian Inova 500 e os deslocamentos químicos estão descritos em unidades de ppm a partir da referência (TMS). Procedimento geral para a síntese dos ácidos cumarino-3-carboxílicos 9a-g A um balão de fundo redondo de 10 mL foram adicionados 2 mL de água destilada, 1,1 mmol do respectivo aldeído e 1 mmol de ácido de Meldrum. A mistura foi mantida sob agitação magnética e aquecimento a 75 ºC até consumo total do ácido de Meldrum, observado por CCD. Após o tempo indicado para cada substrato, o meio reacional foi resfriado à temperatura ambiente, o sólido formado foi filtrado a vácuo e lavado com água destilada. O sólido obtido foi colocado numa estufa a 100 ºC durante 30 minutos para secar. 3-Carboxicumarina 9a: 2h, sólido branco, 180,65 mg, 95%. Pf 188,0-188,8 ºC (Lit.45 191-192 ºC). IV (KBr) 3444, 1712, 1682, 1624, 1558, 1508, 1369, 1323, 1269, 1219, 1134, 1026, 996, 852, 821, 790, 748, 640, 560, 524, 474 cm-1. 6-Cloro-3-carboxicumarina 9b: 3h, sólido branco, 154,97 mg, 69%. Pf 197,4-198,3 ºC (Lit.45 195 ºC). IV (KBr) 3446, 3061,1745, 1728, 1663, 1560, 1479, 1363, 1240, 1207, 1147, 1085, 1029, 961, 912, 879, 836, 802, 736, 677, 597, 543, 460, 433 cm-1. 6-Bromo-3-carboxicumarina 9c: 2h, sólido branco, 260,98 mg, 97%. Pf 209,1-201,2 ºC (Lit.46 195-196 ºC). IV (KBr) 0 3446, 1736, 1716, 1676, 1606, 1556, 1473, 1408, 1355, 1303, 1267, 1244, 1209, 1147, 1008, 970, 883, 819, 802, 758, 742, 665, 605, 559, 516, 455 cm-1. 6-Nitro-3-carboxicumarina 9d: 2h, sólido amarelo, 199,88 mg, 85%. Pf 235 ºC (Lit.46 234-235 ºC). IV (KBr) 3446, 3267, 1739, 1722, 1616, 1570, 1536, 1521, 1477, 1364, 1232, 1207, 1139, 1124, 1093, 1008, 977, 962, 916, 864, 627, 800, 762, 729, 673, 605, 540, 464 cm-1. 8-Metóxi-3-carboxicumarina 9e: 2h, sólido amarelo, 180,55 mg, 82%. Pf 217,8-218,3 ºC (Lit.45 219 ºC). IV (KBr) 3468, 1772, 1676, 1604, 1570, 1479, 1438, 1379, 1276, 1259, 1215, 1195, 1147, 1097, 960, 802, 742, 707, 630, 594, 569, 472, 424 cm-1. 7-Hidróxi-3-carboxicumarina 9f: 2h, sólido amarelo, 181,41 mg, 88%. Pf 261 ºC (Lit.46 261-263 ºC). IV (KBr) 3444, 1712, 1681, 1624, 1558, 1508, 1369, 1323, 1269, 1219, 1134, 1026, 852, 821, 790, 748, 640, 524, 474 cm-1. 6,7-Metilenodióxi-3-carboxicumarina 9g: 2h, sólido amarelo, 203,72 mg, 87%. Pf 276,5-277,0 ºC (Lit.47 263-264 ºC). IV (KBr) 3446, 1776, 1741, 1676, 1604, 1479, 1468, 1379, 1309, 1276, 1259, 1215, 1195, 1147, 1097, 960, 889, 802, 742, 707, 630, 594, 563, 472 cm-1. Síntese da cumarina A um balão de fundo redondo de 5 mL foram adicionados 0,5 mL de piridina e 0,0514 g de 9a (0,27 mmol). A mistura foi mantida sob aquecimento a 90 ºC. Após 72 h a análise por CCD mostrou que a reação estava em equilibrio. Sendo assim, o solvente foi evaporado a vácuo e o resíduo foi purificado por cromatografia em coluna utilizando como eluente hexano/acetato de etila (4:1), levando a obtenção de 0,019 g da cumarina (47%) como um sólido branco. Pf 67 ºC (Lit.48 68-71 ºC). IV(KBr) 1754, 1706, 1670, 1605, 1564, 1517, 1452, 1401, 1397, 1278, 1259, 1177, 1121, 932, 893, 830, 756, 730, 610, 526, 460 cm-1. Síntese da umbeliferona A um balão de fundo redondo de 10 mL contendo 0,2498 g (1,21 mmol) de 3-carboxi-7-hidróxicumarina 9f foram adicionados 7 mL de água destilada. O meio reacional foi aquecido a 90 ºC e mantido sob agitação magnética. Após 2 minutos foram adicionados 1,1560 g (6,08 mmol) de metabissulfito de sódio. A reação permaneceu sob refluxo e agitação por 24 h. Após este tempo a mistura foi resfriada a temperatura ambiente e o solvente evaporado. O resíduo foi purificado por cromatografia em coluna utilizando como eluente clorofórmio/metanol (9:1). As frações que apresentaram a mancha fluorescente foram agrupadas e o solvente evaporado, resultando em 0,0113 g (4%) da umbeliferona como sólido amarelo. Pf 229-230 ºC (Lit.49 230-231 ºC). RMN de 1H (DMSO-D6): δ 10,58 (s, 1H); 7,93 (d, 1H, 9,6 Hz); 7.52 (d, 1H, 8,4 Hz); 6,78 (dd, 1H, 8,4 Hz, 2,4 Hz); 6,71 (d, 1H, 2,4 Hz); 6.20 (d, 1H, 9,6 Hz). Síntese da aiapina MÉTODO 1. A um balão de fundo redondo de 10 mL contendo 0,0556 g (0,2374 mmol) de 3-carboxi-6,7-metilenodióxicumarina 9g foram adicionados 3 mL de água destilada. O meio reacional foi aquecido sob agitação magnética. Após 2 minutos, foram adicionados 0,2515 g (1,32 mmol) de metabissulfito de sódio e a reação permaneceu sob refluxo e agitação por 20 h, até consumo total do ácido de Meldrum. Após este tempo, o solvente foi evaporado e o resíduo purificado por cromatografia em coluna utilizando como eluente hexano/éter etílico (1:1). As frações que apresentaram a mancha fluorescente foram unidas e o solvente evaporado, resultando 0,0221g (49%) da aiapina como sólido incolor. Pf 222-223 ºC (Lit.50 229-230 ºC). IV(KBr) 3055, 2918, 1706, 1633, 1579, 1488, 1417, 1257, 1122, 1039, 941, 883, 838, 730, 609, 551. RMN de 1H (DMSO-D6, 500MHz): δ 7.91 (d, 1H, 9,5Hz); 7,22 (s, 1H); 7.10 (s, 1H); 6,31 (d,1H, 9,5 Hz); 6,16 ( s, 2H). RMN de 13C (DMSO-D6, 125 MHz): δ 160,8; 151,4; 151,2; 144,9; 113,1; 112,9; 105,9; 102,9; 98,4; 79,6. MÉTODO 2. A um balão contendo 0,0303 g (0,13 mmol) da 3-carboxi-6,7-metilenodióxicumarina 9g foram adicionados 3 mL de piridina. A mistura foi mantida sob aquecimento a 100 ºC. Após 5 dias a análise por CCD mostrou que a reação encontrava-se em equilibrio. O solvente foi evaporado e o resíudo purificado por cromatografia em coluna utilizando como eluente hexano/éter etílico (1:1). As frações que apresentaram a mancha fluorescente foram unidas e o solvente evaporado, resultando 0,0040 g (16%) da aiapina como um sólido amarelo. Pf 222-223 ºC. Síntese do sesamol 1040 A uma solução de 10 g (66,6 mmol) do piperonal 11 em 340 mL de DCM foram adicinados 16 mL de H2O2 e 9,4 mL de ácido fórmico. Esta mistura foi mantida sob refluxo por 20 horas. Após este tempo, a reação foi resfriada e foram adicionados 300 mL de NaOH 1,5 mol L-1. A mistura foi agitada por 30 minutos. Em seguida, as fases foram separadas e a orgânica foi evaporada. Ao resíduo obtido foram adicionados 230 mL de metanol e a fase aquosa alcalina. Esta solução foi aquecida por mais 30 minutos e após resfriamento, foi lavada duas vezes com 200 mL de DCM. A fase aquosa foi acidificada até pH 1 com HCl 37% e lavada três vezes com 300 mL de DCM. As fases de DCM foram combinadas e evaporadas, fornecendo 7,2 g (78%) do sesamol 10, Pf 62-64 ºC (Lit.40 62-65 ºC). Síntese do 6-hidróxi-piperonal 1g49 Em um balão de duas bocas de 150 mL acoplado a um condensador conectado a tubo de cloreto de cálcio e, na outra boca, conectado a um sistema de geração do gás HCl, uma mistura de 3,0 g (21,7 mmol) do sesamol 10, 3,83 g (32,6 mmol) de Zn(CN)2, 0,74 g (5,43 mmol) de ZnCl2 e 10 mg de NaCl foi suspensa em 75 mL de éter anidro. A esta mistura foi borbulhado HCl seco por 2 horas. Após 20 minutos a solução ficou verde e houve formação de um sólido também verde. É possível visualizar o consumo do Zn(CN)2 durante o curso da reação. Com aproximadamente 1 hora de adição de HCl, ocorre dissolução de todos os sólidos e um novo sólido verde é formado e o HCl é borbulhado ao meio por mais 1 hora. Após este período, o borbulhamento do gás é interrompido, o éter é decantado e o sólido é lavado duas vezes com 5 mL de éter. A este sólido são adicionados 50 mL de água (ocorre intenso desprendimento de gás) e 3 gotas de H2SO4 concentrado. Este sistema foi submetido a refluxo por 30 minutos. Ao final deste período, o sistema foi resfriado e o sólido filtrado. O sólido foi imediatamente transferido para um erlenmeyer e dissolvido em DCM. Esta solução foi seca com MgSO4, e o agente secante foi separado por filtração e o líquido foi evaporado fornecendo 3,00 g (83%) do 6-hidróxi-piperonal 1g. Pf 121-124 ºC (Lit. 49 121-124 ºC). NOTAS SOBRE ESTA SÍNTESE. 1) Realizar este procedimento em capela com forte exaustão. 2) O HCl seco foi gerado pela adição lenta de 200 mL de HCl (37%) em 200 g de CaCl2 anidro, e antes de chegar ao meio reacional passou por um trap com H2SO4 (98%). 3) A ponta do borbulhador de HCl deve estar no meio da solução, pois o nível do éter diminui no decorrer da reação. 4) A agitação deve ser eficiente, pois o sólido formado na reação adere à bagueta. 5) As mangueiras utilizadas para a passagem do HCl devem ser as menores possíveis e as conexões devem estar envoltas por fita veda-rosca. 6) Os reagentes sólidos devem estar secos e finamente particulados. Síntese do 6-nitro-piperonal 1241 A uma solução, resfriada a -26 ºC, do piperonal (11) em 5 mL de 1,2-dicloroetano, foram adicionados, lentamente, 2,5 mL de HNO3 (90%). Após a adição, a reação foi mantida sob agitação por 6,5 horas a -15 ºC. Ao final deste período, a reação foi vertida em 50 mL de água gelada e lavada três vezes com 20 mL de AcOEt. As fases orgânicas foram agrupadas, secas em MgSO4 e evaporadas para fornecer 12 contaminado com piperonal (11). Após recristalização com CHCl3/hexano foi obtido 1,48 g (95%) do 6-nitro-piperonal 12. Pf 93-94 ºC (Lit.41 93-94 ºC). Síntese do amino-álcool 1342 A uma suspensão resfriada a 0 ºC do nitro-piperonal 12 (0,43 g, 2,2 mmol) com 0,013 g de Ni-Raney em 8 mL de metanol, adicionou-se o NaBH4 (0,2 g, 5,3 mmol) em pequenas porções. Após a adição do NaBH4 a reação terminou em 10 minutos. O Ni-Raney foi filtrado do meio e a solução restante foi diluída com 20 mL de DCM e lavada três vezes com 5 mL de água. Depois de evaporada, a fase orgânica forneceu um sólido impuro, que após recristalização com DCM/hexano, forneceu 0,28 g (75%) de 13. RMN de 1H (DMSO-D6): δ 4,27 (d, 2H); 4,66 (s, 2H); 4,92 (t, 1H); 5,80 (s, 2H); 6,32 (s, 1H); 6,66 (s, 1H). RMN de 13C (DMSO- D6): δ 146,5; 141,3; 137,9; 117,6; 108,18; 99,83; 96,92; 60,74. Síntese do 6-amino-piperonal 14 MÉTODO 1. A uma solução de 13 (0,11 g, 0,67 mmol) foi adicionada, sob forte agitação, uma solução de PDC (0,353 g, 0,94 mmol) em 20 mL de DCM. Após 18 horas de reação, o meio reacional foi filtrado em celite e a solução obtida foi lavada três vezes com 10 mL de água. Depois de evaporada, a fase orgânica forneceu um sólido impuro, que foi recristalizado com DCM/hexano para fornecer 0,067 g (61%) do 6-amino-piperonal 14. MÉTODO 2.43 A uma solução de FeSO4 (3,6 g, 24,5 mmol) em 20 mL de água em ebulição foram adicionados 15 mL de solução aquosa fervente do 6-nitro-piperonal 12 (0,36 g, 1,86 mmol). Em seguida foram adicionados 4 mL de NH4OH concentrado. O meio reacional tornou-se escuro e foi mantido sob aquecimento por 5 minutos. Em seguida, a reação foi filtrada sob vácuo e o resíduo lavado com 50 mL de água quente. Depois de resfriado, o combinado das fases aquosas foi lavado três vezes com 50 mL de DCM. A fase orgânica foi evaporada e o resíduo recistalizado com DCM/hexano para fornecer 0,157 g (57%) do 6-amino-piperonal 14. Pf 104-106 ºC (Lit.43 103-105 ºC). Síntese do 6-hidróxi-piperonal 1g44 A uma suspensão do 6-amino-piperonal 14 (4,8 g, 29,1 mmol) em 140 mL de H2O foram adicionados, sob forte agitação, 4,5 mL de H2SO4 80%. Em seguida, uma solução contendo 2,0 g de NaNO2 em 20 mL de água foi adicionada lentamente. Após 30 minutos a solução foi filtrada e adicionada gota-a-gota em 50 mL de solução saturada fervente de CuSO4. Esta solução foi resfriada, o seu pH foi corrigido até 13 pela adição de NaOH 20% e lavada três vezes com 30 mL de DCM. Em seguida a fase aquosa foi acidificada até pH 1 e extraída três vezes com 30 mL de DCM. O combinado destas últimas frações orgânicas foi evaporado e forneceu 0,45 g (9%) do 6-hidróxi-piperonal 1g.
MATERIAL SUPLEMENTAR Procedimentos selecionados e espectros representativos das substancias estão disponíveis em http://quimicanova.sbq.org.br, na forma de arquivo PDF, com acesso livre.
AGRADECIMENTOS Os autores agradecem o suporte financeiro do Conselho Nacional de Desenvolvimento Científico e Tecnológico - CNPq, Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - CAPES, e Fundação de Amparo à Pesquisa do Estado da Bahia - FAPESB. Também agradecemos à CAPES a bolsa de pós-doutorado de L. L. B. Santana, e ao CNPq a bolsa de mestrado de C. O. Costa, a bolsa PIBIC de C. E. M. Iunes e a bolsa de produtividade em pesquisa de S. Cunha.
REFERÊNCIAS 1. Pozharskii, A. F.; Soldatenkov, A.; Katritzky, A. R.; Heterocycles in Life and Society: An Introduction to Heterocyclic Chemistry and Biochemistry and the Role of Heterocycles in Science, Technology, Medicine and Agriculture, Wiley: Chichester, 1977. 2. Barreiro, E. J.; Quim. Nova 1991, 14, 179. 3. Menos de 10% dos procedimentos dos seguintes manuais, amplamente empregados na segunda metade do século XX, são dedicados à preparação de heterociclos: Mano, E. B.; Seabra, A. O.; Práticas de Química Orgânica, 3 ed., Editora Edgard Blucher LTDA: São Paulo, 1987; Fieser, L. F.; Experiments in Organic Chemistry, 3rd ed., D. C. Heath and Company: Boston, 1955. 4. Stefani, H. A. Introdução à Química de Compostos Heterocíclicos, Guanabara Koogan: Rio de Janeiro, 2009. 5. Soares, B. G.; Souza, N. A.; Pires, D. X.; Química Orgânica: Teoria e Técnicas de Preparação, Purificação e Identificação de Compostos Orgânicos, Ed. Guanabara S.A.: Rio de Janeiro, 1988. 6. Furniss, B. S.; Hannaford, A. J.; Smith, P. W. G.; Tactchell, A. R.; Vogel's Textbook of Practical Organic Chemistry, 5th ed., Longman Scientific & Technical: Singapore, 1989. 7. Pavia, D. L.; Lampman, G. M.; Kriz, G. S.; Engel, R. G.; Introduction to Organic Laboratory Techniques: a Small Scale Approach, Saunders College Publishing: Philadelphia, 1998. 8. Tietze, L.-F., Eicher, T.; Reactions and Syntheses in the Organic Chemistry Laboratory, University Science Books: Mill Valley, California, 1989. 9. Dias, A. G.; da Costa, M. A.; Guimarães, P. I. C.; Guia Prático de Química Orgânica, Vol 2, Síntese Orgânica: Executando Experimentos, Editora Interciência, Rio de Janeiro, 2008. 10. Marques, J. A.; Borges, C. P. F.; Práticas de Química Orgânica, 2 ed., Editora Átomo: Campinas, 2012. 11. Barbosa, T. P.; Diniz Neto, H.; Quim. Nova 2013, 36, 331. DOI: http://dx.doi.org/10.1590/S0100-40422013000200021 12. Konrath, E. L.; Piedade, M.; Eifler-Lima, V. L.; Quim. Nova 2012, 35, 1887. DOI: http://dx.doi.org/10.1590/S0100-40422012000900033 13. Bisol, T. B.; Marques, M. V.; Rossa, T. A.; Nascimento, M. G.; Sá, M. M.; Quim. Nova 2012, 35, 1260. DOI: http://dx.doi.org/10.1590/S0100-40422012000600035 14. Marques, M. V.; Bisol, T. B.; Sá, M. M.; Quim. Nova 2012, 35, 1696. DOI: http://dx.doi.org/10.1590/S0100-40422012000800034 15. Cunha, S.; Santos Filho, R. F.; Riatto, V. B.; Dourado, G. A. A.; Quim. Nova 2013, 36, 190. DOI: http://dx.doi.org/10.1590/S0100-40422013000100032 16. Cunha, S.; de Santana, L. L. B.; Quim. Nova 2012, 35, 642. DOI: http://dx.doi.org/10.1590/S0100-40422012000300036 17. Cunha, S.; Lustosa, D. M.; Conceição, N. D.; Fascio, M.; Magalhães, V.; Quim. Nova 2012, 35, 638. DOI: http://dx.doi.org/10.1590/S0100-40422012000300035 18. Cunha, S.; Beretta, M.; Fascio, M.; Santos, A. O.; Rodrigues Jr., M. T.; Bastos, R. M.; Quim. Nova 2005, 28, 364. 19. Cunha, S.; Lião, L. M.; Bonfim, R. R.; Bastos, R. M.; Monteiro A. P. M.; Alencar, K. S.; Quim. Nova 2003, 26, 425. DOI: http://dx.doi.org/10.1590/S0100-40422003000300022 20. Venugopala, K. N.; Rashmi, V.; Odhav, B. BioMed Research International (2013), doi:10.1155/2013/963248. DOI: http://dx.doi.org/10.1155/2013/963248 PMID: 23586066 21. Holden, M. S.; Crouch, R. D.; J. Chem. Educ. 1998, 75, 1631 Ver também: Bayarri, N.; Estevez, C.; Química Verde: Experimentos de Laboratorio para un Curso Universitario de Química, American Chemical Society: Washington, 2003. DOI: http://dx.doi.org/10.1021/ed075p1631 22. John, E. V. O.; Israelstam, S. S.; J. Org. Chem. 1961, 26, 240. DOI: http://dx.doi.org/10.1021/jo01060a602 23. Deshmukh, M.; Burud, R.; Baldino, C.; Chan, P.; Liu, J.; Synth. Commun. 2003, 33, 3299. DOI: http://dx.doi.org/10.1081/SCC-120023987 24. Maggi, R.; Bigi, F.; Carloni, S.; Mazzacani, A.; Sartori, G.; Green Chem. 2001, 3, 173. DOI: http://dx.doi.org/10.1039/b101822c 25. Teixeira, E. F.; dos Santos, A. P. B.; Bastos; R. S.; Pinto. A. C.; Kümmerle, A. E.; Coelho, R. R.; Quim. Nova 2010, 33, 1603. DOI: http://dx.doi.org/10.1590/S0100-40422010000700032 26. Mendonca, G. F.; Magalhaes, R. R.; Mattos, M. C. S.; Esteves, P. M.; J. Braz. Chem. Soc. 2005, 16, 695. DOI: http://dx.doi.org/10.1590/S0103-50532005000500003 27. Wang, L.; Jing, H.; Bu, X.; Chang, T.; Jin, L.; Liang Y.; Catal. Commun. 2007, 8, 80. DOI: http://dx.doi.org/10.1016/j.catcom.2006.05.018 28. Pinto, A. C.; Silva, B. V.; A Química Perto de Você: Experimentos de Química Orgânica, Sociedade Brasileira de Química: São Paulo, 2012. Este livro pode ser considerado como o primeiro livro brasileiro de experimentos de graduação totalmente dedicado à química verde. 29. Oliveira, C. A.; Souza, A. C. J.; Santos, A. P. B.; Silva, B. V.; Lachter, E. R.; Pinto, A. C.; Rev. Virtual Quim. 2014, 6, 73. 30. Santos, A. P. B.; Gonçalves, I. R. C.; Pais, K. C.; Martinez, S. T.; Lachter, E. R.; Pinto, A. C.; Quim. Nova 2009, 32, 1667. DOI: http://dx.doi.org/10.1590/S0100-40422009000100026 31. Silva, R. B.; Torres, J. C.; Garden, S. J.; Violante, F. A.; Rezende, M. J. C.; da Silva B. V.; Pinto, A. C.; Quim. Nova 2008, 31, 924. DOI: http://dx.doi.org/10.1590/S0100-40422008000100029 32. Bose, P.; Roy, A.; J. Indian Chem. Soc. 1936, 13, 586. 33. Canuto, K. C.; Silveira, e. R.; Bezerra, A. M. E.; Quim. Nova 2010, 33, 662. DOI: http://dx.doi.org/10.1590/S0100-40422010000300034 34. Maes, D.; Vervisch, S.; Debenedetti, S.; Davio, C.; Mangelinckx, S.; Giubellina, N.; De Kimpe, N.; Tetrahedron 2005, 61, 2505. DOI: http://dx.doi.org/10.1016/j.tet.2004.12.061 35. Fillion, E.; Dumas, A.; Kuropatwa, B.; Malhotra, N.; Sitler, T.; J. Org. Chem. 2006, 71, 409. DOI: http://dx.doi.org/10.1021/jo052000t PMID: 16388672 36. Trost, B. M.; Toste, F. D.; Greenman, K.; J. Am Chem. Soc. 2003, 125, 4518. DOI: http://dx.doi.org/10.1021/ja0286573 37. Jafarpour, F.; Jalalimanesh, N.; Olia, M. B. A.; Kashani, A. O.; Tetrahedron 2010, 66, 9508. DOI: http://dx.doi.org/10.1016/j.tet.2010.10.019 38. Mahulikar, P. P.; Mane, R. B.; J. Chem. Res. 2006, 12. 39. Adams, R. Bockstahler, T; J. Am. Chem. Soc. 1952, 74, 5346. DOI: http://dx.doi.org/10.1021/ja01141a038 40. Pansegrau; P. D.; Munson; B. P.; US pat. 5,840,997 1998. (CA 129:343330 ) 41. Murphy, B. P.; J. Org. Chem. 1985, vol. 50, 5875. DOI: http://dx.doi.org/10.1021/jo00350a090 42. Pogorelić, I.; Filipan-Litvić, M.; Merkaš, S.; Ljubić, G.; Cepanec, I.; Litvić, M.; J. Mol. Catal. A: Chem. 2007, 202. 43. Borgert, M. T.; Elder, F. R.; J. Am. Chem. Soc. 1929, 51, 532. DOI: http://dx.doi.org/10.1021/ja01377a025 44. Arnold, R.; Bordwell, F.; J. Am. Chem. Soc. 1942, 64, 2983. DOI: http://dx.doi.org/10.1021/ja01264a076 45. Armstrong, V.; Soto, O.; Valderrama, J. A.; Tapia, R.; Synth Commun. 1988, 18, 717. DOI: http://dx.doi.org/10.1080/00397918808077361 46. Song, A.; Wang, X.; Lam, K.S.; Tetrahedron Lett. 2003, 44, 1775. DOI: http://dx.doi.org/10.1016/S0040-4039(03)00146-1 47. Kenji, F.; Bull. Chem. Soc. Jpn. 1962, 35, 1321. DOI: http://dx.doi.org/10.1246/bcsj.35.1321 48. Dittmer, Donald C.; J. Org. Chem. 2006, 70, 4682. DOI: http://dx.doi.org/10.1021/jo050070u 49. Chatterjee, A.; J. Am. Chem. Soc. 1949, 71, 606. DOI: http://dx.doi.org/10.1021/ja01170a062 PMID: 18112067 50. Demytteaere, J.; van Syngel, K.; Markusse, A. P.; Vervish, S.; Debenedetti, S.; De Kimpe, N.; Tetrahedron. 2002, 58, 2163. DOI: http://dx.doi.org/10.1016/S0040-4020(02)00081-9
#Dedicado ao Professor Angelo da Cunha Pinto, do Instituto de Química da Universidade Federal do Rio de Janeiro, por suas valorosas contribuições à Química Brasileira, em particular no desenvolvimento de experimentos para o ensino de Química Orgânica e formação dos profissionais da Química. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access