
 |
 |
 |
 |
 |
Artigo
|
|
| Utilização do compósito nanoestruturado SiO2/TiO2 na fotodegradação de corantes têxteis com luz solar natural Use of SiO2/TiO2 nanostructured composites in textile dyes and their photodegradation in response to natural sunlight |
|
Camila S. InagakiI; Andrea Eloisa da Silva ReisI; Nayara M. OliveiraI; Vitor H. PaschoalI; Ítalo O. MazaliII; Antonio A. S. AlfayaI,*
IDepartamento de Química, Universidade Estadual de Londrina, CP 10.011, 86057-970 Londrina - PR, Brasil Recebido em 28/02/2015 *e-mail: alfaya@uel.br SiO2/TiO2 nanostructured composites with three different ratios of Si:Ti were prepared using the sol-gel method. These materials were characterized using energy dispersive X-ray fluorescence, Fourier transform infrared spectroscopy, X-ray diffraction, scanning electron microscopy, high-resolution transmission electron microscopy, photoluminescence, Raman with Fourier transform infrared spectroscopy, and the specific surface area. The band gaps of materials were determined by diffuse reflectance spectra, and the values of 3.20 ± 0.01, 2.92 ± 0.02, and 2.85 ± 0.01 eV were obtained as a result of the proportional increases in the amount of Ti within the composite. The materials exhibit only the anatase (TiO2) crystalline phase and have crystalline domains ranging from 4 to 5 nm. The photodegradation process of methylene blue, royal blue GRL, and golden yellow GL dyes were studied with respect to their contact times, pH variations within the solution, and the variations in the dye concentration of the solution in response to only sunlight. The maximum amount of time for the mineralization of dyes was 90 min. The kinetics of the process follows an apparently first order model, in which the obtained rate constant values were 5.72 × 10-2 min-1 for methylene blue, 6.44 × 10-2 min-1 for royal blue GRL, and 1.07 × 10-1 min-1 for golden yellow. INTRODUÇÃO Atualmente há uma grande necessidade de se preservar os recursos naturais do planeta, principalmente os recursos hídricos, os quais são poluídos progressivamente pela contaminação direta dos efluentes industriais.1,2 Um dos setores que mais contribui para a poluição das águas naturais é o setor têxtil, especificamente na etapa de tingimento de tecidos, que envolve uma enorme quantidade de água e o uso de corantes, com características tóxicas e carcinogênicas.3-5 Ao final dos processos de tingimento, as moléculas de corante que não se fixaram à fibra do tecido permanecem em solução e acabam na maioria das vezes sendo descartadas diretamente nos cursos dos rios. Estima-se que 10-15% da produção total de corantes é perdida, provocando além da forte coloração da água, problemas ligados à saúde dos seres que vivem no local e à inviabilidade de utilização desta água para o abastecimento público.6,7 Pelo fato de os corantes serem substâncias resistentes a algumas das técnicas convencionais de tratamento de efluentes industriais, como os processos biológicos, a filtração por membranas, a coagulação, a adsorção e a troca iônica, métodos alternativos estão sendo investigados.8-10 Dentre os processos recentes, os chamados Processos Oxidativos Avançados (POA) têm se mostrado uma alternativa interessante para o tratamento desses efluentes.3,8-12 Os POA são baseados na produção in situ do radical hidroxila, que pelo fato de apresntar um alto potencial padrão de redução (2,73 V vs EPH), ou seja, alta capacidade oxidativa, é utilizado para promover a degradação de diversas moléculas orgânicas.13,14 A fotocatálise heterogênea faz parte dos POA e utiliza basicamente um semicondutor na presença de irradiação UV para a geração de radicais hidroxila in situ.15,16 A principal vantagem deste método é a possibilidade de mineralização dos compostos orgânicos, reduzindo-os a produtos simples como gás carbônico, água e íons inorgânicos.17,18 O TiO2 é o semicondutor mais estudado em fotocatálise heterogênea devido a suas características de baixa toxicidade, insolubilidade em água, alta estabilidade química, custo baixo e band gap igual a 3,2 eV.19-26 No entanto, o TiO2 apresenta alguns problemas quando da sua utilização em fotocatálise. Primeiro, ele é pouco eficiente sob a luz visível e apresenta grande facilidade de recombinação dos elétrons com os buracos fotogerados e, deste modo, a sua eficiência na fotodegradação diminui consideravelmente.27,28 Segundo, o fato do TiO2 ser normalmente um pó muito fino, quando misturado em água forma uma dispersão branca, a qual apresenta um grande espalhamento da radiação incidente e, também, dificulta a sua recuperação por processos de filtração e/ou decantação.29-31 A fim de minimizar os problemas citados anteriormente e explorar mais eficientemente a energia incidente, modificações na composição dos fotocatalisadores têm sido reportadas, como a adição de outros semicondutores, de metais e não metais como dopantes ou suportes para nanopartículas de dióxido de titânio.30-35 Os materiais nanoestruturados também contribuem para melhorar a eficiência fotocatalítica, atuando no aumento da área específica, diminuição da recombinação do par elétron-buraco fotogerado e no aumento do número de sítios ativos da superfície.2,36 Dentre os vários métodos para síntese de materiais nanoestruturados, o processo sol-gel tem se mostrado uma boa opção, pois apresenta uma série de vantagens: (1) pode ser executado em temperatura ambiente; (2) é uma técnica de baixo impacto ambiental; (3) produz compósitos nanoestruturados de alta pureza e (4) possibilita a inserção de elementos de forma altamente dispersa e com elevado controle na morfologia estrutural do material produzido.20,37,38 Sabe-se que alguns métodos para a geração de radicais hidroxila necessitam de uma fonte de energia (lâmpadas UV, por exemplo) para a degradação dos corantes e o uso de oxidantes fortes, tornando o tratamento dos efluentes têxteis economicamente inviável na prática.11,14,20 O grande problema a ser resolvido de forma consistente é o desenvolvimento de novos materiais fotocatalisadores que possam utilizar a luz solar natural de forma eficiente, com baixo custo e que não necessitem de oxidantes fortes adicionais para a sua utilização no tratamento de efluentes industriais contaminados por corantes. Assim, o desenvolvimento de fotocatalisadores que estejam intimamente relacionados com os princípios da química verde e que possam ser utilizados em larga escala são extremamente interessantes do ponto de vista tecnológico. O presente trabalho teve como objetivo preparar o compósito nanoestruturado SiO2/TiO2 pelo método sol-gel, caracterizar e avaliar suas propriedades fotocatalíticas frente aos corantes têxteis: azul de metileno, azul royal GRL e amarelo ouro GL em solução aquosa utilizando apenas a luz solar natural.
PARTE EXPERIMENTAL Síntese do material nanoestruturado SiO2/TiO2, ST O material nanoestruturado SiO2/TiO2 foi sintetizado pelo processo sol-gel desenvolvido por Alfaya e colaboradores com algumas modificações.39 Três materiais foram preparados com diferentes proporções de Si:Ti nomeados STa, STb e STc. A preparação do STa é descrita a seguir. Em um balão foram adicionados 62,0 mL de TEOS - ortossilicato de tetraetila (Acrös Organics, 98%), 62,0 mL de etanol absoluto P.A. (Cinética), 5,7 mL de água (Milli-Q Plus) e 0,4 mL de ácido nítrico concentrado (BIOTEC, 65%). Esta mistura foi mantida em agitação e em refluxo por 2 h. Em seguida, o aquecimento foi desligado e, com o sistema na temperatura ambiente, foram adicionados 144,1 mL de etanol absoluto e 18,7 mL de TBOT - ortotitanato de tetrabutila (Aldrich, 99%) lentamente e com forte agitação. Após a completa adição de TBOT, esta mistura foi mantida em agitação por 2 h na temperatura ambiente. Na última etapa, foram adicionados 18,8 mL de água e 0,7 mL de ácido nítrico concentrado e a mistura foi mantida em agitação por mais 2 h, na temperatura ambiente. Então, a mistura reacional foi transferida para um béquer e mantida a 60 ºC até a total gelatinização do sistema. O gel obtido foi seco em estufa a 110 ºC por 24 h. O material obtido foi triturado e peneirado para separar um lote de partículas com diâmetro entre 0,15 e 0,25 mm. Este material foi submetido a tratamento térmico a 500 ºC por 120 h com fluxo de ar (100 mL min-1). A síntese para obtenção dos materiais STb e STc seguiu o mesmo procedimento de preparação do STa, diferindo apenas nas quantidades de TBOT adicionadas. Para o STb e STc foram adicionados 28,0 e 42,0 mL de TBOT, respectivamente. Caracterização A determinação dos teores de SiO2 e TiO2 nos materiais foi realizada pela técnica de fluorescência de raios-X por energia dispersiva (EDXRF), em um equipamento Shimadzu 720HS Ray-Ny, cujos dados foram tratados pelo método matemático dos parâmetros fundamentais. A área específica (AE) e o volume médio dos poros (Vp) foram determinados pelo uso do equipamento Quantachrome Autosorb 1 (ASAP - Surface Area and Porosity Analyser), sendo os dados tratados pelos métodos de BJH e BET. As amostras foram previamente tratadas a 120 ºC sob vácuo por 6 h. Os espectros de infravermelho com transformada de Fourier (FTIR) dos materiais foram obtidos em um espectrômetro Shimadzu FTIR-8300 com pastilha de KBr a 1% em massa após 240 acumulações. Para a identificação de fases cristalinas presentes nos materiais, foram realizadas análises de difração de raios-X de pó (XRD) em um difratômetro PANalytical X'Pert Pro MPD, em um intervalo de 5º < 2θ < 70º, com passo angular de 0,05º, operando a 30 kV e 20 mA, com monocromador de grafite e radiação CuKα. Os espectros Raman-FTIR dos materiais ST foram obtidos em um equipamento Bruker RAM II, com excitação em 1064 nm, a 500 mW, resolução de 4 cm-1, varredura de 400 a 4000 cm-1 após 260 acumulações. Para o cálculo do band gap (Bg), foram obtidos os espectros de refletância difusa (DRS) dos materiais em um espectrofotômetro Ocean Optics USB2000+ com sonda para backscattering e software integrado Spectra Suite. A função de Kubelka-Munk e o modelo de Tauc foram aplicados para a obtenção dos valores de band gap dos materiais nanoestruturados. Os espectros de fotoluminescência dos materiais ST foram obtidos utilizando-se um espectrofotômetro Ocean Optics mini USB 2000+, com laser de Xe (405 nm), potência de 0,31 mW e filtro de 435 nm. As imagens das partículas dos materiais preparados foram obtidas por microscopia eletrônica de varredura (MEV) utilizando-se um microscópio eletrônico de varredura Phillips FEI Quanta 200, com porta amostra de alumínio, sendo as amostras fixadas com fita de carbono e recobertas com ouro, por meio de um metalizador Balzers MED 020. As imagens de microscopia eletrônica de transmissão em alta resolução (HRTEM) foram realizadas em um microscópio eletrônico JEOL JEM-3010, operando a 300 kV. Estudos de fotodegradação Os corantes utilizados para os testes fotocatalíticos foram o azul de metileno - AM (Basic Blue 9, CAS - 12270-13-2, SYNTH), o azul royal GRL - AR (Basic Blue 41, CAS - 12270-13-2, SIGMA-ALDRICH) e o amarelo ouro GL - AO (Basic Yellow 28, CAS - 54060-92-3), sendo que o último corante foi gentilmente cedido pela Indústria Marisol S.A. de Florianópolis em Santa Catarina. As estruturas das moléculas dos corantes são mostradas na Figura 1.
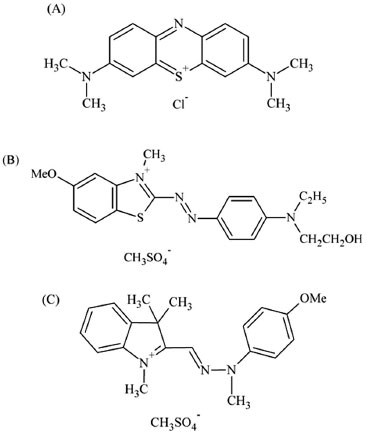 Figura 1. Estrutura dos corantes AM (A), AR (B) e AO (C)
Inicialmente, testes preliminares foram realizados expondo soluções aquosas dos corantes à luz solar natural por 8 h, na ausência de fotocatalisador e na presença de 0,1 g de sílica (Davisil 635 - Sigma-Aldrich) para se avaliar a fotoestabilidade dos corantes. Os testes mostraram que os corantes apresentam uma alta estabilidade à irradiação solar e não foram detectados sinais de fotodegradação dos corantes após este período de exposição solar. Os estudos de fotodegradação foram realizados utilizando-se 15,0 mL de uma solução aquosa dos corantes AM, AR e AO na concentração de 1,0 × 10-5 mol L-1 em várias placas de Petri, na presença de 0,1 g do fotocatalisador, e o conjunto foi exposto à luz solar natural. A irradiação solar foi realizada nas proximidades do Departamento de Química da Universidade Estadual de Londrina - UEL (S: 23º 19' 36,7''/ W: 51º 12' 5,5''), no período de agosto a novembro, entre 9 e 15 h (2500-4000 µW cm-2 para λ= 290-390 nm) medidos por um radiômetro MRU-201 Instrutherm. A cada intervalo de tempo pré-determinado, uma placa do conjunto era retirada da exposição solar, e desta placa era retirada uma alíquota de 3,0 mL para ser analisada em espectrofotômetro UV-Vis, Thermo-Scientific Genesys II, utilizando-se uma cubeta de quartzo de 1,0 cm de caminho ótico. A fotodegradação dos corantes em solução foi monitorada por meio das bandas de absorção 664, 609 e 430 nm dos corantes AM, AR e AO, respectivamente. A porcentagem de remoção da cor dos corantes em solução foi obtida aplicando-se a Equação 1.13 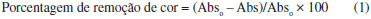 sendo Abso igual ao valor da absorvância da solução inicial e Abs igual ao valor da absorvância após um determinado tempo de irradiação. Para a comprovação do grau de mineralização das moléculas orgânicas, a quantidade de carbono orgânico total (TOC) foi determinada nos extratos, por meio de um equipamento Shimadzu TOC-V CPN. Recuperação do fotocatalisador A reciclagem e reutilização do fotocatalisador STc também foi avaliada, a fim de averiguar alguma perda de eficiência do material. Assim, no presente estudo, o fotocatalisador STc, após a sua utilização nos experimentos de fotocatálise, foi filtrado em papel filtro, lavado exaustivamente com água e transferido para um béquer com água, no qual foi exposto durante 5 horas sob a irradiação solar. Após este tempo o fotocatalisador foi filtrado, lavado com água novamente, seco a 60 ºC e guardado em frasco de plástico escuro para nova utilização.
RESULTADOS E DISCUSSÃO Nesta seção, é apresentada a caracterização dos materiais sintetizados, a fim de investigar a sua estrutura química e morfologia, assim como suas propriedades óticas. Posteriormente, é avaliado o potencial de fotodegradação dos corantes têxteis azul de metileno, azul royal GRL e amarelo ouro GL pelo material STc e a luz solar natural e também os fatores que podem influenciar neste processo, como o pH da solução de corante e a concentração do corante em solução. A cinética do processo de fotodegradação também é avaliada. Caracterização Os teores de SiO2 e TiO2 obtidos por EDXRF nos materiais STa, STb e STc são mostrados na Tabela 1.
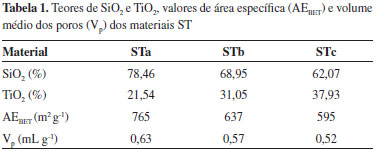
As isotermas de adsorção e desorção de N2 para os materiais Sta, STb e STc são mostradas na Figura 1S - material suplementar on line. De acordo com a classificação da IUPAC,40 os perfis para os materiais sintetizados são característicos de sólidos microporosos. A área específica e o volume médio dos poros dos materiais preparados são apresentados na Tabela 1. Percebe-se pelos valores mostrados na Tabela 1 que conforme a concentração de Ti aumenta na matriz, ocorre a diminuição no volume médio dos poros, assim como na área específica. Isto pode ser explicado pelo fato de a matriz de sílica ser altamente irregular e a inserção de TiO2 provocar um ordenamento na estrutura da sílica amorfa, diminuindo a área específica e o volume médio dos poros.41,42 Um espectro de FTIR dos materiais (Figura 2S - material suplementar on line) foi obtido para confirmar a efetividade do tratamento térmico na remoção de resíduos orgânicos dos precursores utilizados na síntese. O espectro apresenta bandas em torno de 3430 cm-1 e em 1635 cm-1 que podem ser atribuídas ao estiramento e à deformação angular do grupo -OH da molécula de água fisiossorvida.43,44 A banda próxima a 950 cm-1 pode ser atribuída à ligação Si-OH na superfície da matriz44 e, com o aumento da quantidade de Ti nos materiais, essa banda parece se deslocar para comprimentos de onda maiores. Isto pode estar relacionado à interação do Ti com a matriz, com o aparecimento de ligações Si-O-Ti e Si-O-Si. A banda larga centrada em 1080 cm-1 pode ser atribuída ao estiramento assimétrico, a banda de 800 cm-1 ao estiramento simétrico e a banda em 450 cm-1 à deformação angular das ligações Si-O-Si presentes na matriz.44-46 A ausência de bandas referentes às ligações C-H indica que o tratamento térmico a 500 ºC por 120 h foi suficiente para eliminar toda a parte orgânica resultante da síntese de sol-gel por catálise ácida. As imagens de MEV da partícula STc e os respectivos mapeamentos dos elementos Si e Ti são mostrados na Figura 2.
 Figura 2. Imagem de MEV e os mapeamentos dos elementos Si e Ti na partícula do STc
As imagens mostram que os elementos se encontram altamente dispersos pela partícula e nenhuma evidência da presença de aglomerados pode ser observada na magnificação utilizada. O espectro de refletância difusa foi obtido para cada material ST e a partir da aplicação da função de Kubelka-Munk e do modelo de Tauc, baseado na Equação 2,47-49 foram calculados os valores de band gap dos materiais.  Na equação 2 temos: α igual ao coeficiente de absorção da amostra; h igual à constante de Plank; ν igual à frequência; B igual ao fator de desordem e E igual à energia. Fazendo-se o gráfico (f(R) hν)1/2 versus E e a extrapolação da reta dos dados obtidos foi possível determinar os valores de band gap para os materiais ST, como mostrado na Figura 3S (material suplementar on line). Os valores obtidos do band gap (Bg) para os materiais foram: 3,20 ± 0,01, 2,92 ± 0,02 e 2,85 ± 0,01 eV para os materiais STa, STb e STc, respectivamente. Observa-se que quanto maior o teor de SiO2 no material maior será o valor de band gap. Isso pode ser devido ao fato de a sílica exercer um papel dispersante e isolante no material, provocando um efeito de confinamento quântico, ou seja, reduzindo o tamanho das partículas de TiO2 e consequentemente aumentando a energia de separação entre as bandas de valência e de condução.30 Os difratogramas dos materiais ST são mostrados na Figura 4S (material suplementar on line). Os espectros de difração de raios-X dos materiais ST mostram apenas o halo amorfo típico da sílica entre 15º e 30º (2θ), sem evidências de planos cristalinos nos materiais.50 Isso pode reforçar os indícios de que os cristalitos de TiO2 presentes no material são de fato de tamanho muito pequeno e estão bastante diluídos na matriz, não sendo possível identificá-los pelo método de difração de raios-X de pó convencional. Os espectros de Raman-FTIR dos materiais ST mostram a existência da fase anatase do TiO2 como pode ser visto na Figura 5S (material suplementar on line). As bandas em 516 cm-1 e 641 cm-1 podem ser atribuídas aos modos A1g e Eg do TiO2 na fase anatase.51-53 A banda em 960 cm-1 pode ser atribuída ao estiramento das ligações Si-OH na superfície54 dos materiais e, com o aumento do teor de Ti nas amostras, esta banda diminui a sua intensidade. Isto mostra que com o aumento da quantidade de titânio na matriz de sílica ocorrem alterações na superfície dos materiais, diminuindo a quantidade de grupos silanóis. Observa-se ainda que com a maior concentração de Ti no material os picos referentes à fase anatase tornam-se mais intensos, indicando que uma maior quantidade desta fase está presente no material ST. A imagem de HRTEM do material STc é apresentada na Figura 3.
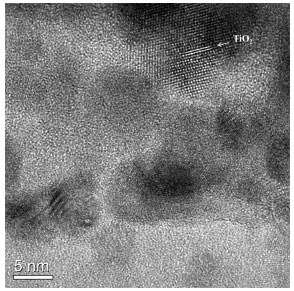 Figura 3. Imagem de HRTEM do material STc
Observa-se a existência de nanocristalitos com domínios entre 4 e 5 nm, confirmando a presença de nanoestruturas cristalinas no material preparado. Pelas distâncias interplanares medidas na imagem foi possível a identificação de um único plano cristalino, o plano 101 da fase anatase de TiO2 (JCPDS: 01-071-1166). Os espectros de fotoluminescência para os materiais ST e para o TiO2 comercial P-25 da Degussa são mostrados na Figura 4.
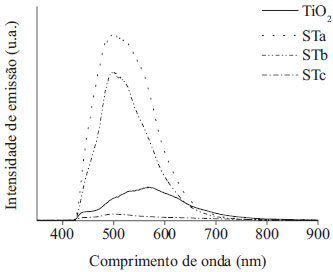 Figura 4. Espectros de emissao de fotoluminescência para os materiais ST e TiO2 (P- 25)
Os espectros indicam que um aumento na concentração de Ti na matriz provoca uma diminuição nas intensidades de fotoluminescência. Isto sugere que os materiais ST possuem propriedades ópticas diferentes dos seus óxidos puros, no caso o material STc apresenta uma intensidade de fotoluminescência menor que o TiO2 (P-25). Assim, o material nanoestruturado STc deve possuir uma menor taxa de recombinação (elétron/buraco) e, portanto, uma melhor eficiência quântica.55 Outro fato importante resultante da observação da Figura 4 é que existem processos quânticos ocorrendo em comprimentos de onda na faixa da luz visível. Isto reforça o interesse na utilização destes materiais em reações fotocatalíticas, pois uma vez que apresentam transições na faixa do visível, há a possibilidade de absorverem também em comprimentos de onda na faixa do visível. Podemos dizer então que estes materiais são capazes de absorver radiações na faixa do UV e do visível da luz solar natural. Estudos de fotodegradação Estudo preliminar Um teste fotocatalítico preliminar foi realizado com o corante azul de metileno (AM) em solução aquosa, para se estabelecer qual dos materiais ST apresentava o melhor desempenho na fotodegradação deste corante usando a luz solar natural. O experimento foi realizado da seguinte forma: 0,1 g dos materiais STa, STb e STc e 15,0 mL de uma solução aquosa de azul de metileno na concentração de 1,0 × 10-5 mol L-1 foram colocados em várias placas de Petri separadamente e estes conjuntos foram expostos à luz solar natural por 30 min. Após a irradiação solar, alíquotas do sobrenadante de cada placa foram retiradas e analisadas pela técnica do UV-Vis, utilizando-se a banda de 664 nm. Os valores obtidos de absorvância foram tratados pela Equação 1 e os resultados obtidos foram 72,6, 89,8 e 91,6% para os fotocatalisadores STa, STb e STc, respectivamente. A maior eficiência fotocatalítica do material STc pode ser atribuída à maior concentração de Ti e ao menor valor de band gap do material. Além disso, com a inserção de TiO2 em uma matriz de sílica, o material pode apresentar ligações do tipo Si-O-Ti-OH na superfície, o que promove o aparecimento de sítios com alta acidez de Bronsted.56 As regiões contendo as ligações Si-O-Ti-OH, por estarem muito próximas aos sítios fotoativos do TiO2, produtores de radicais hidroxila, podem favorecer a degradação das moléculas do corante. Desta forma, os materiais contendo estas ligações mostram-se mais efetivos na oxidação de compostos quanto comparados ao TiO2 e SiO2 isolados.56 Um esquema do mecanismo de fotodegradação do sistema SiO2/TiO2 é mostrado na Figura 5.
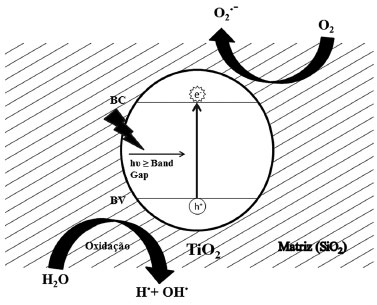 Figura 5. Esquema do mecanismo de fotocatálise no material ST
Fotodegradação dos corantes têxteis Por ter demonstrado o melhor desempenho fotocatalítico, o material STc foi o escolhido para os estudos de adsorção dos corantes têxteis catiônicos azul de metileno (AM), azul royal GRL (AR) e amarelo ouro GL (AO) na concentração de 1,0 × 10-5 mol L-1, 2,0 × 10-5 mol L-1 e 4,0 × 10-5 mol L-1, respectivamente. Neste estudo, foi empregado o mesmo procedimento para os estudos preliminares citados anteriormente, porém mantendo as soluções de corante em contato com o fotocatalisador STc durante 30 minutos no escuro. As quantidades de corante adsorvidas foram iguais a 0,057, 0,133 e 0,108 mmol g-1 para AM, AR e AO, respectivamente. A alta capacidade adsortiva deste material indica que isto pode ser um fator altamente favorável ao bom desempenho fotocatalítico, pois aumenta a difusão dos poluentes adsorvidos para os sítios fotoativos, que podem interagir melhor com as espécies oxidativas fotogeradas na superfície do catalisador e, portanto, serem mineralizados.30,57,58 Sabendo que o fotocatalisador STc adsorve rapidamente as moléculas do corante, os experimentos de irradiação solar foram conduzidos sem a etapa prévia de adsorção, pois a total saturação da superfície do catalisador pelas moléculas do corante poderia prejudicar a interação da luz com os sítios ativos, diminuindo a eficiência da fotodegradação. A seguir, o perfil de fotodegradação dos três corantes em solução aquosa em relação ao tempo de irradiação solar, a influência do pH da solução e concentração inicial do corante são apresentados na Figura 6.
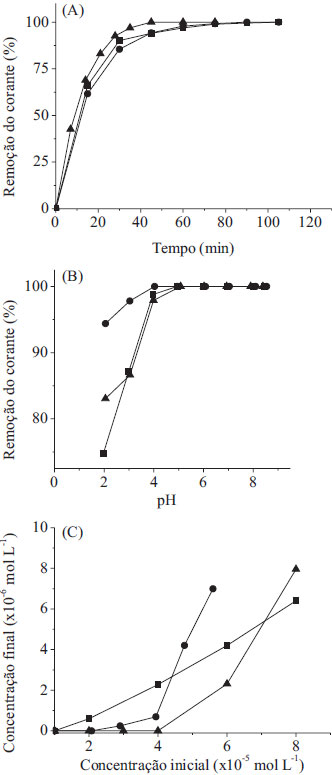 Figura 6. Fotodegradaçao dos corantes AM (■), AR (●) e AO (▲) em soluçao aquosa em funçao do tempo de exposiçao solar (A); variando-se o pH da soluçao de 2,0 a 8,5 e com o tempo fixo de 90 min para o AM e AR e 45 min de irradiaçao para AO (B) e o efeito da concentraçao inicial do corante, com tempo fixo de 90 min para o AM e AR e 45 min de irradiaçao para o AO (C) utilizando em todos os casos o fotocatalisador STc
Os corante AM e AR apresentaram um tempo de fotodegradação de 90 min e o corante AO um tempo de 45 min, como pode ser observado na Figura 6A. Características inerentes ao corante como estrutura das moléculas, natureza dos grupos funcionais, absortividade molar, solubilidade e pureza dos reagentes são fatores que podem atuar diretamente na fotodegradação desses compostos, resultando neste perfil de degradação. Os sobrenadantes, após os tempos de irradiação solar correspondentes à total degradação de cada corante, foram analisados por TOC, pelo método da combustão catalítica a alta temperatura, e o resultado obtido foi negativo para carbono orgânico, indicando que o resultado do UV-Vis pode ser utilizado neste caso como o final da fotodegradação das moléculas orgânicas de corante. Foi observado que o fotocatalisador não apresentava qualquer coloração após os tempos de irradiação solar, usados para a total degradação das moléculas dos corantes em solução. Desta forma, quando determinamos por UV-Vis o final da fotodegradação, a análise por TOC do extrato confirma a total mineralização do corante nestas condições experimentais pelo fotocatalisador STc. Efeito do pH A influência do pH da solução aquosa do corante no desempenho fotocatalítico é mostrada na Figura 6B. Para o estudo da influência do pH da solução aquosa de corante os tempos de irradiação solar foram fixos, sendo de 90 minutos para o AM e AR e de 45 minutos para o AO. De acordo com a Figura 6B, a fotodegradação é mais efetiva para valores de pH acima de 5,0 para os corantes AO e AM e acima de 4,0 para o corante AR. O valor do pH das soluções dos corantes, sem ajustes adicionais, é de 5,9. O perfil do comportamento da fotodegradação dos corantes em relação ao pH da solução indica que em valores menores de 4,0 (para AR) e de 5,0 (para AO e AM) existe uma forte competição entre os prótons da solução e as moléculas dos corantes catiônicos pelos sítios ativos da superfície do fotocatalisador, diminuindo assim a concentração das moléculas dos corantes sobre a superfície do fotocatalisador e consequentemente diminuindo a eficiência fotocatalítica.59,60 Em meio básico o processo de fotodegradação é mais eficiente, devido, provavelmente, ao fato das hidroxilas presentes no meio poderem reagir com os buracos fotogerados (h+) na superfície do fotocatalisador e produzir mais radicais hidroxila, conforme a Equação 3.61 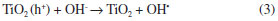 As reações em meio ácido proporcionam a formação de radicais hidroxila de forma bem mais lenta e em menor quantidade, como representado nas Equações 4 a 6.61 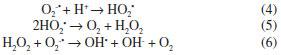 Efeito da variação da concentração inicial do corante Os estudos da variação da concentração inicial dos corantes também foram realizados fixando-se o tempo de irradiação solar. Para a solução de AM e AR o tempo de irradiação solar utilizado foi de 90 minutos e para as soluções de AO foi de 45 minutos. As concentrações das soluções dos corantes variaram de 1,0 × 10-5 a 8,0 × 10-5 mol L-1 para AM, de 1,0 × 10-5 a 5,6 × 10-5 mol L-1 para AR e de 1,0 × 10-5 a 8,0 × 10-5 mol L-1 para AO. De acordo com a Figura 6C, quanto maior a concentração inicial do corante menor a taxa de degradação nos tempos fixos utilizados. Considerando que a superfície do catalisador possui características adsorventes (sítios ácidos), um aumento da concentração dos corantes em solução provoca a diminuição da geração de radicais hidroxila na superfície dos catalisadores, uma vez que todos os sítios estão ocupados por moléculas de corante resultando, portanto, em menor eficiência fotocatalítica. Além disso, as moléculas de corante podem absorver a radiação UV e funcionar como uma barreira para a irradiação solar do fotocatalisador, consequentemente causando uma redução na produção de radicais como OH• e O2•.18,62 Estudo cinético Em estudos da cinética de fotodegradação de corantes, é comumente aceito que este processo obedeça a cinética de pseudo-primeira ordem.7,12,33,57,63 O modelo cinético de primeira ordem aparente pode ser representado pelo gráfico ln(C0/C) versus o tempo de exposição solar, onde C0 é a concentração inicial do corante e C é a concentração do corante após um determinado tempo de irradiação.33 Desta forma, a fim de verificar se este tipo de cinética se aplica ao presente caso, foram aplicados os modelos cinéticos de primeira ordem, segunda ordem e primeira ordem aparente aos dados experimentais. Os modelos de primeira ordem e de segunda ordem não apresentaram correlação com os dados do processo de fotodegradação dos corantes, somente o modelo de primeira ordem aparente. O gráfico ln(C0/C) versus t é mostrado na Figura 5S (material suplementar on line). Os coeficientes de correlação linear (R2) e as constantes de velocidade são mostrados na Tabela 1S (material sumplementar on line). Os valores da constante de velocidade obtidos foram 5,72 × 10-2 min-1 para o AM, 6,44 × 10-2 min-1 para o AR × 1,07 x 10-1 min-1 para o AO. Isto sugere que a absortividade molar dos corantes pode influenciar diretamente na cinética de fotodegradação, pois o corante AO que possui a menor absortividade molar dentre os corantes utilizados, como mostrado na Tabela 1S (material suplementar on line), é o corante que se degrada mais rapidamente (45 min). Recuperação e reutilização do fotocatalisador Um dos aspectos mais importante nos processos de fotodegradação é a facilidade de recuperação e a possibilidade de reutilização do fotocatalisador. Verificamos que o fotocatalisador STc pode ser reutilizado mais de 50 vezes sem perda significativa da sua eficiência fotocatalítica. A comparação dos tempos de contato necessários para a degradação total do corante AM no primeiro ciclo de utilização do material com o tempo de contato do ciclo número 50 demonstrou uma perda de eficiência menor do que 2%. Isto demonstra que o TiO2 na matriz de sílica é bastante estável, não sofrendo lixiviação do material STc.
CONCLUSÃO Os materiais SiO2/TiO2 (ST) foram sintetizados de forma simples e fácil pelo método sol-gel e os produtos obtidos apresentaram uma alta área específica com os elementos Si e Ti altamente e uniformemente dispersos por todo o material. Os materiais ST, por XRD de pó, não apresentam indicações de fases cristalinas, mesmo após o tratamento térmico de 500 ºC por 120 h. Os materiais ST por análise de HRTEM apresentaram domínios cristalinos na faixa entre 4 e 5 nm e a única fase cristalina identificada foi a anatase do TiO2. A fotodegradação dos corantes AM, AR e AO em solução aquosa utilizando o fotocatalisador STc e a luz solar natural, sem o uso de oxidantes fortes adicionais, resultou em total mineralização dos corantes. Fotocatalisadores do tipo STc apresentam grande potencial tecnológico prático para o tratamento final de efluentes industriais contendo corantes têxteis, pois não necessitam de oxidantes adicionais e muito menos de energia elétrica para a geração de radiação na fase do UV, apenas a luz solar natural.
MATERIAL SUPLEMENTAR As figuras citadas como material suplementar podem ser acessadas em http://quimicanova.sbq.org.br, na forma de arquivo PDF, com acesso livre.
AGRADECIMENTOS Os autores agradecem à CAPES (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior) e ao CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico) pelas bolsas concedidas. Aos Laboratórios Multiusuários de Difração de raios-X e de Microscopia Eletrônica e Microanálise da UEL (Universidade Estadual de Londrina) pelas análises. Ao Laboratório de Física Aplicada (Grupo de ótica e optoeletrônica) da UEL pelos espectros de fotoluminescência e também à Profa. Dra. F. Sato do Departamento de Física da UEM (Universidade Estadual de Maringá) pelos espectros de Raman-FTIR.
REFERÊNCIAS 1. Ahmed, S.; Rasul, M. G.; Martens, W. N.; Brown, R.; Hashib, M. A.; Water, Air, Soil Pollut. 2011, 215, 3. DOI: http://dx.doi.org/10.1007/s11270-010-0456-3 2. Qu, X.; Alvarez, P. J. J.; Li, Q.; Water Res. 2013, 47, 3931. DOI: http://dx.doi.org/10.1016/j.watres.2012.09.058 PMID: 23571110 3. Körbahti, B. K.; Artut, K.; Geçgel, C.; Özer A.; Chem. Eng. J. 2011, 173, 677. DOI: http://dx.doi.org/10.1016/j.cej.2011.02.018 4. Abdelaal, M. Y.; Mohamed, R. M.; J. Alloys Compd. 2013, 576, 201. DOI: http://dx.doi.org/10.1016/j.jallcom.2013.04.112 5. Kamboh, M. A.; Solangi, I. B.; Sherazi, S. T. H.; Memon, S.; Desalination 2011, 268, 83. DOI: http://dx.doi.org/10.1016/j.desal.2010.10.001 6. Zermane, F.; Cheknane, B.; Basly, J. P.; Bouras, O.; Baudu, M.; J. Colloid Interface Sci. (2013), doi:10.1016/j.jcis.2012.12.038 DOI: http://dx.doi.org/10.1016/j.jcis.2012.12.038 PMID: 23332940 7. Oliveira, D. F. M.; Batista, P. S.; Miller Jr, P. S.; Velani, V.; França, M. F.; Souza, D. R.; Machado, A. E. H.; Dyes Pigm. 2011, 92, 563. DOI: http://dx.doi.org/10.1016/j.dyepig.2011.06.007 8. Dotto, G. L.; Vieira, M. L. G.; Gonçalves, J. O.; Pinto, L. A. A.; Quim. Nova 2011, 34, 1193. DOI: http://dx.doi.org/10.1590/S0100-40422011000700017 9. Li, X.; Zhu, J.; Li, H.; Appl. Catal., B 2012, 123, 174. DOI: http://dx.doi.org/10.1016/j.apcatb.2012.04.009 10. Gümüs, D.; Akbal, F.; Water, Air, Soil Pollut. 2011, 216, 117. DOI: http://dx.doi.org/10.1007/s11270-010-0520-z 11. Chong, M. N.; Jin, B.; Chow, C. W. K.; Saint, C.; Water Res. 2010, 44, 2997. DOI: http://dx.doi.org/10.1016/j.watres.2010.02.039 PMID: 20378145 12. Rauf, M. A.; Ashraf, S. S.; Chem. Eng. J. 2009, 151, 10. DOI: http://dx.doi.org/10.1016/j.cej.2009.02.026 13. Frade, T.; Gomes, A.; Pereira, M. I. S.; Lopes, A.; Ciríaco, L.;Quim. Nova 2012, 35, 30. DOI: http://dx.doi.org/10.1590/S0100-40422012000100006 14. Nogueira, R. F. P.; Trovó, A. G.; Silva, M. R. A.; Villa, R. D.; Oliveira, M. C.; Quim. Nova 2007, 30, 400. DOI: http://dx.doi.org/10.1590/S0100-40422007000200030 15. Devi, L. G.; Kavitha, R.; Appl. Catal., B 2013, 140, 559. DOI: http://dx.doi.org/10.1016/j.apcatb.2013.04.035 16. Soutsas, K.; Karayannis, V.; Poulios, I.; Riga, A.; Ntampegliotis, K.; Spiliotis, X.; Papapolymerou, G.; Desalination 2010, 250, 345. DOI: http://dx.doi.org/10.1016/j.desal.2009.09.054 17. Nakamura, L. K. O.; Vaz, J. M.; Quim. Nova 2011, 34, 1173. DOI: http://dx.doi.org/10.1590/S0100-40422011000700013 18. Mahvi, A. H.; Ghanbarian, M.; Nasseri, S.; Khairi, A.; Desalination 2009, 239, 309. DOI: http://dx.doi.org/10.1016/j.desal.2008.04.002 19. Gözmen, B.; Turabik, M.; Hesenov, A.; J. Hazard. Mater. 2009, 164, 1487. DOI: http://dx.doi.org/10.1016/j.jhazmat.2008.09.075 PMID: 18995958 20. Behnajady, M. A.; Eskandarloo, H.; Modirshahla, N.; Shokri, M.; Desalination 2011, 278, 10. DOI: http://dx.doi.org/10.1016/j.desal.2011.04.019 21. Sakulkhaemaruethai, S.; Sreethawong, T.; Int. J. Hydrogen Energy 2011, 36, 6553. DOI: http://dx.doi.org/10.1016/j.ijhydene.2011.03.005 22. Ramirez, A. M.; Demeestere, K.; Belie, N.; Mäntylä, T.; Levänen, E.; Building and Environment 2010, 45, 832. DOI: http://dx.doi.org/10.1016/j.buildenv.2009.09.003 23. Prestes, T. H.; Gibbon, D. O.; Lansarin, M. A.; Moro, C. C.; Quim. Nova 2010, 33, 798. DOI: http://dx.doi.org/10.1590/S0100-40422010000400006 24. Bessegato, G. G.; Santos, V. P.; Lindino, C. A.; Quim. Nova 2012, 35, 332. DOI: http://dx.doi.org/10.1590/S0100-40422012000200019 25. Silva, W. L.; Lansarin, M. A.; Moro, C. C.; Quim. Nova 2013, 36, 382. DOI: http://dx.doi.org/10.1590/S0100-40422013000100026 26. Maeda, M.; Hirota, K.; Appl. Catal., A 2006, 302, 305. DOI: http://dx.doi.org/10.1016/j.apcata.2006.01.032 27. Mourão, H. A. J. L.; Mendonça, V. R.; Malagutti, A. R.; Ribeiro, C.; Quim. Nova 2009, 32, 2181. DOI: http://dx.doi.org/10.1590/S0100-40422009000800032 28. Han, C.; Pelaez, M.; Likodimos, V.; Kontos, A. G.; Falaras, P.; O'Shea, K.; Dionysiou, D. D.; Appl. Catal., B 2011, 107, 77. DOI: http://dx.doi.org/10.1016/j.apcatb.2011.06.039 29. Qourzal, S.; Barka, N.; Tamimi, M.; Assabbane, A.; Nounah, A.; Ihlal, A.; Ait-Ichou, Y.; Mater. Sci. Eng., C 2009, 29, 1616. DOI: http://dx.doi.org/10.1016/j.msec.2008.12.024 30. Meng, X.; Qian, Z.; Wang, H.; Gao, X.; Zhang, S.; Yang, M.; J. Sol-Gel Sci. Technol. 2008, 46, 195. DOI: http://dx.doi.org/10.1007/s10971-008-1677-4 31. Yin, B.; Wang, J.-T; Wei, X.; Long, D.-H.; Qiao, W.-M.; Ling, L.-C.; New Carbon Mater. 2013, 28, 47. DOI: http://dx.doi.org/10.1016/S1872-5805(13)60064-5 32. Yang, G.; Yan, Z.; Xiao, T.; Appl. Surf. Sci. 2012, 258, 8704. DOI: http://dx.doi.org/10.1016/j.apsusc.2011.12.027 33. Lu, N.; Zhao, Y.; Liu, H.; Guo, Y.; Yuan, X.; Xu, H.; Peng, H.; Qin, H.; J. Hazard. Mater. 2012, 199, 1. DOI: http://dx.doi.org/10.1016/j.jhazmat.2011.08.070 PMID: 22115842 34. Choi, J.; Park, H.; Hoffmann, M. R.; J. Phys. Chem. C 2010, 114, 783. DOI: http://dx.doi.org/10.1021/jp908088x 35. Xu, J.-J.; Chen, M.-D.; Fu, D.-G; Trans. Nonferrous Met. Soc. China 2011, 21, 340. DOI: http://dx.doi.org/10.1016/S1003-6326(11)60719-X 36. Macwan, D. P.; Dave, P. N.; Chaturvedi, S.; J. Mater. Sci. 2011, 46, 3669. DOI: http://dx.doi.org/10.1007/s10853-011-5378-y 37. Wang, D.; Bierwagen, G. P.; Prog. Org. Coat. 2009, 64, 327. DOI: http://dx.doi.org/10.1016/j.porgcoat.2008.08.010 38. Debecker, D. P.; Hulea, V.; Mutin, P. H.; Appl. Catal., A 2013, 451, 192. DOI: http://dx.doi.org/10.1016/j.apcata.2012.11.002 39. Alfaya, A. A. S.; Gushikem, Y.; Castro, S. C.; Chem. Mater. 1998, 10, 909. DOI: http://dx.doi.org/10.1021/cm970679e 40. Sing, K. S. W.; Everett, D. H.; Haul, R. A. W.; Moscou, L.; Pierotti, R. A.; Rouquerol, J.; Siemieniewska, T.; Pure & Appl. Chem. 1985, 57, 603. 41. Kim, Y. K.; Kim, E. Y.; Whang, C. M.; Kim, Y. H.; Lee, W. I.; J. Sol-Gel Sci. Technol. 2005, 33, 87. DOI: http://dx.doi.org/10.1007/s10971-005-6693-z 42. Jung, K. Y.; Park, S. B.; J. Photochem. Photobiol., A 1999, 127, 117. DOI: http://dx.doi.org/10.1016/S1010-6030(99)00151-3 43. Li, G.; Liu, F; Zhang, Z.; J. Alloys Compd. 2010, 493, L1. DOI: http://dx.doi.org/10.1016/j.jallcom.2009.12.086 44. Yuan, Q.; Li, N.; Jinchun, T.; Li, X.; Wang, R.; Zhang, T.; Shao, C.; Sens. Actuators, B 2010, 149, 413. DOI: http://dx.doi.org/10.1016/j.snb.2010.06.036 45. Chun, H.; Yizhong, W.; Hongxiao, T.; Appl. Catal., B 2001, 30, 277. DOI: http://dx.doi.org/10.1016/S0926-3373(00)00237-X 46. Zhang, H.; Qiao, Y.; Zhang, X.; Fang, S.; J. Non-Cryst. Solids 2010, 356, 879. DOI: http://dx.doi.org/10.1016/j.jnoncrysol.2010.01.003 47. Murphy, A. B.; Sol. Energy Mater. Sol. Cells 2007, 91, 1326. DOI: http://dx.doi.org/10.1016/j.solmat.2007.05.005 48. Štengl, V.; Králová, D.; Int. J. Photoenergy (2011), doi:10.1155/2011/532578. DOI: http://dx.doi.org/10.1155/2011/532578 49. Müllerová, J.; Prušáková, L; Netrvalová, M.; Vavruňková, V.; Šutta, P.; Appl. Surf. Sci. 2010, 256, 5667. DOI: http://dx.doi.org/10.1016/j.apsusc.2010.03.022 50. Strauss, M.; Maroneze, C. M.; Silva, J. M. S.; Sigoli, F. A.; Gushikem, Y.; Mazali, I O.; Mater. Chem. Phys. 2011, 126, 188. DOI: http://dx.doi.org/10.1016/j.matchemphys.2010.11.041 51. Sedpho, S.; Wongratanaphisan, D.; Mangkorntong, P.; Mangkorntong, N.; Choopun, S.; CMU. J. Nat. Sci. Special Issue on Nanotechnology 2008, 7, 99. 52. Zhang, W. F.; He, Y. L.; Zhang, M. S.; Yin, Z.; Chen, Q.; J. Phys. D: Appl. Phys. 2000, 33, 912. DOI: http://dx.doi.org/10.1088/0022-3727/33/8/305 53. Silva, S. S.; Magalhães, F.; Sansiviero, M. T. C.; Quim. Nova 2010, 33, 85. DOI: http://dx.doi.org/10.1590/S0100-40422010000100025 54. Liu, Z.; Davis, R. J.; J. Phys. Chem. 1994, 98, 1253. DOI: http://dx.doi.org/10.1021/j100055a035 55. Long, M.; Cai, W.; Cai, J.; Zhou, B.; Chai, X.; Wu, Y.; J. Phys. Chem. B 2006, 110, 20211. DOI: http://dx.doi.org/10.1021/jp063441z PMID: 17034198 56. Anderson, C.; Bard, A. J.; J. Phys. Chem. B 1997, 101, 2611. DOI: http://dx.doi.org/10.1021/jp9626982 57. Ong, S-A.; Min O-M.; Ho, L-N.; Wong, Y-S.; Environ. Sci. Pollut. Res. 2013, 20, 3405. DOI: http://dx.doi.org/10.1007/s11356-012-1286-1 58. Subash, B.; Krishnakumar, B.; Swaminathan, M.; Shanthi, M.; Langmuir 2013, 29, 939. DOI: http://dx.doi.org/10.1021/la303842c PMID: 23234536 59. Shimizu, N.; Ogino, C.; Dadjour, M. F.; Murata, T.; Ultrason. Sonochem. 2007, 14, 184. DOI: http://dx.doi.org/10.1016/j.ultsonch.2006.04.002 PMID: 16753327 60. Djokić, V.; Vujović, J.; Marinković, A.; Petrović, R.; Janaćković, D.; Onjia, A.; Mijin, D.; J. Serb. Chem. Soc. 2012, 77, 1747. DOI: http://dx.doi.org/10.2298/JSC121015130D 61. Mijin, D.; Radivojević, J.; Jovančić, P.; CI&CEQ 2007, 13, 33. DOI: http://dx.doi.org/10.2298/CICEQ0701033M 62. Clausen, D. N.; Takashima, K.; Quim. Nova 2007, 30, 1896. DOI: http://dx.doi.org/10.1590/S0100-40422007000800019 63. Marugán, J.; López-Muñoz, M-J.; Grieken, R. V.; Aguado, J.; Ind. Eng. Chem. Res. 2007, 46, 7605. DOI: http://dx.doi.org/10.1021/ie070093u |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access