
 |
 |
 |
 |
 |
Artigo
|
|
| Síntese de N-glicosilsulfonamidas derivadas de d-glicose e N-acetilglicosamina Synthesis of d-glucose- and N-acetylglucosamine-based N-glycosylsulfonamides |
|
Lucas L. FrancoI; Mário C. BrandaoII; José D. S. FilhoI; Ricardo J. AlvesII,*
IDepartamento de Química, Instituto de Ciências Exatas, Universidade Federal de Minas Gerais, 31270-901 Belo Horizonte - MG, Brasil Recebido em 13/03/2015 *e-mail: ricardodylan@farmacia.ufmg.br Herein, we report the synthesis of β-N-glycosylsulfonamides derivatives of D-glucose and N-acetylglucosamine using conventional methods. We also describe a procedure that allows the preparation of these compounds in good yields without the anomerization of the intermediate glycosylamines. This method includes the intermediates obtained from the less reactive 1- and 2-naphthalenesulfonyl chlorides. INTRODUÇÃO A importância dos carboidratos como fonte de energia e como componentes estruturais da parede celular de diversos organismos é, há muito, reconhecida.1 Atualmente, é sabido que os carboidratos são, também, encontrados como constituintes de diversas proteínas (glicoproteínas) e lípides (glicolípides), presentes em ambientes intra- e extracelulares. Tais compostos são denominados, genericamente, glicoconjugados. A participação dos glicoconjugados em processos de reconhecimento e adesão celular é de grande importância em diversas condições fisiológicas e patológicas, tais como manutenção da integridade de tecidos, rolamento de leucócitos em processos inflamatórios, metástase, infecções.1 Diferentes enzimas e proteínas transportadoras estão envolvidas na biossíntese e processamento de oligo- e polissacarídeos como amido, glicogênio (fontes de energia), ácido teicóico, peptideoglicanas (constituintes de parede celular bacteriana), quitina, 1,3- e 1,6-α-d-glicanas, d-mananas (constituintes de parede celular fúngica), gangliosídeos (componentes de membrana celular de células). Assim, a interferência no funcionamento das enzimas e proteínas transportadoras tem profunda influência na biologia dos organismos afetados, com implicações que podem ter importância terapêutica. Tal interferência pode ser ocasionada por defeitos genéticos, geralmente relacionados à não expressão ou expressão de forma defeituosa da enzima ou proteína transportadora.1,2 Outra forma de interferência nesse processo é com o uso de inibidores enzimáticos ou dos transportadores. Nesse sentido, a síntese e avaliação de derivados de carboidratos como inibidores enzimáticos ou como inibidores do transporte de carboidratos, principalmente d-glicose, tem sido objeto de pesquisa de diversos grupos.2-8 N-glicosilssulfonamidas, derivados sacarídicos em que a hidroxila anomérica é substituída por um grupo sulfonamido, têm se apresentado como uma classe de compostos bioativos.3-8 Além disso, sua utilização como bioisósteros de N-glicosilamidas em peptídeos e proteínas também tem sido estudadas.6 A síntese dessa classe de substâncias é de grande interesse e tem sido relatada por diversos grupos, inclusive o nosso.3-8 No presente trabalho relatamos a síntese de N-glicosilssulfonamidas derivadas de d-glicose e d-N-acetilglicosamina, dois dos principais monossacarídeos encontrados em oligo- ou polissacarídeos e glicoconjugados de interesse biológico e farmacológico.1 Especificamente, descreve-se a síntese das substâncias 1-20, cujas estruturas são mostradas na Figura 1. As substâncias 11-20, em que os carboidratos apresentam os grupos hidroxila livres, são inibidores enzimáticos potenciais, ao passo que os derivados peracetilados 1-10, precursores sintéticos daqueles, seriam pró-fármacos, isto é, substâncias inativas per se que seriam ativadas in vivo por processos enzimáticos, no caso, pela ação de esterases. De fato, há vários relatos na literatura em que derivados peracetilados de carboidratos apresentam atividade in vivo superior à dos derivados correspondentes desacetilados.9-11 Isto tem sido atribuído ao fato de que os derivados acetilados, mais lipofílicos, atravessariam mais facilmente as membranas celulares e, uma vez no interior das células seriam convertidas nos derivados hidroxilados ativos pela ação de esterases.
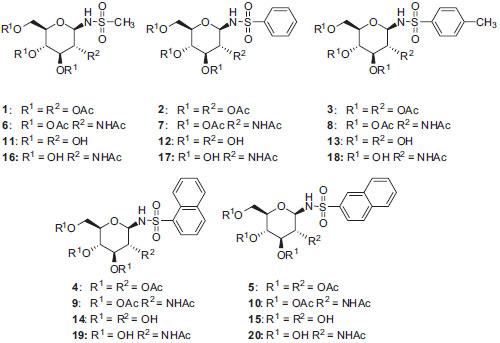 Figura 1. N-glicosilsulfonamidas derivadas de d-glicose e N-acetilglicosamina
Neste trabalho foi planejada a síntese dos compostos 1-20 que estão apresentados na Figura 1, os derivados 1-10 são precursores das substâncias 11-20.
RESULTADOS E DISCUSSÃO Inicialmente planejou-se a obtenção das N-glicosilsulofonamidas 1-10 por uma metodologia já utilizada pelo grupo no trabalho de Butera e colaboradores,7 denominado Método 1, conforme esquema de síntese representado na Figura 2.
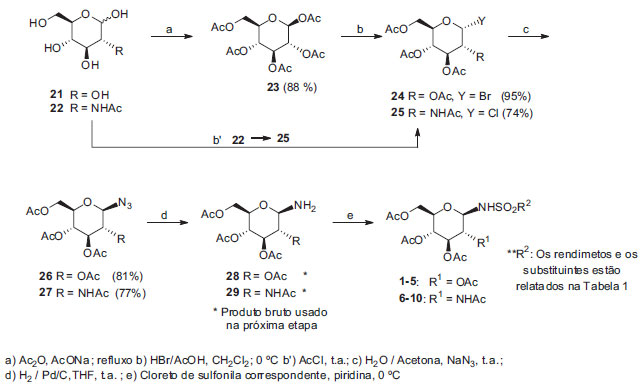 Figura 2. Esquema de síntese das N-glicosilsulfonamidas 1-10 pelo método 1
Método 1 Os tempos de reação e respectivos rendimenos são mostrados na Tabela 1.
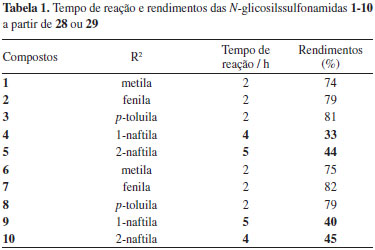
Pode-se observar que para a obtenção de 4, 5, 9 e 10 foram necessárias quatro a cinco horas de reação, tempos superiores aos dos demais derivados. Além disso, esses compostos foram obtidos com rendimentos inferiores a 50%. Já é relatada na literatura a baixa reatividade de cloretos de 1-naftalenossulfonila, por questões estéricas.12 Como nesse trabalho os derivados 5 e 10 foram obtidos com maior tempo de reação e baixos rendimentos e o reagente cloreto de 2-naftalenossulfonila tem um menor impedimento estérico em relação ao cloreto de 1-naftalenossulfonila, também é proposto que a baixa reatividade desses derivados é devida, em parte, a efeitos eletrônicos, pela possiblidade de formação de um híbrido de ressonância contendo um carbocátion benzílico. Essa baixa reatividade dos cloretos de naftalenossulfonila permitiu a anomerização de 28 e 29 durante as reações e por isso as N-glicosilsulfonamidas 4, 5, 9 e 10 foram obtidas como misturas anoméricas α:β (~1:3), conforme observado por RMN de 1H da Figura 3A. A recristalização permitiu a obtenção dos derivados desejados na forma anomérica pura (β), como mostrado no espectro da Figura 3B, mas com rendimentos significativamente inferiores aos dos demais derivados.
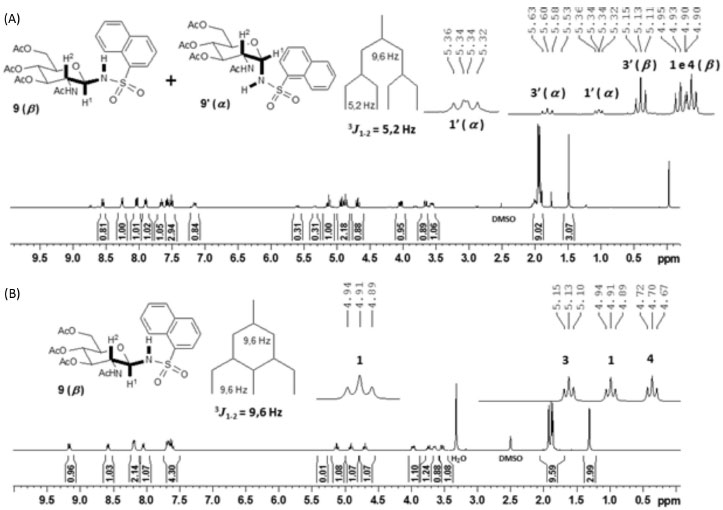 Figura 3. A. Espectro de RMN de 1H da mistura anomérica α:β (~1:3) de 9; B. Espectro de RMN de 1H de 9 na forma anomérica pura (β) (400 MHz, DMSO-d6)
Com a finalidade de melhorar os rendimentos das N-glicosilssulfonamidas naftalênicas realizou-se uma nova tentativa de síntese de acordo com o trabalho relatado por Colinas e colaboradores,13 que consiste na obtenção de N-glicosilsulfonamidas por meio da reação de uma sulfonamida com o respectivo derivado peracetilado de d-glicose, d-galactose, d-manose, entre outros, catalisada por BF3.Et2O. Esse método, denominado Método 2, foi avaliado, nesse trabalho, a partir dos derivados peracetilados de d-glicose e N-acetilglicosamina com a p-toluenossulfonamida e 2-naftalenossulfonamida. O esquema de síntese está representado na Figura 4.
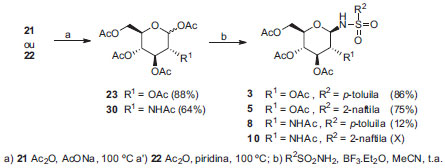 Figura 4. Esquema de síntese para a obtençao de N-glicosilsulfonamidas usando como catalizador BF3.Et2O
Método 2 Os derivados de d-glicose 3 e 5 foram obtidos com rendimentos de 87% e 75% respectivamente, mas o derivado 8 foi obtido com rendimento de 12% e o composto 10 não foi obtido por esse método. Os reagentes p-toluenosulfonamida e 2-naftalenosulfonamida foram recuperados em grandes proporções em ambos os casos (75% e 91% respectivamente). A proposta para explicar a ineficiência desse método para derivados de N-acetilglicosamina é a participação do grupo acetamido de C-2, que poderia levar à formação de um íon oxazolínio, pouco reativo.14 Após elaboração e purificação foi obtido o produto de hidrólise em C-1, conforme apresentado no esquema de síntese da Figura 5.
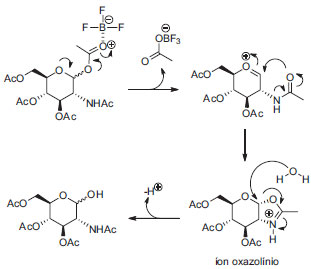 Figura 5. Mecanismo proposto para degradaçao de 30
Embora esse método tenha uma grande eficiência para obtenção de N-glicosilssulfonamidas em geral e tenha permitido a obtenção de 5 com rendimento satisfatório, não se mostrou eficiente para derivados de N-acetilglicosamina. Considerando que a baixa reatividade dos cloretos de naftalenossulfonila levou à anomerização das glicosilaminas 28 e 29 pelo Método 1, com a formação de misturas anoméricas, foi planejada uma alteração no referido método. A modificação, denominada Método 3, consistiu na conversão de 28 e 29 nos sais de amônio correspondentes 31 e 32 a partir da redução dos respectivos derivados 26 e 27, na presença de ácido p-toluenossulfônico, seguida da reação daqueles com os respectivos cloretos de sulfonila em diclorometano, com adição gradual de trietilamina. Os sais de amônio 31 e 32 são obtidos como anômeros β, puros, tanto pelo fato de os precursores azido serem de configuração β bem como pelo fato de que a protonação previne a anomerização. Além disso, sais de amônio preferem a posição β, pelo efeito anomérico reverso.13 A adição gradual de trietilamina levaria à formação de pequenas quantidades de amina livre, que reagiria mais rapidamente com os cloretos de sulfonila, proporcionalmente em excesso no meio. Com essa modificação finalmente foi possível obter N-glicosilsufonamidas naftalênicas na forma anômérica β com bons rendimentos. Como esperado, as sulfonamidas 3 e 8 também foram obtidas com rendimentos comparáveis àqueles obtidos no Método 1. O esquema de síntese correspondente está representado na Figura 6.
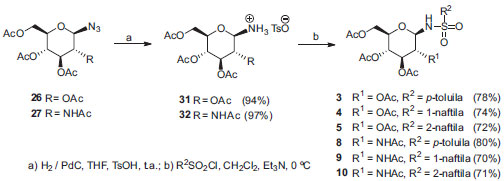 Figura 6. Esquema de síntese para obtençao de N-glicosilssufonamidas pelo Método 3
Método 3 Por fim todos os compostos obtidos 1-10 foram convertidos nas N-glicosilsulfonamidas 11-20 utilizando-se o método de Zémplen,15-17 conforme mostrado no esquema da Figura 7.
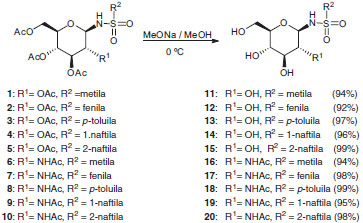 Figura 7. Esquema de síntese para obtençao das N-glicosilssulfonamidas 11-20
As N-glicosilssulfonamidas 11-20 foram purificadas por recristalização com rendimentos que variaram de 92% a 99%.
CONCLUSÃO Foram sintetizadas vinte N-glicosilsulfonamidas derivadas de d-glicose e N-acetilglicosamina, dez peracetiladas 1-10 e os respectivos correspondentes desacetilados 11-20, 12 delas são inéditas: 4, 5, 7, 9, 10, 12, 14, 15, 17, 18, 19, 20. Pelo Método 1 todas as substâncias planejadas foram obtidas, mas com baixos rendimentos para os derivados naftalênicos (4, 5, 9 e 10). Embora o Método 2 tenha possibilitado a obtenção de N-glicosilsulfonamidas com um número reduzido de etapas, ele não se mostrou eficiente para derivados de N-acetilglicosamina. Um novo procedimento, denominado no presente trabalho Método 3, resultante de modificação do Método 1, permitiu a obtenção das N-glicosilssulfonamida naftalênicas com rendimentos significativamente superiores àqueles obtido com o Método 1, sendo, portanto, um novo procedimento para a obtenção de glicosilsulfonamidas.
PARTE EXPERIMENTAL As faixas de fusão foram determinadas em aparelho MQAPF 301. Os espectros no infravermelho foram obtidos em aparelho Spectrum One, Perkin-Elmer com sistema ATR. Os valores de poder rotatório específico, [α]D, foram medidos em polarímetro ADP220 Bellinghan à temperatura de 20 ºC. Os espectros de RMN de 1H e de 13C foram registrados em aparelhos Bruker Avance DPX-200 e Bruker Avance DPX-400. Como referência interna foi utilizado o tetrametilsilano (TMS) ou o sinal do solvente. A evolução das reações foi acompanhada por cromatografia em camada delgada de sílica (CCD) com 0,25 mm de espessura de sílica (sílica gel 60G Merck) utilizando-se como revelador vapor de iodo, solução etanólica de ninidrina 0,3% p/v ou solução etanólica de ácido sulfúrico 15% v/v. As purificações por cromatografia em coluna de sílica (CCS) foram realizadas com sílica gel 60 (0,063-0,200 mm / 70-230 mesh Merck). Os reagentes utilizados neste trabalho foram adquiridos das empresas Sigma-Aldrich ou Merck. Os solventes utilizados durante esse trabalho foram obtidos comercialmente das empresas: F-Maia, Synth e Merck. Todos os solventes utilizados neste trabalho foram previamente destilados e tratados segundo procedimentos descritos na literatura.18,19 Os compostos 21-31 foram preparados conforme procedimentos descritos na literatura.7,8,12,20-27 1,2,3,4,6-penta-O-acetil-β-d-glicopiranose (23)7,25,26 Em um gral de vidro foram triturados 4 g de AcONa (48,76 mmol) e 5 g de d-glicose (27,74 mmol). Transferiu-se o material para um balão de fundo rendondo de 250 mL e adicionaram-se 25 mL de Ac2O (264,47 mmol). A mistura foi mantida sob agitação magnética e aquecimento (100 ºC) por 3 horas. O monitoramento da reação foi feito por CCD (Hexano 6:4 AcOEt). O produto bruto foi vertido em um béquer contendo cerca de 20 g de gelo picado e mantido sob agitação magnética por 1 hora. Foi observada a formação de um precipitado branco que foi filtrado e recristalizado em EtOH. Foram obtidos 9,38 g de 23 (88%). Brometo de 2,3,4,6-tetra-O-acetil-β-d-glicopiranosila (24)7 Em um balão de fundo redondo 250 mL foram solubilizados 4 g (10 mmol) de 23 em 25 mL de CH2Cl2, e, em seguida, adiconou-se uma solução previamente preparada de HBr em AcOH (25% v/v), gota a gota, a 0 ºC. A solução foi mantida sob agitação magnética a 0 ºC por 5 horas. O monitoramento da reação foi feito por CCD (Hexano 7:3 AcOEt). Após observação do consumo completo de 23 verteu-se o material em um béquer contendo cerca de 20 g de gelo picado e foi feita três extrações com 20 mL de CH2Cl2. A fase orgânica foi lavada com duas frações de 20 mL de solução saturada de NaHCO3 e duas frações de 20 mL de água destilada. Em seguida a fase orgânica foi secada com Na2SO4, filtrada e concentrada em evaporador rotatório. Foram obtidos 4 g de 24 (95%) na forma de um sólido branco. Cloreto de 2-acetamido-1,2-didesoxi-3,4,6-tri-O-acetil-α-d-glicopiranosila (25)23 Em um balão de fundo redondo 50 mL foram adicionados 2 g de 21 (9 mmol) sob atmosfera de nitrogênio a -15 ºC. Com auxílio de seringa graduada foram adicionados 5 mL de cloreto de acetila (70 mmol) e a mistura foi mantida sob agitação magnética à temperatura ambiente por 48 h. O sistema foi aberto e adicionaram-se 20 mL de CH2Cl2 e, em seguida, a fase orgânica foi extraída com duas frações de 15 mL de H2O destilada, três frações de 15 mL de solução saturada de NaHCO3. A fase orgânica resultante foi secada com Na2SO4, filtrada e concentrada em evaporador rotatório até a obtenção de 10% do volume do solvente (cerca de 4 mL). Verteram-se cerca de 20 mL de Et2O sobre a solução e foi observada a formação de um precipitado branco. O sistema foi vedado e mantido em repouso no freezer por cerca de 12 horas. Após a filtração, foram obtidos 2,44 g de 25 (74%) na forma de um sólido branco. 2,3,4,6-tetra-O-acetil-1-azido-1-desoxi-β-d-glicopiranose (26) e 2-acetamido-3,4,6-tri-O-acetil-1-azido-1,2-didesoxi-β-d-glicopiranose (27)7,12 Em um balão de fundo redondo 100 mL foram adicionados 0,64 g de azida de sódio (9,6 mmol) em 8 mL de água destilada e adicionaram-se uma solução contendo 4,8 mmol de 24 ou 25 em 10 mL de acetona. Manteve-se a mistura sob agitação magnética à temperatura ambiente por cerca de 3 horas. O monitoramento da reação foi feito por CCD (Hexano 6:4 AcOEt). Após observação do consumo do material de partida a acetona foi removida sob fluxo de ar e o precipitado branco formado foi filtrado e lavado com água gelada. Foram obtidos 1,48 g de 26 (81%) e 1,31 g de 27 (77%) na forma de um sólido branco. 2,3,4,6-tetra-O-acetil-1-desoxi-β-d-glicopiranosilamina (28) e 2-acetamido-3,4,6-tri-O-acetil-1,2-didesoxi-β-d-glicopiranosilamina (29)7 Em um balão de fundo redondo de 100 mL foram adicionados 2 mmol de 26 ou 27 e 10% p/p de Pd-C em 5 mL de THF anidro. A mistura foi mantida sob agitação magnética à temperatura ambiente em atmosfera de hidrogênio por cerca de 3 horas. O monitoramento da reação foi feito por CCD (AcOEt 100%). Após observação do consumo do material de partida o material foi filtrado e o THF foi removido em evaporador rotatório. O sólido branco obtido (28 ou 29) foi diretamente usado na etapa seguinte. 2-acetamido-2-desoxi-1,3,4,6-tetra-O-acetil-d-glicopiranose (30)14 Em um balão de fundo redondo de 250 mL foram adicionados 5 g de 21 (22,61 mmol) em 40 mL de piridina anidra. Em seguida foram adicionados 38 mL de Ac2O (401 mmol) e a mistura foi mantida sob agitação magnética e aquecimento (100 ºC) por cerca de 5 horas. O monitoramento da reação foi feito por CCD (AcOEt 100%). O material foi vertido em uma cápsula de porcelana e a piridina foi removida sob fluxo de ar comprimido. À pasta acastanhada escura resultante foram adicionados 30 mL de solução HCl 6 mol L-1 e extraiu-se com duas frações de 25 mL de CH2Cl2. A fase orgânica foi lavada com três frações de 20 mL água destilada e duas frações de 20 mL solução saturada de NaHCO3, secada com Na2SO4, filtrada e concentrada em evaporador rotatório. Foram obtidos 3,5 g de 30 (64%) na forma de um sólido branco. p-toluenossulfonato de de 2,3,4,6-tetra-O-acetil-1-desoxi-β-d-glicopiranosilamônio (31) e p-toluenossulfonato de 2-acetamido-3,4,6-tri-O-acetil-1,2-didesoxi-β-d-glicopiranosilamônio (32)7 Em um balão de 100 mL foram adicionados 2 mmol de 26 ou 27, 10% p/p de Pd-C e 2,2 mmol de TsOH em 5 mL de THF anidro. A mistura foi deixada sob agitação magnética à temperatura ambiente em atmosfera de hidrogênio por cerca de 3 horas. O monitoramento da reação foi feito por CCD (AcOEt 100%). Após observação do consumo do material de partida o material foi filtrado e o THF foi removido em evaporador rotatório. O sólido branco obtido (31 ou 32) foi diretamente usado na etapa seguinte. Procedimento geral do Método 17 Em balão de fundo redondo de 50 mL foram solubilizados 0,28 mmol de 28 ou 29 e 0,84 mmol do cloreto de sulfonila correspondente (RSO2Cl) em 3 mL de piridina anidra. O material foi mantido sob agitação magnética a 0 ºC (banho de gelo) por 2-5 h (monitoramento por CCD). Após o consumo completo do marerial de partida foi adicionada uma solução de HCl 6 mol L-1, gota a gota, até pH = 1 a 0 ºC. Para os produtos 2, 3, 7 e 8 foi observada a formação de um precipitado branco que foi filtrado. Para os produtos 1, 4, 5, 6, 9 e 10 fez-se a extração da fase aquosa com três frações de 15 mL de CH2Cl2 e a fase orgânica resultante foi secada com Na2SO4, filtrada e concentrada em evaporador rotatório. Os produtos brutos obtidos foram purificados por recristalização em metanol para 2, 3, 4, 5, 7, 8, 9 e 10 e coluna cromatográfica para 1 e 6 (Hexano / AcOEt 20-100%). Os rendimentos individuais estão relatados na descrição de cada substância. Procedimento geral do Método 213 Em um balão de fundo redondo de 50 mL foram solubilizados 0,28 mmol de 23 ou 30 e 0,56 mmol da sulfonilamida adequada (RSO2NH2) em 3 mL de MeCN anidra. O material foi mantido sob agitação magnética a 0 ºC e foram adicionados, gota a gota, 0,6 mL de BF3.Et2O. Em seguida, manteve-se agitação magnética à temperatura ambiente por 24 h. Após 24 h, removeu-se o solvente e foram adicionados 20 mL de CH2Cl2. Lavou-se a fase orgânica com três frações de 10 mL de solução saturada de NaHCO3 e duas frações de 10 mL de água destilada. A fase orgânica resultante foi secada com Na2SO4, filtrada e concentrada em evaporador rotatório. O produto bruto obtido foi purificado por rectristalização em metanol para 2, 3, 4, 5, 7, 8, 9 e 10 e coluna cromatográfica para 1 e 6 (Hexano / AcOEt 20-100%). Os rendimentos individuais estão relatados na descrição de cada substância. Procedimento geral do Método 3 Em balão de fundo redondo de 50 mL foram solubilizados 0,28 mmol 28 ou 29 e 0,84 mmol de cloreto de sulfonila correspondente (RSO2Cl) em 3 mL de THF anidro. A mistura foi mantida sob agitação magnética a 0 ºC (banho de gelo). A cada 30 minutos foi adicionada uma solução de 0,1 mmol de Et3N em 0,5 mL de THF, totalizando 0,4 mmol de Et3N no intervalo de 2 horas. O material foi mantido sob agitação magnética a 0 ºC por cerca de 3 horas adicionais no total de 5 horas. Após observação do consumo do material de partida, o solvente foi removido em evaporador rotatório e foram adicionados 20 mL de CH2Cl2. Extraiu-se essa fase orgânica com três frações de 20 mL de água destilada. Em seguida, a fase orgânica foi secada com Na2SO4, filtrada e concentrada em evaporador rotatório. O produto bruto obtido foi purificado por coluna cromatográfica (Hexano / AcOEt 20-100%). Os rendimentos individuais estão relatados na descrição de cada substância. N-(2,3,4,6-tetra-O-acetil-β-d-glicopiranosil)metanossulfonamida (1) Sólido branco, rendimento: 74%, FM: C15H23O11NS, MM: 425,1 g mol-1, F.F. 137,9-140,8 ºC, IV. (υmax, cm-1) 3226 (N-H), 2949 (C-H, alifático), 1746 (C=O, éster), 1043 (C-O). RMN de 1H (δ DMSO-d6, 400 MHz): 8,45 (d; 3J1-NH = 9,8 Hz, N-H), 5,36 (t; 3J2-3= 8 Hz, H-3), 4,95 (t; 3J1-2= 9,6 Hz, H-1), 4,85-4,79 (m; H-2 e H-4), 4,14-4,05 (m; H-5, H-6 e H-6'), 2,96 (s; H-7), 1,99 (s; OCOCH3). RMN de 13C (δ DMSO d6, 100 MHz): 169,95-168,97 (OCOCH3), 81,63 (C-1), 72,64 (C-3), 71,70 (C-5), 69,97 (C-4), 67,95 (C-6), 52,64 (C-2), 42,75 (C-7), 20,35 (COCH3). [α]D20 +10,2 (c 1,7, CHCl3); (lit.: [α]D20 +10,3 c 1,7, CHCl3).28 N-(2-3,4,6-tetra-O-acetil-β-d-glicopiranosil)benzenossulfonamida (2) Sólido branco, rendimento: 79%, FM: C20H25O11NS, MM: 487,11 g mol-1, FF: 165,5-167,7 ºC, IV. (υmax, cm-1) 3285 (N-H), 3065 (C-H, aromático), 2995 (C-H, alifático), 1738 (C=O, éster), 1031 (C-O). RMN de 1H (δ CDCl3, 400 MHz): 7,88 (d; 3J8-9 = 8,7 Hz, H-8), 7,57 (t; 3J10-9 = 8,7 Hz, H-10), 7,50 (t, 3J9-10 = 8,7 Hz, H-9), 5,74 (d; 3J1-NH = 9,8 Hz, N-H), 5,26 (t; 3J3-2 = 9,2 Hz, H-3), 4,98 (t, 3J4-3 = 9,2 Hz, H-4) 4,88 (t; 3J1-2= 9,2 Hz, H-1), 4,82 (t; 3J2-1= 9,2 Hz, H-2), 4,11 (dd; 2J6-6' = 12,2 Hz, H-6), 3,90 (dd; 2J6-6'= 12,2 Hz, H-6'), 3,69 (ddd; 2J5-6 = 3,8 Hz, 2J5-6' = 2,8 Hz, H-5), 2,00 (s; OCOCH3). RMN de 13C (δ DMSO-d6, 100 MHz): 171,00-169,74 (OCOCH3), 141,43 (C-7), 133,20 (C-10), 129,20 (C-8), 127,23 (C-9), 83,05 (C-1), 73,61 (C-3), 72,69 (C-4), 70,25 (C-2), 68,28 (C-5), 61,81 (C-1), 20,79 (COCH3). [α]D20 +29,2 (c 2,5, CHCl3); (lit.:[α]D20 +29,2, c 2,5, CHCl3).28 N-(2-3,4,6-tetra-O-acetil-β-d-glicopiranosil)-p-toluenossulfonamida (3) Sólido branco, rendimento: 81%, FM: C21H27O11NS, MM: 501,13 g mol-1, FF: 162,9-163,5 ºC, IV. (υmax, cm-1) 3279 (N-H, aromático), 3041 (C-H, alifático), 2941 (C-H, alifático), 1740 (C=O, éster), 1038 (C-O). RMN de 1H (δ acetona-d6, 400 MHz): 7,73 (d; 3J8-9= 8,2 Hz, H-8), 7,52 (d; 3JNH-1 = 9,7 Hz, N-H), 7,33 (d; 3J8-9= 8,2 Hz, H-9), 5,27 (t; 3J3-2 = 9,7 Hz, H-3), 4,97 (t; 3J1-2= 9,7 Hz, H-1), 4,87 (t; 3J2-3= 9,7 Hz, H-2), 4,84 (t; 3J3-4= 9,7 Hz, H-4), 4,02 (dd; 2J6-6'= 12,2 Hz, H-6), 3,90 (ddd; 2J5-6 = 3,9 Hz, 2J5-6' = 2,5 Hz, H-5), 3,80 (dd; 2J6-6'= 12,2 Hz, H-6'), 2,36 (s; H-11), 1,87 (s; OCOCH3). RMN de 13C (δ DMSO-d6, 100 MHz): 170,33-169,66 (OCOCH3), 143,80 (C-7), 140,38 (C-10), 129,93 (C-8), 127,64 (C-9), 83,12 (C-1), 73,67 (C-3), 73,52 (C-5), 71,03 (C-4), 68,87 (C-2), 62,41 (C-6), 21,15 (C-11), 20,26 (COCH3). [α]D20 +26,2 (c 2,5, CHCl3); (lit.:[α]D20 +29,6 ,c 2,5, CHCl3; [α]D19 +26,2 (c 2,5, CHCl3 ).28,29 N-(2-3,4,6-tetra-O-acetil-β-d-glicopiranosil)-1-naftalenossulfonamida (4) Sólido branco, rendimento: 74%, FM: C24H27O11NS, MM: 537,13 g mol-1, FF: 144,3-144,9 ºC, IV. (υmax, cm-1) 3270 (N-H), 3033 (C-H, aromático), 2958 (C-H, alifático), 1743 (C=O, éster), 1034 (C-O). RMN de 1H (δ CDCl3, 400 MHz): 8,55 (d, 3J8-9 = 8,4 Hz, H-8), 8,26 (d; 3J10-9 = 8,6 Hz, H-10), 8,04 (d, 3J12-13 = 8,4 Hz, H-12), 7,90 (d, 3J15-14 = 8,5 Hz, H-15), 7,66 (t, 3J14-15 = 8,5 Hz, H-14), 7,57 (t, 3J9-8 = 8,4 Hz, H-9), 7,53 (t, 3J13-12 = 8,4 Hz, H-13), 7,13 (d, 3JNH-1 = 9,8 Hz, NH), 5,13 (t; 3J3-2= 9,3 Hz, H-3), 4,93 (t; 3J1-2 = 9,3 Hz, H-1), 4,87 (t, 3J4-3 = 9,4 Hz, H-4), 4,68 (t; 3J2-3 = 9,3 Hz, H-2), 4,03 (dd; 2J6-6'= 12,1 Hz, H-6), 3,66 (dd; 2J6'-6= 12,1 Hz, H-6'), 3,56 (ddd, 3J5-6 = 3,8 Hz, 3J5-6' = 2,1 Hz, H-5), 1,92 (s; OCOCH3). RMN de 13C (δ DMSO-d6, 100 MHz): 170,51-169,46 (OCOCH3), 163,16 (C-7), 134,37 (C-16), 134,12-124,18 (C-8-15), 82,58 (C-1), 73,19 (C-3), 72,85 (C-5), 69,93 (C-4), 67,93 (C-2), 61,47 (C-6), 20,33 (COCH3). [α]D20 +14,7 (c 1,0, CHCl3). N-(2-3,4,6-tetra-O-acetil-β-d-glicopiranosil)-2-naftalenossulfonamida (5) Sólido branco, rendimento: 72%, FM: C24H27O11NS, MM: 537,13 g mol-1, FF: 159,5-161,9 ºC, IV. (υmax, cm-1) 3356 (N-H), 3072 (C-H, aromático), 2898 (C-H, alifático), 1742 (C=O, éster), 1049 (C-O). RMN de 1H (δ CDCl3, 400 MHz) 7,97-7,75 (m; H-8, H-11, H-13 e H-14), 7,67-7,61 (m, H-9 e H-12), 5,72 (d; 3JNH-1 = 9,9 Hz, NH), 5,26 (t; 3J3-2 = 9,2 Hz, H-3), 4,99-4,86 (m; H-1, H-2 e H-4), 4,08 (dd; 2J6-6'= 12,2 Hz, H-6), 3,84 (dd; 2J6'-6 = 12,2 Hz, H-6'), 3,70 (ddd, 3J5-6 = 4 Hz, 3J5-6' = 2,9 Hz, H-5), 1,95 (s; OCOCH3). RMN de 13C (δ DMSO-d6, 100 MHz): 170,99-169,66 (OCOCH3), 138,21 (C-7), 135,11-122,52 (C-10-16), 83,09 (C-1), 73,68 (C-3), 72,71 (C-5), 70,39 (C-2), 68,33 (C-4), 61,79 (C-6), 20,73 (COCH3). [α]D20 +18,3 (c CHCl3). N-(2-acetamido-3,4,6-tri-O-acetil-2-desoxi-β-d-glicopiranosil)metanossulfonamida (6) Sólido branco, rendimento: 75%, FM: C15H24O10N2S, MM: 424,12 g mol-1, F.F. 215,5-216,8 ºC, IV. (υmax, cm-1) 3326 (N-H), 3269 (N-H), 2945 (C-H, alifático), 1739 (C=O, éster), 1658 (C=O, amida), 1039 (C-O). RMN de 1H (δ DMSO-d6, 400 MHz): 8,29 (sl; N-H (C-1)), 8,00 (d; 3J1-NH= 9 Hz, N-H (C-2)), 5,13 (t; 3J3-2 = 9,8 Hz, H-3), 4,77 (t; 3J1-2 = 9,8 Hz, H-1 e H-2), 4,11 (dd; 2J6-6' = 11,8 Hz, H-6), 4,07 (dd; 2J6'-6 = 11,8 Hz, H-6'), 3,83-3,78 (m; H-2 e H-5), 2,94 (s; H-7), 1,95 (s; OCOCH3), 1,88 (s; NHCOCH3). RMN de 13C (δ DMSO-d6, 100 MHz): 170,41 (OCOCH3), 169,77 (NHCOCH3), 83,19 (C-1), 73,51 (C-3), 72,41 (C-4), 68,88 (C-5), 62,28 (C-6), 52,65 (C-2), 43,25 (C-7), 23,12 (NHCOCH3), 20,84 (COCH3). [α]D20 +31,4 (c 1,0, CHCl3). N-(2-acetamido-3,4,6-tri-O-acetil-2-desoxi-β-d-glicopiranosil)benzenossulfonamida (7) Sólido branco, rendimento: 82%, FM: C20H26O10N2S, MM: 486,13 g mol-1, F.F. 173,9-177,1 ºC, IV. (υmax, cm-1) 3301 (N-H), 3279 (N-H), 3017 (C-H, aromático), 2944 (C-H, alifático), 1742 (C=O, éster), 1658 (amida), 1031 (C-O). RMN de 1H (δ acetona-d6, 400 MHz): 7,86 (d; 3J8-9 = 8,2 Hz, H-8), 7,66-7,57 (m, H-9, N-H (C-1) e N-H (C-2)), 5,27 (t; 3J3-2= 9,7 Hz, H-3), 5,00 (t; 3J1-2= 9,7 Hz, H-1), 4,88 (t; 3J4-3= 9,7 Hz, H-4), 4,08 (dd; 3J6-6'= 12,2 Hz, H-6), 3,96 (t; 3J2-3= 9,7 Hz, H-2), 3,81 (dd; 2J6'-6 = 12,2 Hz, H-6'), 3,80 (ddd, 3J5-6 = 3,7 Hz, 3J5-6' = 1,9 Hz, H-5), 2,00 (s; OCOCH3 e NHCOCH3). RMN de 13C (δ DMSO-d6, 100 MHz): 170,99 (OCOCH3), 169,77 (NHCOCH3), 143,36 (C-7), 132,96 (C-10), 129,40 (C-8), 127,62 (C-9), 84,21 (C-1), 73,56 (C-3), 73,52 (C-5), 69,40 (C-4), 62,65 (C-6), 53,69 (C-2), 22,65 (NHCOCH3), 20,42 (COCH3). [α]D20 +11,4 (c 1,0, CHCl3). N-(2-acetamido-3,4,6-tri-O-acetil-2-desoxi-β-d-glicopiranosil)-p-toluenossulfonamida (8) Sólido branco, rendimento: 80%, FM: C15H23O11NS, MM: 500,15 g mol-1, F.F. 203,1-205,5 ºC, IV. (υmax, cm-1) 3280, 2946, 1741, 1661, 1031, 981. RMN de 1H (δ acetona-d6, 400 MHz): 7,78 (d; 3J8-9= 7,9 Hz, H-8), 7,51 (d; 3JNH-1 = 9,8 Hz, N-H (C-1)), 7,35 (d; 3J9-8 = 7,9 Hz, H-9), 7,23 (d; 3JNH-2 = 9,7 Hz, N-H (C-2)), 5,26 (t; 3J3-2 = 9,7 Hz, H-3), 4,97 (t; 3J1-2 = 9,7 Hz, H-1), 4,88 (t; 3J4-3 = 9,7 Hz, H-4), 4,03 (dd; 2J6-6' = 11,9 Hz, H-6), (dd; 2J6'-6 = 11,9 Hz, H-6), 3,81 (ddd; 3J5-6 = 3,9 Hz, 3J5-6' = 1,8 Hz, H-5), 2,40 (s; H-11), 1,97 (s; OCOCH3), 1,71 (s, NHCOCH3). RMN de 13C (δ DMSO-d6, 100 MHz): 171,01 (OCOCH3), 169,83 (NHCOCH3), 143,58 (C-7), 140,65 (C-10), 129,85 (C-8), 127,68 (C-9), 84,31 (C-1), 73,43 (C-3 e C-5), 69,39 (C-4), 62,63 (C-6), 53,78 (C-2), 22,67 (C-11), 21,17 (NHCOCH3), 20,42 (COCH3). [α]D20 +31,4 (c 0,59, THF); (lit.:[α]D20 +31,3 (c 0,59, THF).30 N-(2-acetamido-3,4,6-tri-O-acetil-2-desoxi-β-d-glicopiranosil)-1-naftalenossulfonamida (9) Sólido branco, rendimento: 70%, FM: C24H28O10N2S, MM: 536,15 g mol-1, F.F. 201,1-202,9 ºC, IV. (υmax, cm-1) 3247, 3224, 2956, 1733, 1661, 1046, 981. RMN de 1H (δ CDCl3, 400 MHz): 9,16 (d; J1-NH = 9,6 Hz, N-H (C-1)), 8,61 (d; 3J8-9 = 7,7 Hz, H-8), 8,33 (sl; H-10 e H-12), 8,14 (t; 3J14-15 = 7,8 Hz, H-14), 7,89-7,58 (m; N-H (C-2), H-9, H-13 e H-15), 5,13 (t; 3J3-4= 9,7 Hz, H-3), 4,91 (t; 3J1-2 = 9,6 Hz, H-1), 4,70 (t; 3J3-4 = 9,7 Hz, H-4), 3,96 (dd; 3J6-6' = 12,5 Hz, H-6), 3,72 (t; 3J2-3 = 9,7, H-2), 3,72-3,64 (m; H-5),3,53 (dd; 3J6'-6 = 12,5 Hz, H-6'), 1,88 (s, OCOCH3), 1,31 (s; NHCOCH3). RMN de 13C (δ CDCl3, 100 MHz): 169,79-169,04 (OCOCH3 e NHCOCH3), 137,00 (C-7), 133,66 (C-16), 133,60 (C-11), 128,65-124,36 (C-8 -C-10 e C12 - C-15), 82,06 (C-1), 72,86 (C-3), 71,83 (C-5), 68,15 (C-4), 61,50 (C-6), 52,64 (C-2), 22,13 (NHCOCH3), 20,27 (COCH3). [α]D20 +38,9 (c 1,0, CHCl3). N-(2-acetamido-3,4,6-tri-O-acetil-2-desoxi-β-d-glicopiranosil)-2-naftalenossulfonamida (10) Sólido branco, rendimento: 71%, FM: C24H28O10N2S, MM: 536,15 g mol-1, F.F. 209,4-210,9 ºC, IV. (υmax, cm-1) 3322, 3069, 2954, 1742, 1655, 1030, 964. RMN de 1H (δ CDCl3, 400 MHz): 8,96 (sl; N-H (C-1)), 8,48 (s; H-16), 8,12 (d; 3JNH-2 = 8 Hz, N-H (C-2)), 8,06-8,01 (m; H-8 e H-13), 7,91 (d; 3J11-12 = 7,7 Hz, H-11), 7,85 (d; 3J14-13 = 7,6 Hz, H-14), 7,70-7,62 (m; H-9 e H-12), 5,15 (t; 3J3-4 = 9,5 Hz, H-3), 4,94 (sl; H-1), 4,68 (t; 3J4-3 = 9,5 Hz, H-4), 3,96 (dd; 2J6-6' = 12,2 Hz, H-6'), 3,77-3,72 (m; H-2 e H-5), 3,64 (dd; 2J6'-6 = 12,2 Hz, H-6'), 1,90 (s, OCOCH3), 1,64 (s, NHCOCH3). RMN de 13C (δ CDCl3, 100 MHz): 169,71 (OCOCH3), 169,19 (NHCOCH3), 139,34 (C-7), 134,06- -122,67 (C-8-C-16), 82,53 (C-1), 72,93 (C-3), 71,86 (C-5), 68,16 (C-4), 61,63 (C-6), 52,56 (C-2), 22,51 (NHCOCH3), 20,14 (COCH3). [α]D20 +35,2 (c 1,0, CHCl3). Procedimento geral para obtenção de 11-2015-17 Em um balão de fundo redondo de 50 mL foram adicionados 0,5 mmol da N-glicosilssulfonamida (1-10) em 2 mL de uma solução previamente preparada de MeONa 1 mol L-1 em MeOH anidro a 0 ºC e manteve-se sob agitação magnética a 0 ºC por cerca de 30 minutos. Após observação por CCD do consumo completo do material de partida promoveu-se a neutralização do meio com resina ácida Amberlite IRA 120, (pH = 7). Em seguida a resina foi removida por filtração e a solução foi concentrada em evaporador rotatório. O produto bruto obtido foi recristalizado em isopropanol. O rendimento para obtenção de 11-20 variou de 92% a 99%. N-(β-d-glicopiranosil)metanossulfonamida (11) Sólido branco, rendimento: 94%, FM: C7H15O7NS, MM: 257,06 g mol-1, F.F. 194,6-197 ºC, IV. (υmax, cm-1) 3425, 3099, 2904, 1003, 975. RMN de 1H (δ DMSO-d6, 400 MHz): 7,96 (d; 3J1-NH = 9,4 Hz, N-H (C-1)), 5,00-4,00 (m, 2xOH) 4,89 (d; 3J = 5,4 Hz, OH), 4,50 (t; 3J1-NH = 9,4 Hz, H-1), 4,21 (t, 3J3-4 = 8,3 Hz, H-3), 3,69 (t; 3J2-3 = 9,4 Hz, H-2), 3,38-3,03 (m; H-4-H-6 e OH), 2,98 (s, H-7). RMN de 13C (δ DMSO-d6, 100 MHz): 84,66 (C-1), 78,23 (C-3), 77,43 (C-5), 72,18 (C-4), 70,12 (C-2), 61,09 (C-6), 43,28 (C-7); [α]D20 +8,3 (c 0,5, MeOH). N-(β-d-glicopiranosil)benzenossulfonamida (12) Sólido branco, rendimento: 92%, FM: C12H17O7NS, MM: 319,07 g mol-1, F.F. 189,0-189,6 ºC, IV. (υmax, cm-1) 3396, 3258, 3174, 2925, 1007, 878. RMN de 1H δ DMSO-d6, 400 MHz): 8,48 (d; 3JNH-1 = 9,8 Hz, N-H (C-1)), 7,89 (d; 3J8-9 = 7,7 Hz, H-8), 7,55 (t; 3J10-9 = 7,7 Hz, H-10), 7,52 (d; 3J9-8 = 7,7 Hz, H-9), 4,97-4,83 (m, 3xOH), 4,37 (t; 3J1-2 = 8,2 Hz, H-1), 4,06 (sl, OH), 3,48-3,00 (m; H-2-H-6). RMN de 13C (δ DMSO-d6, 100 MHz): 142,84 (C-7), 131,90 (C-10), 128,50 (C-8), 126,80 (C-9), 84,76 (C-1), 78,01 (C-3), 77,55 (C-5), 72,25 (C-4), 69,82 (C-2), 60,82 (C-6). [α]D20 +9,7 (c 0,5, MeOH). N-(β-d-glicopircanosil)-p-toluenossulfonamida (13) Sólido branco, rendimento: 97%, FM: C13H19O7NS, MM: 333,09 g mol-1, F.F. 199,2-201,5 ºC, IV. (υmax, cm-1) 3285, 3263, 2889, 1015, 979. RMN de 1H (δ DMSO-d6, 400 MHz): 8,43 (sl; N-H (C-1)), 7,76 (d; 3J8-9 = 8,1 Hz, H-8), 7,29 (d; 3J9-8 = 8,1 Hz, H-9), 5,04-4,92 (m, 3xOH), 4,34 (d; 3J1-2 = 9 Hz, H-1), 4,16 (sl; OH), 3,09-2,99 (m, H-2-H-6), 2,35 (s; H-11). RMN de 13C (δ DMSO-d6, 100 MHz): 142,20 (C-7), 140,13 (C-10), 129,10 (C-8), 127,05 (C-9), 84,88 (C-1), 78,15 (C-3), 78,15 (C-5), 72,37 (C-4), 69,90 (C-2), 60,93 (C-6), 21,10 (C-11). [α]D22 +6,8º (c 1,45, H2O); [α]D20 +6,9 (c 1,45, H2O).29 N-(β-d-glicopiranosil)-1-naftalenossulfonamida (14) Sólido branco, rendimento: 96%, FM: C16H19O7NS, MM: 369,09 g mol-1, F.F. 186,2-186,5 ºC, IV. (υmax, cm-1) 3285, 3270, 2889, 1015, 979. RMN de 1H (δ DMSO-d6, 400 MHz): 8,84 (d; 3J8-9 = 7,6 Hz, H-8), 8,67 (d; 3J10-9 = 7,6 Hz, H-10), 8,35 (d; (d; 3JNH-1 = 9,1 Hz, NH), 8,16 (d; 3J15-14 = 7,8 Hz, H-15), 7,68-7,58 (m; H-9, H-12, H-13 e H-15), 4,99 (d; 3J = 5,5 Hz, 2xOH), 4,82 (d; 3J = 5,7 Hz, OH), 4,40 (t, 3J1-NH = 9,1 Hz, H-1), 3,92 (sl, OH), 3,61-2,97 (m; H-2-H-6). RMN de 13C (δ DMSO-d6, 100 MHz): 138,77 (C-7), 134,16-124,95 (C-8-C-16), 84,99 (C-1), 78,28 (C-3), 78,06 (C-5), 72,80 (C-4), 70,01 (C-2), 61,02 (C-6); [α]D20 +16,4 (c 0,5, MeOH). N-(β-d-glicopiranosil)-2-naftalenossulfonamida (15) Sólido branco, rendimento: 99%, FM: C16H19O7NS, MM: 369,09 g mol-1, F.F. 189-190,5 ºC, IV. (υmax, cm-1) 3556, 3500, 2897, 1036, 1003. RMN de 1H (δ DMSO-d6, 400 MHz): 8,58 (sl; N-H (C-1)), 8,55 (s; H-16), 8,13 (d; 3J8-9 = 8 Hz, H-8), 8,04-7,99 (m; H-11 e H-13), 7,94 (d; 3J14-13 = 7,8 Hz, H-14), 7,67-7,62 (m; H-9 e H12), 5,01 (sl; 2xOH), 4,94 (sl; OH), 4,45 (d; 3J1-2 = 9 Hz, H-1), 4,28 (sl; OH), 3,44-3,01 (m; H-2-H-6). RMN de 13C (δ DMSO-d6, 100 MHz): 140,00 (C-7), 133,99-123,16 (C-8-C-16), 84,79 (C-1), 78,10 (C-3), 77,55 (C-5), 72,29 (C-4), 69,79 (C-2), 60,70 (C-6); [α]D20 +16,5 (c 0,5, MeOH). N-(2-acetamido-2-desoxi-β-d-glicopiranosil)metanossulfonamida (16) Sólido branco, rendimento: 94%, FM: C9H18O7N2S, MM: 298,08 g mol-1, F.F. 193,5-194-2 ºC, IV. (υmax, cm-1) 3250, 2884, 1646, 1024, 972. RMN de 1H (δ DMSO-d6, 400 MHz): 7,84 (d; 3JNH-1 = 8,9 Hz, NH (C-1)), 7,81 (d; 3JNH-2 = 9,1 Hz, NH (C-2), 5,05-5,02 (m; 2xOH), 4,59 (t; 3J1-NH = 8,9 Hz, H-1), 4,34 (t; 3J3-2 = 8,8 Hz, H-3), 3,71 (dd; 2J6-6' = 3,7 Hz, H-6), 3,51-3,06 (m; H-2, H-4-H-6), 2,98 (s; H-7), 1,82 (s; NHCOCH3). RMN de 13C (δ DMSO-d6, 100 MHz): 169,64 (NHCOCH3), 83,77 (C-1), 78,53 (C-3), 74,78 (C-4), 70,79 (C-5), 61,27 (C-6), 54,36 (C-2), 43,13 (C-7), 23,15 (NHCOCH3); [α]D20 +21,5 (c 0,5, MeOH). N-(2-acetamido-2-desoxi-β-d-glicopiranosil)benzenossulfonamida (17) Sólido branco, rendimento: 98%, FM: C14H20O7N2S, MM: 360,1 g mol-1, F.F. 189,7-199,4 ºC, IV. (υmax, cm-1) 3300, 3273, 2932, 1649, 1024, 957. RMN de 1H (δ DMSO-d6, 400 MHz): 8,37 (sl; NH (C-1)), 7,56 (m; NH (C-2) e H-8), 7,69 (t; 3J10-9 = 8 Hz, H-10), 7,30 (d; 3J8-9 = 8 Hz, H-9), 5,03 (sl; OH), 4,48 (d, 3J1-2 = 9,2 Hz, H-1), 4,27 (sl; OH), 3,43-3,04 (m, H-2-H-6), 1,66 (s; NHCOCH3). RMN de 13C (δ DMSO-d6, 100 MHz): 170,16 (NHCOCH3), 142,44 (C-7), 132,26 (C-10), 128,89 (C-8), 126,84 (C-9), 83,71 (C-1), 78,29 (C-3), 74,53 (C-4), 70,25 (C-5), 60,86 (C-6), 54,90 (C-2), 23,0q (NHCOCH3); [α]D20 +27,6 (c 0,5, MeOH). N-(2-acetamido-2-desoxi-β-d-glicopiranosil)-p-toluenossulfonamida (18) Sólido branco, rendimento: 99%, FM: C15H22O7N2S, MM: 374,11 g mol-1, F.F. 199,5-202,2 ºC, IV. (υmax, cm-1) 3250, 2907, 2843, 1651, 1025, 906. RMN de 1H (δ piridina-d5, 400 MHz): 10,24 (sl; NH (C-1)), 9,18 (d; 3JNH-2 = 8 Hz, NH (C-2)) 8,19 (d; 3J8-9 = 7,7 Hz, H-8), 7,19 (d; 3J9-8 = 7,7 Hz, H-9), 5,40 (d; 3J1-2 = 8,1 Hz), 4,67-3,88 (m, H-2-H-6), 2,13 (s; H-11), 1,88 (s; NHCOCH3). RMN de 13C (δ piridina-d5, 100 MHz): 171,46 (NHCOCH3), 142,54 (C-7), 140,66 (C-10), 129,41 (C-8), 127,37 (C-9), 85,53 (C-1), 79,58 (C-3), 76,44 (C-4), 71,77 (C-5), 62,03 (C-6), 56,27 (C-2), 22,92 (C-11), 20,84 (NHCOCH3); [α]D20 +36,6 (c 0,5, MeOH). N-(2-acetamido-2-desoxi-β-d-glicopiranosil)-1-naftalenossulfonamida (19) Sólido branco, rendimento: 95%, FM: C18H22O7N2S, MM: 410,11 g mol-1, F.F. 144,6-145,5 ºC, IV. (υmax, cm-1) 3278, 3217, 2908, 1651 1036, 1003. RMN de 1H (δ DMSO-d6, 400 MHz): 8,84 (sl; NH (C-1)), 8,23 (d; 3J8-9 = 7,4 Hz, H-8), 7,93-7,88 (m; H-10 e H-12), 7,52-7,35 (m; H-9-H-13-H-15 e NH C-2)), 5,07 (sl; OH), 4,79 (sl, OH), 4,25 (d; 3J1-2 = 9,8 Hz, H-1), 4,08 (sl; OH), 3,43-2,91 (m, H-2-H-6), 1,55 (s; NHCOCH3). RMN de 13C (δ DMSO-d6, 100 MHz): 169,06 (NHCOCH3), 133,73-124,42 (C-7-C-16), 83,44 (C-1), 78,06 (C-3), 74,19 (C-4), 70,09 (C-5), 60,60 (C-6), 55,05 (C-2), 22,23 (NHCOCH3); [α]D20 +30,2 (c 0,5, MeOH). N-(2-acetamido-2-desoxi-β-d-glicopiranosil)-2-naftalenossulfonamida (20) Sólido branco, rendimento: 98%, FM: C16H19O7NS, MM: 369,09 g mol-1, F.F. 185,3-188,5 ºC, IV. (υmax, cm-1) 3263, 3363, 2841, 1650,1026, 905. RMN de 1H (δ DMSO-d6, 400 MHz): 8,47 (s; H-16), 8,41 (d; 3JNH-1 = 8,6 Hz, NH (C1)), 8,14 (d; 3J8-9 = 7,8 Hz), 8,05-8,00 (m; H-11 e H-13), 7,86 (d; 3JNH-2 = 8,5 Hz, NH (C-1)), 7,76 (d; 3J14-12 = 7,7 Hz, H-14), 7,61-7,55 (m; H-9 e H-12), 4,93 (d; 3J = 5,2 Hz, 2xOH), 4,57 (d; 3J = 5,1 Hz, OH), 4,21 (t; 3J1-NH = 8,6 Hz, H-1), 3,64-3,06 (m; H-2-H-6), 1,62 (s; NHCOCH3). RMN de 13C (δ DMSO-d6, 100 MHz): 169,74 (NHCOCH3), 139,44 (C-7), 134,02-122-91 (C-8-C-16), 83,62 (C-1), 78,22 (C-3), 74,38 (C-4), 70,21 (C0-5), 60,75 (C-6), 54,81 (C-2), 22,80 (NHCOCH3); [α]D20 +30,9 (c 0,5, MeOH).
AGRADECIMENTOS Os autores agradecem ao Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), à Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG) e à Coordenação de Aperceiçoamento de Pessoal de Nível Superior (CAPES) pelo apoio financeiro que permitiu a realização desse trabalho.
REFERÊNCIAS 1. Nelson D. L.; Michael, L. C.; Lehninger Principles of Biochemistry, 4th ed., W. H. Freeman and company: New York, 2006. 2. Horsch, M.; Mayer, C.; Sennhauser, U.; Rast, D. M.; Pharmacol. Ther. 1997, 76, 187. DOI: http://dx.doi.org/10.1016/S0163-7258(97)00110-1 PMID: 9535180 3. Nogueira, C. M.; Parmanhan, B. R.; Farias P. P.; Corrêa, A. G.;Revista Virtual de Química 2009, 1, 149. DOI: http://dx.doi.org/10.5935/1984-6835.20090017 4. Colinas P. A.; Curr. Org. Chem. 2012, 16, 1670. DOI: http://dx.doi.org/10.2174/138527212800840892 5. Crespo, R.; Bravo M. G.; Colinas P. A.; D. Bravo R. D.;Bioorg. Med. Chem. Lett. 2010, 20, 6469. DOI: http://dx.doi.org/10.1016/j.bmcl.2010.09.052 PMID: 20888767 6. Srivastava, A.; Varghese, B.; Loganathan, D.; Chem. Eur. J. 2013, 19, 17720. DOI: http://dx.doi.org/10.1002/chem.201302018 7. Butera, A. P.; Souza Filho, J. D.; Carvalho, D. T.; Figueiredo, R. C.; Faria, L. C. A.; Nunes, M. A.; Prado, M. A. F.; Alves, R. J.; Andrade, M. H. G.; Silva, K. T. S.; Quim. Nova 2007, 30, 1267. DOI: http://dx.doi.org/10.1590/S0100-40422007000500040 8. Ribeiro, J. P.; Carvalho, D. T.; André, S.; Cañada, F. J.; Alves, R. J.; Gabius, H. J.; Barbero, J. J.; C. R. Chim. 2011, 14, 96. DOI: http://dx.doi.org/10.1016/j.crci.2010.03.013 9. Liardini, M. C. F.; Oliveira, M. M; Sampaio, M. R. P.; J. Med. Chem. 1975, 18, 1159. DOI: http://dx.doi.org/10.1021/jm00245a027 10. Barthel, S. R.; Antonopoulos, A.; Laurent, F. C.; Schaffer, L.; Hernandez, G.; Patil, S. A.; North, S. J.; Dell, A.; Matta, K. L.; Neelamegham, S.; Haslam, S. M.; Dimitroff, C. J.; J. Biol. Chem. 2011, 289, 21717. DOI: http://dx.doi.org/10.1074/jbc.M110.194597 11. Malicdan, M. C. V.; Nogushi, S.; Tokutomi, T.; Goto, Y.; Nonaka, I.; Hayashi, Y. K.; Nishino, I.;J. Biol. Chem. 2012, 287, 2689. DOI: http://dx.doi.org/10.1074/jbc.M111.297051 PMID: 22157763 12. Hough, L.; Taha, M. I.;J. Chem. Soc. 1956, 10, 2042. DOI: http://dx.doi.org/10.1039/jr9560002042 13. Colinas, P. A.; Témpera, C. A.; Rodríguez, O. M.; Bravo, R. D.; Synthesis 2009, 24, 4143. 14. Manger, I. D.; Rademacher, T. W.; Dwek, R. A.; Biochemistry 1992, 31, 10724. DOI: http://dx.doi.org/10.1021/bi00159a012 PMID: 1420188 15. Perrin, C. L.; Pure Appl. Chem. 1995, 67, 719. 16. Collins, P.; Ferrier, R.; Monossacharides: Their chemistry and their roles in natural products, John Wiley & Sons: Chichester, 1995. 17. Figueiredo, R.C.; Meyer, N. B.; Prado, M. A. F.; Alves, R. J.; Rojo, R.; Quim. Nova 2009, 32, 2128. DOI: http://dx.doi.org/10.1590/S0100-40422009000800026 18. Ferreira, V. F.; Quim. Nova 1992, 15, 348. 19. Williams, D. B. G.; Lawton, M.; J. Org. Chem. 2010, 75, 8351. DOI: http://dx.doi.org/10.1021/jo101589h PMID: 20945830 20. Vogel, A. I.; Química Orgânica: Análise Orgânica Qualitativa, 4th ed., Rio de Janeiro: Guanabara Koogan, 1992. 21. Burrfield, D. R.; Lee, K. W.; J. Org. Chem. 1977, 42, 3061. 22. Kur'yanov, V.O.; Chupakhina, T. A.; Zemlyakov, A. E.; Chirva, V. Y.; Shishkin, O. V.; Shishkina, S. V.; Kamalov, G. L; Russ. J. Bioorg. Chem. 2005, 31, 460. DOI: http://dx.doi.org/10.1007/s11171-005-0063-z 23. Heidlas, J. E.; Lees, W. J.; Pale, P.; Whitesides, G. M;J. Org. Chem. 1992, 57, 146. DOI: http://dx.doi.org/10.1021/jo00027a029 24. Shen, T. Y.; Li, J. P.; Dorn, C. P.; Ebel, D.; Bugianesi, R.; Fecher, R.; Carbohydr. Res. 1972, 23, 87. DOI: http://dx.doi.org/10.1016/S0008-6215(00)81580-3 25. Pearson, W. A.; Spessard, G. O.;J. Chem. Educ. 1975,52, 814. DOI: http://dx.doi.org/10.1021/ed052p814 26. Szilagy, L.; Gyorgyodeák, Z. A.;Carbohydr. Res. 1985, 143, 21. DOI: http://dx.doi.org/10.1016/S0008-6215(00)90692-X 27. Chupakina, T. A.; Kur'yanov, V. O.; Chirva, V. Y.; Grigorash, R. Y.; Kotlyar, S. A.; Kamalov, G. L.;Russ, J. Bioorg. Chem. 2004,30, 334. 28. Burckhardt, H.;Chem. Ber. 1952, 85, 8. 29. Burckhardt, H.;Justus Liebigs Ann. Chem. 1957, 605, 182. DOI: http://dx.doi.org/10.1002/jlac.19576050123 30. Vipin, K.; Org. Biomol. Chem. 2007, 5, 3847. DOI: http://dx.doi.org/10.1039/b712841j |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access