
 |
 |
 |
 |
 |
Educação
|
|
| Interações fármaco-receptor: aplicações de técnicas computacionais em aula prática sobre a evolução dos inibidores da enzima conversora de angiotensina Drug-receptor interactions: in silico approaches applied to experimental classes regarding the evolution of angiotensin converting enzyme inhibitors |
|
Maurício Temotheo TavaresI; Marina Candido PrimiI; Michelle Carneiro PolliII; Elizabeth Igne FerreiraI; Roberto Parise-FilhoI,*
IDepartamento de Farmácia, Faculdade de Ciências Farmacêuticas, Universidade de São Paulo, Av. Prof. Lineu Prestes nº 580, 05508-000 São Paulo - SP, Brasil Recebido em 23/02/2015 *e-mail: roberto.parise@usp.br Teaching classes and events regarding the molecular aspects of drug-receptor interactions is not an easy task. The ligand stereochemistry and the spatial arrangement of the macromolecular targets highly increase the complexity of the process. In this context, the use of alternative and more playful approaches could allow students to gain a more thorough understanding of this important topic in medicinal chemistry. Herein, we describe a practical teaching approach that uses computational strategies as a tool for drug-receptor interaction studies performed for angiotencsin converting enzyme inhibitors (ACEi). Firstly, the students learn how to find the crystallographic structure (enzyme-ligand complex). Then, they proceed to the treatment of crude crystallographic data. Thereafter, they learn how to analyze the positioning of the drug on the active site of the enzyme, looking for regions related to the molecular recognition. At the end of the study, students can summarize the molecular requirements for the interaction and the structure-activity relationships of the studied drugs. INTRODUÇÃO Contemplar todos os aspectos moleculares envolvidos na interação entre um fármaco e seu receptor biológico é, certamente, um dos maiores desafios no ensino da Química Farmacêutica Medicinal aos graduandos em Farmácia. Conhecer a importância dos fatores estereoquímicos e eletrônicos nas interações fármaco-receptor (F-R) faz com que o estudante adquira arsenal cognitivo suficiente para entender e propor relações entre a estrutura química do fármaco e sua atividade biológica (REA).1,2 Desenvolver no estudante a habilidade de raciocinar criticamente a respeito da estrutura do fármaco e a intensidade da resposta biológica que este provoca não é uma tarefa fácil. Porém, o indivíduo que possui tal capacidade é capaz de compreender a importância de determinados grupos funcionais para a melhoria da afinidade de um fármaco estruturalmente otimizado por seu receptor biológico. Neste sentido, estratégias didáticas que fazem uso de recursos tecnológicos e audiovisuais podem contribuir para o processo de ensino-aprendizagem, tornando-o lúdico e permitindo ao aluno vivenciar as interações F-R de forma totalmente singular.3,4 É verdade que o ensino convencional das interações F-R, que faz uso de recursos estritamente expositivos, possui extrema importância na formação intelectual do indivíduo, porém, este tipo de abordagem nem sempre é capaz de atingir o aluno com a mesma intensidade de uma aplicação experimental.5 A possibilidade de um estudante explorar tridimensionalmente o bolsão de interação de um fármaco com o provável receptor eleva a capacidade deste em compreender todos os aspectos e variáveis inerentes à formação do complexo F-R. Como exemplo destas variáveis, pode ser citada a necessidade de o fármaco estar em sua conformação bioativa para exercer sua atividade biológica.6 Vale mencionar que esta consiste em conformação de menor energia, como forma de aproximação da que ocorre no organismo. A existência de bancos de dados de estruturas proteicas e a disponibilidade de programas de química computacional, ambos gratuitos, permitem que estes recursos e ferramentas sejam explorados em prol do ensino químico-farmacêutico.5,6 Uma vez determinada a disposição espacial do complexo F-R, seja por cristalografia de raios-X, por ressonância magnética nuclear (RMN) ou crio-microscopia eletrônica (Cryo-EM), é possível identificar pontos de interação, a natureza destas interações e as regiões que são exploradas pelo ligante (fármaco). Assim, podem ser traçadas correlações (aos níveis molecular e eletrônico) de diferentes estruturas que exploram o mesmo ambiente molecular.7,8 Os inibidores da enzima conversora de angiotensina (iECA) constituem-se em classe terapêutica para o tratamento da hipertensão arterial sistêmica (HAS). Amplamente estudada e com REA estabelecida, a classe dos iECA envolve série de fármacos utilizados na terapêutica, a exemplo têm-se o captopril (1), o enalapril (2) e o ramipril (3) (Figura 1). A história do desenvolvimento destes fármacos, abordada extensivamente nos livros didáticos da área de Química Farmacêutica Medicinal, ilustra com exatidão a importância de se explorar as interações entre o ligante e seu sítio de interação, bem como os benefícios obtidos ao explorar essas interações.9,10
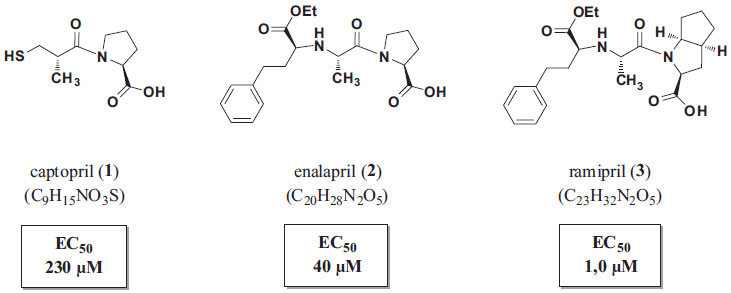 Figura 1. Captopril, enalapril e ramipril, iECAs utilizados na terapêutica com seus respectivos valores de concentração efetiva 50% (EC50)11
O mecanismo de ação associado a esta classe de fármacos reside na habilidade destas moléculas em se ligar ao sítio catalítico da enzima, impedindo, assim, a conversão de angiotensina I em angiotensina II.12 A enzima conversora de angiotensina (ECA) é uma metaloproteinase responsável por hidrolisar a angiotensina I (um decapeptídeo inativo), convertendo-a em angiotensina II (um octapeptídeo ativo) (Figura 2), gerando como principais respostas fisiológicas o aumento da resistência vascular periférica, a secreção renal de aldosterona e a redução dos níveis séricos de bradicinina, que culminam no aumento da pressão arterial do indivíduo.13 A atividade catalítica da ECA é dependente de zinco II (Zn2+) e as bases químicas necessárias ao reconhecimento molecular de seu substrato estão associadas a uma sequência tripeptídica específica presente na angiotensina I. Esta sequência tripeptídica é capaz de realizar coordenação com o íon Zn2+ e sofrer um ataque nucleofílico, que resulta na clivagem do substrato decapeptídico (Figura 2A).14
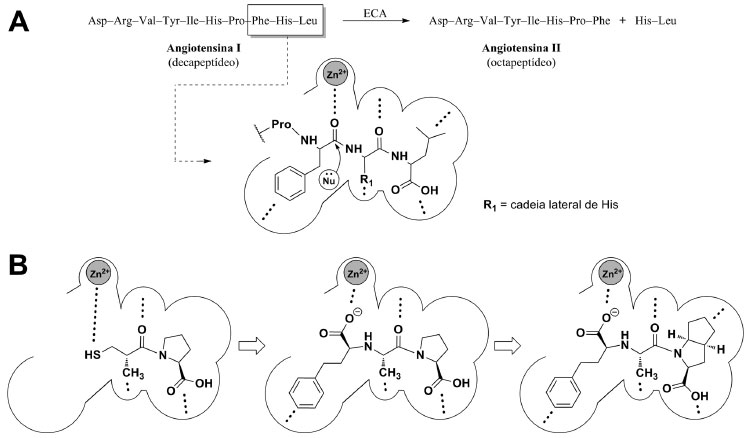 Figura 2. (A) Reconhecimento molecular da tríade peptídica Phe-His-Leu pela ECA. As linhas tracejadas indicam regiões onde há interação entre o substrato e a enzima. (B) Interação dos fármacos captopril, enalaprilato (forma ativa do enalapril) e ramiprilato (forma ativa do ramipril), respectivamente, com o sítio ativo da ECA. Observa-se o aumento do número de interações em um dos bolsões da enzima, exploradas em cada um dos fármacos
O desenvolvimento dos iECAs, como enalapril (2) e ramipril (3), cada um a sua época, ilustra a otimização estrutural efetuada na estrutura do captopril (1) e a capacidade de exploração dos bolsões acessórios de interação, o que se reflete nas EC50 reduzidas de ambos, bem como na redução dos efeitos adversos associados ao captopril (1).10,11,15 A Figura 2B apresenta a evolução estrutural obtida no planejamento destes fármacos, do ponto de vista molecular. Atualmente, são conhecidas as estruturas cristalográficas do complexo de cada um desses fármacos com a ECA.16,17 Este fato permite que a exploração das interações intermoleculares fármaco-receptor biológico seja realizada experimentalmente, fazendo com que o estudante tenha a oportunidade de conhecer tridimensionalmente a fase farmacodinâmica de ação de fármacos. A disciplina de Química Farmacêutica (4 créditos) tem sido ministrada aos estudantes do quinto semestre do curso de Farmácia da Universidade São Francisco (USF), em paralelo à disciplina de Planejamento e Síntese de Fármacos (4 créditos). Por todo o quinto semestre, os temas abordados em ambas as disciplinas são totalmente integrados, objetivando a melhor fixação dos conteúdos pelo corpo discente. Após análise criteriosa do processo ensino-aprendizagem acerca das "Interações Fármaco-Receptor", a estratégia pedagógica aqui apresentada foi cuidadosamente planejada e inserida no plano de atividades da disciplina de Química Farmacêutica da USF e tem sido aplicada sistematicamente como método alternativo à metodologia puramente expositiva. Esta estratégia pode ser abordada em 4 horas/aula, ou então dividida em duas aulas de 2 horas cada. De forma mais específica, relacionam-se os objetivos a serem alcançados pelos discentes, com a aplicação desta estratégia:
Sugere-se, inicialmente, introduzir aos alunos a fundamentação teórica sobre o tema "Interações Fármaco-Receptor" para, posteriormente, torná-los aptos a avaliar estas interações experimentalmente. Os eventos desta atividade prática estão subdivididos em:
Esta abordagem didática contempla, de forma significativa, os aspectos teóricos e práticos relevantes ao aprendizado das interações F-R, permitindo aos alunos a compreensão do mecanismo de ação dos fármacos e sua relação com o planejamento de novas moléculas bioativas. Tendo-se em vista que esta é área estreitamente relacionada à inovação, da qual carece o País, busca-se aprimorar a formação dos futuros profissionais farmacêuticos que se interessem em atuar neste campo, não só no âmbito acadêmico, como, também, na área de Pesquisa e Desenvolvimento (P&D) de indústrias farmacêuticas, especialmente, as indústrias farmoquímicas nacionais.
PARTE EXPERIMENTAL Esta abordagem foi planejada de forma a abranger os conceitos do tema de forma progressiva, de acordo com a evolução das etapas práticas aqui descritas. O tutorial completo do procedimento prático, descrito neste artigo, segue amplamente detalhado e ilustrado no material suplementar. Aquisição, análise, manipulação e tratamento dos complexos cristalográficos Aquisição Desenvolvido em 1971, o Protein Data Bank (PDB) consiste em um repositório mundial de livre acesso para arquivos sobre a disposição tridimensional (3D) de macromoléculas, como proteínas, enzimas e cadeias de ácidos nucleicos. Estas macromoléculas podem estar isoladas ou complexadas a micromoléculas, como substratos, cofatores enzimáticos e/ou fármacos.18 A partir do PDB (http://www.rcsb.org), os alunos aprendem a realizar pesquisas de estruturas proteicas, observando o modo de registro de cada estrutura depositada no banco de dados, denominado código PDB, geralmente composto por quatro algarismos alfanuméricos. Neste momento, os estudantes são orientados a inserir na barra de pesquisa o código PDB de cada um dos complexos que serão utilizados no decorrer da aula, sendo eles: 1UZF16 (ECA complexada com o captopril, Figura 3); 1UZE16 (ECA complexada com o enalaprilato); e 2X9217 (ECA complexada com o ramiprilato). Na página principal de cada um dos co-cristais, os estudantes são orientados a buscar e salvar em seus computadores os arquivos-texto contendo as coordenadas cartesianas de cada complexo cristalográfico, clicando no ícone "Download Files" e, em seguida, sobre "PDB File (Text)", como ilustra a Figura 4. Geralmente o arquivo é salvo com o nome CÓDIGO.pdb, no caso dos arquivos utilizados neste estudo: 1UZF.pdb, 1UZE.pdb e 2X92.pdb.
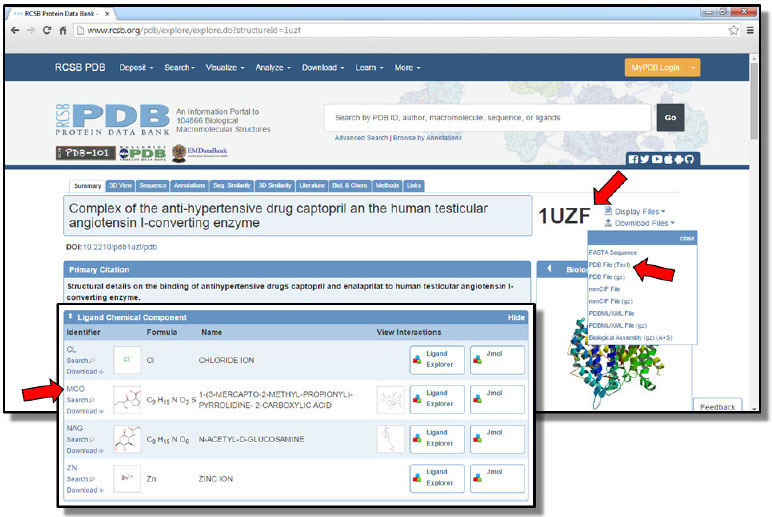 Figura 3. Página principal do co-cristal captopril-ECA (código PDB: 1UZF). As setas indicam o código PDB, o arquivo texto das coordenadas cartesianas do complexo e a identificação do ligante captopril, respectivamente19
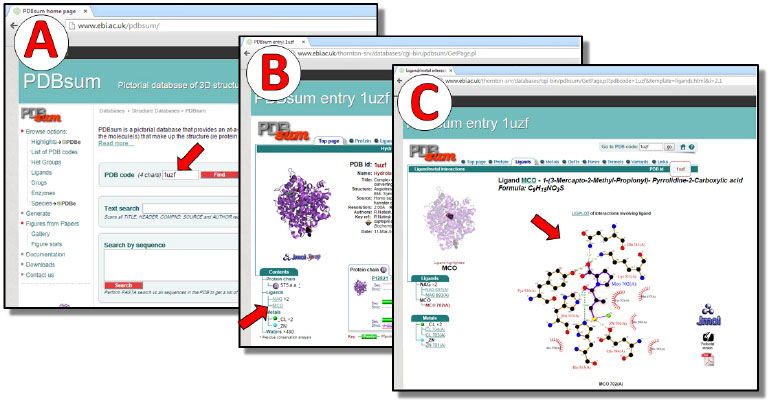 Figura 4. Análise dos complexos no PDBsum. (A) página inicial. (B) página contendo os dados do complexo captopril-ECA (código PDB: 1UZF). (C) diagrama LIGPLOT gerado para o complexo captopril-ECA. As setas vermelhas indicam, respectivamente, o local para inserçao do código PDB e o subcódigo do fármaco captopril (MCO) que, ao ser selecionado, gera o diagrama representativo da interaçao22-24
Análise A análise dos complexos inicia-se na própria página principal dos co-cristais no PDB. Nela os alunos movimentam a barra de rolagem até visualizar o campo "Ligand Chemical Component", em que identificam o subcódigo atribuído a cada micromolécula presente no co-cristal, momento em que devem anotar a identificação referente ao fármaco presente no complexo. Os fármacos de cada complexo utilizado nesta metodologia são identificados pelos subcódigos: MCO (captopril, Figura 3), EAL (enalaprilato) e X92 (ramiprilato). Esta identificação é importante para a análise do bolsão de interação do fármaco no PDBsum. O PDBsum (http://www.ebi.ac.uk/pdbsum/) é um banco de dados ilustrado, que compila informações de estruturas 3D depositadas no PDB, tornando mais intuitiva a visualização e a interpretação das mesmas.20 Neste é possível que o estudante visualize de forma bidimensional (2D) quais aminoácidos da macromolécula compõem o bolsão de ligação do fármaco (ou micromolécula), bem como os aminoácidos deste bolsão, que efetivamente estabelecem as interações F-R com o ligante. Por meio do PDBsum os alunos podem observar a natureza dos aminoácidos existentes, por exemplo, no bolsão catalítico da ECA e avaliar a complementariedade existente entre macro e micromolécula. Na página inicial do PDBsum, os estudantes são orientados a inserir o código PDB do co-cristal que desejam analisar, neste caso os códigos 1UZF (Figura 4A), 1UZE e 2X92, e então clicar em "Find". Com a abertura da nova página, contendo as informações gráficas do complexo, os alunos são orientados a clicar sobre o subcódigo referente ao fármaco em questão, neste caso, MCO (captopril, Figura 4B), EAL (enalaprilato) e X92 (ramiprilato). A próxima janela apresenta um diagrama 2D LIGPLOT,21 gerado automaticamente a partir das informações coletadas do complexo proteína-ligante (Figura 4C). No diagrama LIGPLOT são indicados os pontos de interação entre a micromolécula e todos os aminoácidos do seu sítio de ligação, a distância interatômica de cada interação (apenas para ligações de hidrogênio) e o tipo de interação realizada, sendo representadas principalmente por:
Com a visualização dos diagramas LIGPLOT de cada fármaco complexado à ECA, os estudantes são orientados a observar e anotar o número de cada aminoácido presente no diagrama, que juntos constituem o bolsão de interação (Tabela 1). Esta atividade é necessária para as etapas subsequentes da aula, além de se mostrar como valiosa estratégia pedagógica, que introduz aos alunos as interações F-R dos inibidores da ECA, ao nível molecular.
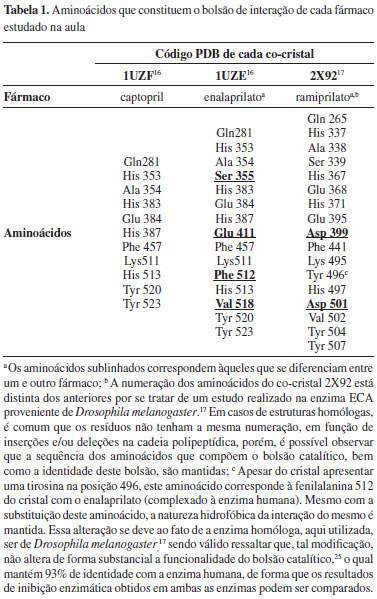
Manipulação e tratamento dos co-cristais O objetivo desta etapa da atividade é apresentar aos estudantes os aspectos tridimensionais relevantes para que ocorra interação entre a macromolécula e o ligante em questão. A manipulação dos arquivos texto dos cristais (arquivo CÓDIGO.pdb, salvo no computador pelos alunos anteriormente) inicia-se no programa WebLabViewer Lite 4.2,26 um programa de livre acesso e disponível para o uso acadêmico-educacional (http://www.scalacs.org/TeacherResources/). O WebLabViewer Lite possui uma interface bastante intuitiva, de fácil manuseio, e pode ser utilizado em determinados estudos de química teórica. Fazendo-se uso deste programa, é possível realizar diversas análises como a visualização espacial de micro e macromoléculas, gerar mapas de superfície molecular, determinar a estereoquímica de compostos, além de medir ângulos e distâncias interatômicas entre regiões da mesma molécula, ou de moléculas distintas. Neste momento, recomenda-se que, por meio de um dos cristais (sugerimos o co-cristal do captopril, 1UZF), seja demonstrado aos alunos as diferenças existentes entre micro e macromolécula, destacando as formas de representação dos ligantes (linhas, modelo de Corey-Pauling-Koltun (CPK) e bastões), das estruturas proteicas (folhas, hélices, tubos e pranchas), bem como a estereoquímica do fármaco e a superfície de potencial eletrostático, tanto da enzima quanto do ligante. Esta abordagem segue amplamente descrita no material suplementar. Ao abrir o arquivo no programa, os estudantes podem visualizar a disposição de todos os átomos presentes no cristal, os que compõem a enzima, aqueles que compõem os ligantes (fármaco, íon Zn2+ e demais moléculas artefatos de cristalização), e também moléculas de água (indicadas por pontos vermelhos). Após as demonstrações preliminares sugeridas anteriormente, e para melhor visualização do bolsão de interação, os alunos são orientados a limpar a estrutura dos três arquivos salvos (1UZF.pdb, IUZE.pdb e 2X92.pdb), ou seja, excluir todos os aminoácidos que não realizam interação com os ligantes, os artefatos de cristalização (tomando cuidado para não se excluir o íon Zn2+) e as moléculas de água. É válido ressaltar que, para estes cristais, não há a participação de moléculas de água intermediando ligações de hidrogênio entre ligante e macromolécula, de forma que todas podem ser removidas sem causar prejuízos à condução do estudo. Em situações em que há o recrutamento de moléculas de água para a formação do complexo fármaco-receptor, este pode ser verificado no diagrama LIGPLOT, previamente utilizado na etapa de Análise dos co-cristais. Após este tratamento, é possível visualizar os fármacos juntamente aos aminoácidos que compõem o bolsão catalítico e que, efetivamente, realizam algum tipo de interação com os mesmos (listados na Tabela 1). A Figura 5 apresenta as estruturas tratadas, que poderão ser encaminhadas aos estudos de correlação das interações entre fármaco e receptor biológico.
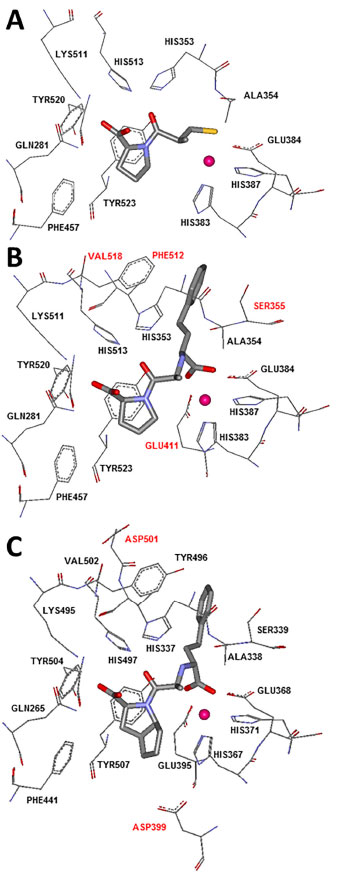 Figura 5. Em bastão as estruturas do captopril (A), enalaprilato (B) e ramiprilato (C), com seus respectivos aminoácidos do bolsão catalítico (representados em linhas), após o tratamento das estruturas cristalográficas. Em magenta está representado o íon Zn2+. Em preto os aminoácidos do bolsão. Em vermelho os aminoácidos que se diferenciam nas interações com cada fármaco
Estudos de correlação das interações dos fármacos com a macromolécula Uma vez as estruturas tratadas, os estudantes são orientados a avaliar as distâncias interatômicas entre as regiões de cada fármaco que interagem com a enzima, sendo estas interações do tipo:
A Figura 6A apresenta as medidas de distância interatômica em cada região de interação do captopril com os resíduos de aminoácidos da ECA (de acordo com o diagrama LIGPLOT). Subsequentemente, os alunos são instruídos a gerar o mapa de potencial eletrostático dos aminoácidos que compõem o bolsão catalítico da ECA e incentivados a rotacionar a estrutura, identificando as regiões de maior e menor densidade eletrônica (Figura 6B). Neste momento, o docente pode discorrer sobre a natureza destes resíduos de aminoácidos e as consequências que os mesmos causam na distribuição eletrônica do bolsão.
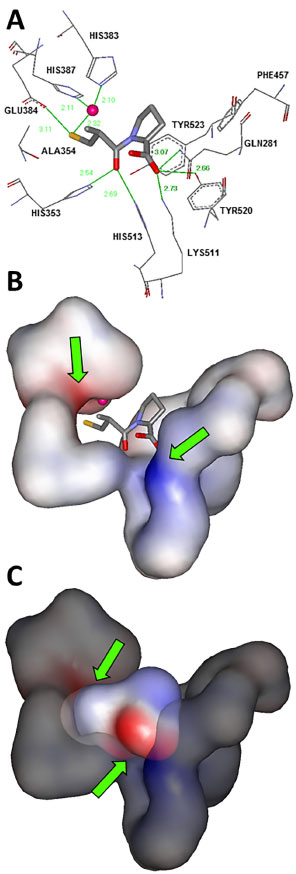 Figura 6. (A) Distâncias interatômicas em Å das regiões de interação do captopril com a ECA. (B) As setas indicam as regiões com elevada densidade eletrônica (em vermelho) e com baixa densidade eletrônica (em azul) da ECA. (C) As setas indicam a complementariedade eletrônica existente entre o captopril e o bolsão catalítico da ECA. Em magenta está representado o íon Zn2+
Em seguida, os estudantes são orientados a gerar os mapas de potencial eletrostático para cada fármaco e sobrepô-los àqueles gerados previamente para os respectivos bolsões (Figura 6C). Tendo sido gerados os mapas, o professor pode então apontar os aspectos relevantes sobre a afinidade e complementariedade existente entre micro e macromolécula, para que assim possa ser formado o complexo F-R, havendo a interação e por sua vez, o efeito biológico esperado. É válido ressaltar aos estudantes que a fase farmacodinâmica de fármacos, como o próprio nome indica, se trata de um evento biológico, dependente de diversos fatores como, por exemplo, fatores termodinâmicos e estado de solvatação. Os estudantes devem estar cientes de que estas variáveis não são consideradas em visualizações de estruturas estáticas, como as co-cristalografadas, mas que se fazem presentes em sistemas biológicos. Análise comparativa entre os complexos e interpretação dos resultados Com os mapas gerados, é possível observar que os fármacos enalaprilato e ramiprilato são capazes de interagir com bolsões acessórios não envolvidos na interação com o captopril (Figura 7). Esta habilidade pode ser diretamente associada à afinidade que estas moléculas possuem pelo bolsão catalítico da enzima. Como o incremento de afinidade é diretamente proporcional à potência dos fármacos, o fato de o enalaprilato e o ramiprilato interagirem em loci adicionais da macromolécula justifica a habilidade destes em produzir a ação esperada em concentrações reduzidas, se comparadas às concentrações terapêuticas do captopril. Isso pode ser comprovado pelos valores de EC50 de cada um desses fármacos. Na medida em que o captopril possui EC50 = 230 µmol L-1, o enalapril (convertido a enalaprilato in vivo) apresenta potência quase 6 vezes superior (EC50 = 40 µmol L-1), enquanto o ramiprilato (bioativado a partir do ramipril), potência 40 vezes superior (EC50 = 1 µmol L-1).11
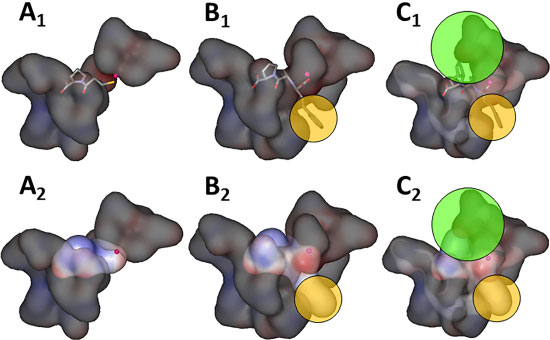 Figura 7. Disposição do captopril no sítio ativo da ECA (A1) e a complementariedade existente entre as superfícies de potencial eletrostático do mesmo com o sítio catalítico (A2). Disposição do enalaprilato no sítio ativo da ECA (B1) e a complementariedade existente entre as superfícies de potencial eletrostático do complexo (B2). O círculo amarelo indica uma nova região de interação, explorada pelo enalaprilato. Disposição do ramiprilato no sítio ativo da ECA (C1) e a complementariedade existente entre as superfícies de potencial eletrostático do complexo (C2). O círculo amarelo indica a região adicional de interação já explorada pelo enalaprilato. O círculo verde indica a nova região de interação, explorada apenas pelo ramiprilato. O íon Zn2+ está representado em magenta
Com o perfil comparativo apresentado na Figura 7, pode-se discutir durante a aula os aspectos positivos que determinadas modificações moleculares conferem a arcabouços bioativos. Neste exemplo, a introdução do grupo feniletil à estrutura do enalaprilato permitiu que ele realizasse interações de Van der Waals e hidrofóbicas com a Serina-355, com a Fenilalanina-512 e, também, com a Valina-518, aminoácidos não envolvidos na interação com o captopril. O mesmo raciocínio se aplica ao sistema bicíclico inserido na estrutura do ramiprilato. Esta estratégia permitiu a interação deste fármaco com a cadeia lateral do Aspartato-399 da enzima, aumentando ainda mais a afinidade do ramiprilato pela enzima. Merece ser realçado, em relação à REA dos iECA, o fato de o enalaprilato e o ramiprilato serem pró-fármacos27 convertidos em suas estruturas ativas no organismo do paciente, e que são pertencentes à subclasse dos iECA dicarboxílicos. Esta subclasse dos iECA possui característica bem peculiar, que é responsável por conferir grande aumento de afinidade aos seus representantes. A função carboxílica destes fármacos, inserida em substituição ao grupo sulfidrila do captopril, mimetiza o estado de transição do substrato da enzima quando este sofre o ataque nucleofílico. Vale mencionar que a função carboxilato é bioisiostérica do intermediário tetraédrico peptídico.10 A natureza multidisciplinar desta abordagem prática permite que os alunos tenham contato com temas que, por ventura, possam ser abordados ao longo do semestre letivo, de maneira que "Latenciação de Fármacos", "Modificações Moleculares", "Inibidores Enzimáticos", "Análogos do Estado de Transição" e "Estereoquímica" são assuntos intimamente correlacionados com os temas aqui apresentados, e que podem ser apontados durante a execução desta atividade, enriquecendo ainda mais o conteúdo da aula.
CONCLUSÕES As atividades práticas de análise das interações F-R, envolvendo práticas computacionais, têm sido executadas com êxito pelos alunos. A abordagem aqui apresentada revela-se estratégia de baixo custo agregado, capaz de ser aplicada à disciplina de Química Farmacêutica Medicinal nos diferentes cursos de Farmácia oferecidos pelas universidades. É importante ressaltar que esta estratégia didática pode ser aplicada para fármacos de diferentes classes terapêuticas, bastando para isso que existam os co-critais dos mesmos, com seus respectivos alvos biológicos. Como sugestão, esta abordagem pode ser adaptada aos antagonistas de receptores adrenérgicos beta-2, como o alprenolol (código PDB: 3NYA),28 comparando-os frente ao complexo do receptor junto à epinefrina, seu agonista endógeno (código PDB: 4LDO).29 A estratégia descrita neste trabalho inclui diversas etapas, como a aquisição dos complexos proteicos co-cristalografados, análise preliminar do bolsão de interação, análise das interações realizadas e análise comparativa da disposição espacial dos fármacos, correlacionando-as com suas respectivas repostas biológicas. Em geral, a primeira reação dos estudantes ao visualizar os complexos tridimensionais no programa WebLabViewer Lite 4.226 revela certo receio dos mesmos em utilizar a ferramenta. Porém, com a condução das etapas iniciais, estes mesmos estudantes desenvolvem rápida intimidade com os recursos do programa e passam a efetuar as tarefas de forma intuitiva e bastante eficiente. Os relatórios elaborados pelos alunos após a aplicação da estratégia aqui apresentada revelam a capacidade desta abordagem em agregar conhecimento aos mesmos de forma lúdica, sendo comuns relatos que enaltecem a capacidade do método em transferir uma visão mais abrangente sobre o mecanismo de ação e interação de fármacos em nível molecular. Ressalte-se a capacidade do experimento de permitir aos estudantes a visualização tridimensional do complexo micro-macro molécula. As avaliações aplicadas no término do semestre letivo da disciplina revelam que, em média, 91% dos alunos consideram a estratégia efetiva para o seu aprendizado, sendo que 88% deles gostariam que esta abordagem fosse aplicada em outras oportunidades e 65% dos estudantes consideram o programa utilizado bastante intuitivo e de fácil manuseio. Apenas 9% dos alunos consideram que a estratégia atingiu parcialmente ou não foi capaz de contribuir para com o seu aprendizado. Dessa forma, observa-se que a integração das técnicas experimentais, que fazem uso de recursos tecnológicos e audiovisuais, ao ensino convencional da Química Farmacêutica Medicinal traz diversos benefícios. A aplicação dessas técnicas como instrumento didático aprimora a compreensão dos aspectos inerentes à formação do complexo F-R pelo estudante, tornando o ensino atrativo e interessante.
MATERIAL SUPLEMENTAR O tutorial completo do procedimento prático descrito neste trabalho segue amplamente detalhado e ilustrado no material suplementar, disponível em http://quimicanova.sbq.org.br, na forma de arquivo PDF, com acesso livre.
REFERÊNCIAS 1. Huggins, D. J.; Sherman, W.; Tidor, B.; J. Med. Chem. 2012, 55, 1424. DOI: http://dx.doi.org/10.1021/jm2010332 PMID: 22239221 2. Pan, A. C.; Borhani, D. W.; Dror, R. O.; Shaw, D. E.; Drug Discovery Today 2013, 18, 667. DOI: http://dx.doi.org/10.1016/j.drudis.2013.02.007 PMID: 23454741 3. Barreiro, E. J.; Rodrigues, C. R.; Quim. Nova 1997, 20, 1. 4. Roberts, J. R.; Hagedorn, E.; Dillenburg, P.; Patrick, M.; Herman, T.; Biochem. Mol. Biol. Educ. 2005, 33, 105. DOI: http://dx.doi.org/10.1002/bmb.2005.494033022426 PMID: 21638554 5. Carvalho, I.; Pupo, M. T.; Borges, A. D. L.; Quim. Nova 2003, 26, 428. DOI: http://dx.doi.org/10.1590/S0100-40422003000300023 6. Verli, H.; Barreiro, E. J.; Quim. Nova 2005, 28, 95. DOI: http://dx.doi.org/10.1590/S0100-40422005000700018 7. Billeter, M.; Perspect. Drug Discovery Des. 1995, 3, 151. DOI: http://dx.doi.org/10.1007/BF02174472 8. Santos, R. N.; Andricopulo, A. D.; Braz. J. Phys. 2013, 43, 268. DOI: http://dx.doi.org/10.1007/s13538-013-0149-7 9. Stewart, J. M.; Ferreira, S. H.; Greene, L. J.; Biochem. Pharmacol. 1971, 20, 1557. DOI: http://dx.doi.org/10.1016/0006-2952(71)90284-X PMID: 4355304 10. Lemke, T. L.; Williams, D. A.; Roche, V. F.; Zito, S. W.; Foye's Principles of Medicinal Chemistry, 7th ed., Lippincott Williams & Wilkins: Philadelphia, 2008. 11. Kirsten, R.; Nelson, K.; Kirsten, D.; Heintz, B.; Clin. Pharmacokinet. 1998, 34, 457. DOI: http://dx.doi.org/10.2165/00003088-199834060-00003 PMID: 9646008 12. Anthony, C. S.; Masuyer, G.; Sturrock, E. D.; Acharya, K. R.; Curr. Med. Chem. 2012, 19, 845. DOI: http://dx.doi.org/10.2174/092986712799034950 PMID: 22214449 13. Brown, N. J.; Vaughan, D. E.; Circulation 1998, 97, 1411. DOI: http://dx.doi.org/10.1161/01.CIR.97.14.1411 PMID: 9577953 14. De Souza, M. C.; Franco, C. H. J.; Pinheiro, C. B.; Diniz, R.; Polyhedron 2014, 81, 290. DOI: http://dx.doi.org/10.1016/j.poly.2014.06.025 15. Patrick, G. L.; An Introduction to Medicinal Chemistry, 4th ed., Oxford University Press: Oxford, 2008. 16. Natesh, R.; Schwager, S. L. U.; Evans, H. R.; Sturrock, E. D.; Acharya, K. R.; Biochemistry 2004, 43, 8718. DOI: http://dx.doi.org/10.1021/bi049480n PMID: 15236580 17. Akif, M.; Georgiadis, D. Mahajan, A.; Dive, V.; Sturrock, E. D.; Isaac, R. E.; Acharya, K. R.; J. Mol. Biol. 2010, 400, 502. DOI: http://dx.doi.org/10.1016/j.jmb.2010.05.024 PMID: 20488190 18. Berman, H. M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T. N.; Weissig, H.; Shindyalov, I. N.; Bourne, P. E.; Nucleic Acids Res. 2000, 28, 235. DOI: http://dx.doi.org/10.1093/nar/28.1.235 PMID: 10592235 19. http://www.rcsb.org/pdb/explore/explore.do?structureId=1uzf, acessada em Maio 2015. 20. Laskowski, R. A.; Hutchinson, E. G.; Michie, A. D.; Wallace, A. C.; Jones, M. L.; Thornton, J. M.; Trends Biochem. Sci. 1997, 22, 488. DOI: http://dx.doi.org/10.1016/S0968-0004(97)01140-7 PMID: 9433130 21. Wallace, A. C.; Laskowski, R. A.; Thornton, J. M.; Protein Eng. 1995, 8, 127. DOI: http://dx.doi.org/10.1093/protein/8.1.31 PMID: 7630882 22. http://www.ebi.ac.uk/pdbsum/, acessada em Maio 2015. 23. http://www.ebi.ac.uk/thornton-srv/databases/cgi-bin/pdbsum/GetPage.pl, acessada em Maio 2015. 24. http://www.ebi.ac.uk/thornton-srv/databases/cgi-bin/pdbsum/GetPage.pl?pdbcode=1uzf&template=ligands.html&l=2.1, acessada em Maio 2015. 25. Coates, D.; Isaac, R. E.; Cotton, J.; Siviter, R.; Williams, T. A.; Shirras, A.; Corvol, P.; Dive, V.; Biochemistry 2000, 39, 8963. DOI: http://dx.doi.org/10.1021/bi000593q PMID: 10913309 26. WebLab Viewer Lite - Molecular Visualization Software 4.2, Accelrys Inc., ACS-Southern California Section, San Diego-CA, USA, 2001. 27. Parise-Filho, R.; Polli, M. C.; Filho, S. B.; Garcia, M.; Ferreira, E. I.; Braz. J. Pharm. Sci. 2010, 46, 393. DOI: http://dx.doi.org/10.1590/S1984-82502010000300003 28. Wacker, D.; Fenalti, G.; Brown, M. A.; Katritch, V.; Abagyan, R.; Cherezov, V.; Stevens, R. C.; J. Am. Chem. Soc. 2010, 132, 11443. DOI: http://dx.doi.org/10.1021/ja105108q PMID: 20669948 29. Ring, A. M.; Manglik, A.; Kruse, A. C.; Enos, M. D.; Weis, W. I.; Garcia, K. C.; Kobilka, B. K.; Nature 2013, 502, 575. DOI: http://dx.doi.org/10.1038/nature12572 PMID: 24056936 |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access