
 |
 |
 |
 |
 |
Nota Técnica
| Detection and quantification of phytochemical markers of Ilex paraguariensis by liquid chromatography |
|
Rodrigo M. C. PintoI; Bruna M. LemesI; Acácio A. F. ZielinskiII; Traudi KleinI; Fernado de PaulaIII; Airton KistIV; Anna S. F. MarquesV; Alessandro NogueiraII; Ivo M. DemiateII; Flávio L. BeltrameI,*
IDepartamento de Ciências Farmacêuticas, Universidade Estadual de Ponta Grossa, 84030-900 Ponta Grossa - PR, Brasil Recebido em 07/03/2015 *e-mail: flaviobeltra@gmail.com Ilex paraguariensis (yerba-mate) is used as a beverage, and its extract requires adequate quality control methods in order to guarantee quality and safe use. Strategies to develop and optimize a chromatographic method to quantify theobromine, caffeine, and chlorogenic acid in I. paraguariensis extracts were evaluated by applying a quality by design (QbD) model and ultra high-performance liquid chromatography (UHPLC). The presence of these three phytochemical markers in the extracts was evaluated using UHPLC-MS and was confirmed by the chromatographic bands in the total ion current traces (m/z of 181.1 [M+H]+, 195.0 [M+H]+, and 353.0 [M-H]-, respectively). The developed method was then transferred to a high-performance liquid chromatography (HPLC) platform, and the three phytochemical markers were used as external standards in the validation of a method for analyses of these compounds in extracts using a diode array detector (DAD). The validated method was applied to quantify the chlorogenic acid, caffeine, and theobromine in the samples. HPLC-DAD chromatographic fingerprinting was also used in a multivariate approach to process the entire data and to separate the I. paraguariensis extracts into two groups. The developed method is very useful for qualifying and quantifying I. paraguariensis extracts. INTRODUCTION Ilex paraguariensis A. St. - Hil., (Aquifoliaceae) is a species which is popularly known as yerba-mate. It is native to South America and is commonly found in Brazil, Paraguay, Argentina and Uruguay.1 The aerial parts of this plant are used as raw material to prepare teas and other typical stimulating drinks such as "chimarrao" and "tererê".2 However, this species and its extracts have also been increasingly used (both nationally and internationally), as a raw material or as extracts in the production of medicines, cosmetics, dyes, drinks and dietary supplements.3 The Brazilian industry produces more than 200 million tons of yerba-mate per year, and part of this production is exported to other countries such as Chile, the United States, France and Spain.4 These exports are due to the interest of the international market in natural products; yerba-mate contains a great variety of stimulant, antioxidant, healing, digestive, anti-adipogenic, antimicrobial, and anti-inflammatory properties, which are assigned to xanthines and phenolic compounds.5-10 Studies have proved some of the biological activities of this plant, but few studies have evaluated the quality of the raw material or extracts used to prepare many of the food or pharmaceutical products derived from yerba-mate. The first edition of the Brazilian Pharmacopeia describes the botanical characterization of I. paraguariensis and suggests that the quantification of caffeine (phytochemical marker) by the gravimetric method should be used as a test to prove the authenticity and quality of the plant species.11 Recent studies have utilized chromatographic methods to analyze yerba-mate. Dugo et al.12 successfully applied high-performance liquid chromatography (HPLC) coupled to mass spectrometry (MS) in one and bidimensional analyses to determine 26 substances in aqueous and methanolic extracts of this plant. Dartora et al.13 conducted studies using ultra high-performance liquid chromatography (UHPLC) to quantify the xanthines and phenolic compounds in yerba-mate extracts and they observed the influence of processing on the chemical composition of the samples. Bravo, Goya and Lecumberry14 characterized the phenolic fraction of yerba-mate by liquid chromatography coupled to a mass spectrometer (LC-MS) and they detected caffeoylquinic and dicaffeoylquinic acids as the major components, as well as identifying other glycoside flavonoids in the material. Peixoto et al.15 validated LC-MS analyses to determine the saponins in unripe fruits of I. paraguariensis, and detected substances of different polarities, which suggested the use of this method for quality control. The chemical quality control of herbal ingredients is very challenging because most plants used as raw material or as extracts are native, and are collected from different regions. Moreover, the constituents responsible for the desired therapeutic activities are unknown and they are not found in a single compound. They are normally all the chemical compounds which are present in the preparation (phytocomplex), and it is this phytocomplex that is responsible for these actions. For these reasons, regulatory agencies have strongly recommended the selection and analysis of phytochemical markers to help in the quality control of herbal products.16,17 Similarly, the use of chromatographic fingerprinting to evaluate the complexity and the variability of chemical components is recommended to improve quality control information.18,19 Traditionally, in order to obtaining the chromatographic fingerprint and an adequate analytical method, the optimization of chromatographic conditions begins with an exploratory gradient and then some chromatographic parameters (injection volume, organic modifier, stationary phase, gradient range, and slope) are manually evaluated by the analyst to optimize the evaluation of the matrix.20 Fortunately, computer-assisted development utilizing design of experiments (DOE) has been incorporated within the LC method development and, together with quality by design (QbD) principles this has helped greatly in the development of chromatographic methods. The QbD strategy employs a systematic and scientific approach focused on the quality of products, and knowledge about the process or product. This strategy provides sufficient understanding to establish a design space, which is the multidimensional combination and interaction of input variables and process parameters that have been demonstrated to provide quality assurance.21 QbD principles can be applied to the development of the LC method to obtain better methods, with insight regarding the interactions of the main factors that can affect the quality of the separation of compounds. The use of statistical DOE is necessary to obtain knowledge about the method with the least amount of effort and experiments. Using DOE it is possible to determine the group of key experiments that would be necessary to construct an empirical model that will relate the effects of the instrumental, physical and chemical factors that affect the separation, and also to model the response surface. In other words, it is possible to find the region of the response surface that meets the requirements of the method - the design space.22 DOE and QbD principles have two drawbacks: one is the number of experiments that are necessary to obtain the design space, and the second is the necessity that the chromatographer has a certain level of experience in DOE. However, with the introduction of UHPLC technologies, the time necessary to obtain the results has been reduced and the development of DOE-QbD software has allowed the entire process to be automated, connecting the software to the UHPLC instrument through an intuitive interface, which facilitates the use of DOE in the development of LC and creates a powerful strategy to develop fast and reliable LC methods. For example, Bianchini, Castellano and Kaufman23 used experimental designs to develop a simple and rapid HPLC method for the simultaneous determination of different impurities in pridinol mesylate. They proved the robustness, precision and selectivity of the method, and also its usefulness for quality control. To contribute to the evaluation of the quality and authenticity of products sold as yerba-mate in the Brazilian market a study for qualification and quantification was evaluated, developed, validated and applied.
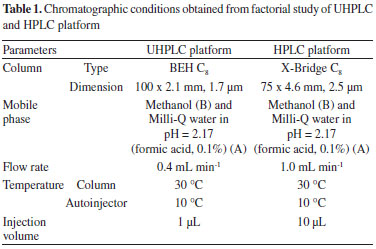
EXPERIMENTAL Plant material One plant sample of I. paraguariensis used for the industrial production of yerba-mate was obtained in Ivai, Paraná, Brazil (coordinates: - 24º 56' 8.29",-50º 53' 24.54"). It was identified (a voucher [number 19414] was deposited in the Herbarium of the State University of Ponta Grossa) and was nominated as a reference sample (sample 2). The aerial parts of this species were processed using the procedure used in the yerba-mate industry. Six other raw samples (aerial parts, samples 1, 3-7) were sold as yerba-mate in the Brazilian commercial market and were purchased for this study. The particle size of all the samples was standardized using a 2.5 mm sieve. Extraction procedure and sample preparation The dried materials were used to prepare the hydroalcoholic and aqueous extracts. The hydroalcoholic extracts were prepared by macerating 100 g of samples in 500 mL of water:ethanol (20:80, v/v), for 10 days with occasional stirring (daily). The aqueous extracts were prepared by infusing 25 g of samples with 500 mL of water at 80 ºC for 30 minutes. Both extracts were filtered and the solvents were removed under reduced pressure at 40 ºC. They were then freeze-dried, yielding ± 4.2% (w/w) for the aqueous extracts and ± 10.5% (w/w) for the hydroalcoholic extracts, respectively, and stored under refrigeration (2-8 ºC until the moment of use). Chemicals Methanol (MeOH) and acetonitrile (ACN) were HPLC-grade (J. T. Baker, PA, USA); formic acid was analytical grade (Synth, SP, Brazil). Water was purified using a Millipore Milli-Q system (Millipore, SP, Brazil) and it was used for all the experiments. The standards of chlorogenic acid (95% purity), theobromine (99% purity), and caffeine (99% purity) were purchased from Sigma-Aldrich (MO, USA). Instrumentation The UHPLC analyses were recorded in an Acquity UPLC H-ClassTM system (Milford, MA, USA) equipped with quaternary solvent delivery pump, auto sampler, and diode array detector (DAD), Empower 3 chromatography data software (Waters Corporation, Milford, MA) and Fusion AE QbD-DOE based LC method development software (S-Matrix Corporation, Eureka, Ca). The HPLC analyses were recorded in an Alliance 2695 HPLC system (Milford, MA, USA) composed of a quaternary pump, an oven, an on-line degasser, an auto injector and a 2998 diode array detector (Waters 2998-DAD) (Waters Corporation, Milford, MA). The UHPLC/MS system consisted of an Acquity UPLC I-ClassTM chromatograph and a Waters Xevo TQ-STM spectrometer (Waters Corporation, Milford, MA), operating in the ESI mode. The MS experiment setup and data acquisition were conducted using Mass lynx (V. 3.5) software. Standards and samples Preparation of standards A stock solution of each standard was prepared in water:MeOH (20:80, v/v, volumetric flask), at a concentration of 0.5 mg mL-1 for caffeine and 0.2 mg mL-1 for theobromine and chlorogenic acid (each). From this stock solution, the concentrations of external standards were prepared by diluting aliquots in the appropriate volume of solvent with calibrated micropipettes. Following the same procedure, the stock solution was also used to prepare the three quality control solutions in three different concentrations. The samples were filtered in PES membranes with a pore size of 0.22 µm (syringe filters - K18-230, Kasvi, PR, Brazil). Preparation of samples The lyophilized extracts (1 mg) were accurately weighed and diluted with an appropriate volume (1 mL) of water:MeOH (20:80, v/v), using calibrated micropipettes. The samples were filtered with PES membranes with a pore size of 0.22 µm (syringe filters - K18-230, Kasvi, PR, Brazil). Factorial design for obtaining the UHPLC chromatographic fingerprints This study was carried out in two stages. The first stage involved screening, followed by the optimization of the chromatographic fingerprint. Briefly, in the screening analyses, qualitative factors were evaluated, such as the column, organic modifier, mobile phase, pH range and gradient time; this totaled 45 experiments. This information is provided in the supplementary material (SM) (Table 1S). Subsequently, a second stage (optimization) was performed, where quantitative factors were analyzed while the selected variables of the screening were fixed (Table 2S in the SM). Thirty-nine (39) experiments were performed during this stage. The data were processed using the Acquity UPLCTM H-Class Method Development System and the criteria used for the selection of the best method were the highest number of peaks with a resolution higher than 1.5 and asymmetry of peak < 2. The defined conditions were adequate for the UHPLC-MS and HPLC-DAD analyses. UHPLC-MS analyses For the ESI detection, the following conditions were applied: a source temperature of 150 ºC; ESI capillary of 3 KV and a cone voltage of 40 V; drying gas (N2) flow of 800 L h-1; and temperature of 500 ºC, argon as collision (3.43 x10-3 mBar). The column was a Waters Acquity BEH C8 1.7 µm particle size (100 mm x 2.1 mm i.d., Waters, SP, Brazil), adjusted to a temperature of 40 ºC; the mobile phase was composed of 0.1% formic acid in water (v/v, pH=2.17) (A), and acetonitrile (B); the flow rate was 0.4 mL.min-1; the injection volume was 1 µL and the gradient elution applied was 5% of B for 1.55 min; 5.0-70.0% of B for 12.05 min; maintained for 1.0 min at 70.0%; 70.0-90.0% of B for 1 min. All the parameters were adjusted for unitary mass resolution. HPLC-DAD analyses The separations were carried out using an X-Bridge HPLC C8 2.5 µm particle size (75 mm x 4.6 mm i.d., Waters, SP, Brazil) column. A binary gradient elution at a flow rate of 1.0 mL min-1 was employed, using an aqueous solution of 0.1% formic acid (v/v, pH=2.17) as solvent A and methanol as solvent B (gradient: 0-5% of B for 2.62 min; 5.0-32.5% of B for 12.13 min; 32.5-90.0% of B for 1 min). An isocratic condition (90% of B was maintained for 4.86 min before performing the reverse gradient to 5% of B for 0.22 min. In order to condition the column, 5% of B was maintained for 7.25 min until a new injection was performed. The injected volume was 10 µL and the detection was obtained by DAD (200-400 nm). The oven and auto-injector temperatures were 30 ºC and 10 ºC, respectively and the chromatograms were recorded at 280 nm. This chromatographic condition was applied in the validation procedure and in the application of the method for the quantification of the main phytochemical markers. Evaluation of precision of instrument injection The precision of the instrument injection was evaluated by the relative standard deviation (RSD %) of retention times and the peak areas of the samples injected. Multivariate analyses Peak alignment Due to intrinsic chromatographic conditions, sometimes slight variations in the obtained data may result in a situation where the peaks are not in the same column as the matrix.24 Studies have demonstrated the importance of this mathematical treatment of chromatographic data.25 Consequently, the correlation optimized warping (COW) algorithm was employed.26 The peaks of sample 2 were adopted as reference and the other samples were aligned to them. PCA analyses The principal component analyses (PCA) were performed using Matlab v.7.5 (MathWorks Inc., Natick, USA) software and the multivariate method was used to analyze the results. A matrix composed of samples (n = 21) and responses (n = 5,700) was built, totaling 119,700 data points. The results obtained for each parameter were adopted as columns and the extract samples were used as rows. PCA was applied to separate the extracts obtained from the yerba-mate samples (n = 21) according to their chromatographic fingerprints. Sample similarities were calculated on the basis of the Euclidean distance and the incremental method was used to form and suggest groups of similar samples.27 Quantitative assay External standard curve High-performance liquid chromatographic-diode array was used to validate the method.28 Using the appropriate standard solution of caffeine, theobromine and chlorogenic acid, aliquots were added in vials to provide the following calibration solutions: 0.08, 0.24, 0.27, 0.30, 0.33, 0.36 and 0.40 µg mL-1 for caffeine; 0.1, 0.40, 0.44, 0.48, 0.52, 0.56 and 0.70 µg mL-1 for theobromine; and 0.04, 0.16, 0.18, 0.20, 0.22, 0.24 and 0.36 µg mL-1 for chlorogenic acid. The calibration standards were prepared in triplicate. The calibration curve was drawn by plotting the peak area against the concentration of the compound. Standard addition curve Aliquots of the appropriate standard solutions of caffeine, theobromine and chlorogenic acid were added to a constant amount of yerba-mate standard extract (1 mL). The samples were prepared in triplicate and were processed as described above. A calibration curve was calculated by plotting the peak area against the concentration of the added compound. Recovery, intermediate precision and accuracy The relative recovery was examined using three quality control solutions prepared in water:MeOH (20:80, v/v) at 0.15, 0.25 and 0.35 µg mL-1 for caffeine; 0.02, 0.05 and 0.06 µg mL-1 for theobromine; and 0.10, 0.25 and 0.30 µg mL-1 for chlorogenic acid. The peak area ratios of the three extracted control solutions at each concentration were compared with the three standard solutions to derive a percentage recovery. The accuracy, and inter and intra-day variability of the method were evaluated by replicate analyses using the same three concentrations that were used for the recovery experiment. Three samples of each concentration were prepared and analyzed on three non-consecutive days. The accuracy of the method was evaluated by back calculation and tested by using blind unknowns at two different concentrations, which were prepared by a different analyst. Selectivity A diode array detector (DAD) was used to identify the markers and also to check the peak purity of the analytical run of the extracts. The retention factors of the standards were also used for the identification of the analytical run. Limits of detection and quantification The limit of detection (LOD) was calculated by taking a signal-to-noise ratio of three as criterion, while the acceptance criterion for the limit of quantification (LOQ) was the signal-to-noise ratio of ten, with coefficient of variation (CV) and accuracy for three extracted samples below 20% variability. Analyses of samples One reference sample and six other commercial samples were analysed (triplicate) by the chromatographic method to obtain and compare the fingerprints and to quantify the phytochemical markers. Statistical analyses All analyses were performed in triplicate and the quantitative data were presented as mean and standard deviation. The mean of each group (hydroalcoholic and aqueous) was calculated and the differences between these groups was tested by independent samples using Student's t-test (p < 0.05).
RESULTS AND DISCUSSION Traditionally, the development of chromatographic methods has been performed by studying the chromatographic characteristics observed in an exploratory run. This approach normally varies the concentration of the organic solvent by time, e.g. 5-100% over one or two hours.20 The analyst uses a visual inspection of the chromatograms to decide whether it is necessary to optimize other parameters that can contribute to chromatographic resolution, such as pH, organic modifier, stationary phase, gradient range, and slope. The development of methods using UHPLC technology have more advantages that HPLC technology, i.e. they are faster, have a higher power resolution and provide a reduction in the amount of sample, solvent and development costs; consequently they provide many of the benefits of green chemistry.29 To obtain experiments that will provide more knowledge about the interaction of the main factors which are responsible for the quality of the separation, QbD principles with DOE are necessary. In our study we used a user-friendly QbD-DOE software that interfaced with the LC equipment and supported the chromatographer in all stages of the experimental design. The adopted criteria were the highest number of peaks with a resolution higher than 1.5 and asymmetry of peak < 2. These criteria were chosen to identify a chromatogram that could identify the greatest amount of chemical information present in the extract (fingerprint), and which also had good enough resolution to detect and quantify the main phytochemical markers of I. paraguariensis. Initially, the qualitative factors selected for screening were the column, mobile phase pH range, gradient time and organic modifier. The experiments totaled 45 runs and the results showed the best response when using a BEH C8 column, acid pH (pH=2.4), 12 min of gradient time and methanol as organic modifier for the analyzed matrix (Table 1S in the SM, condition 33). After the screening study, the critical factors that influenced the selectivity of the method were evaluated. The experimental design, which was obtained through 39 experiments, showed the effect of each factor in the response (Table 2S in the SM). Different responses may be selected during this stage, depending on the objective of the study.21 Different regression models, (linear, quadratic and cubic) were tested during both stages. The cubic model was adopted in this study because it showed the greatest r2 value and it did not present lack of fit (p < 0.05). During the optimization stage, the following regression equation was obtained (Equation 1). 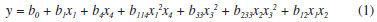 where y is the number of peaks (Rs > 1.5); b0 is the constant; b1 is the flow rate; b2 is the final concentration of organic modifier; b3 is the temperature; and b4 is the pH. The multiplication among coefficients represents the interactions and the x values are the experimental parameters applied to each factor. The cubic regression model presented a r2 value of 0.9544, showing the correlation between the evaluated parameters and the response. The coefficients were subjected to analysis of variance (ANOVA) and the highest statistical significances were found for b1, b4 and b33 (p<0.001), indicating their importance in the modulation of the y response. One way to graphically interpret the interaction between the factors is by means of a three-dimensional graph, which is called response surface, and which plots the factors two by two according to the response. Figure 1 demonstrates the negative effect of the flow and of the concentration of the organic modifier on the number of peaks. Although crude extracts contain a lot of compounds (secondary metabolites) that can lead to a inadequate separation of these compounds in an adequate time analysis, in our study the optimized condition that were obtained made it possible to achieve a higher number of peaks with resolution greater than 1.5. These were obtained in conditions of low flow rate and a final organic level of up to 55%. Aproximately 17 peaks were found in the optimum flow condition of the chromatographic pump, pH and the final level of the organic solvent (Table 2S in the SM, condition 14).
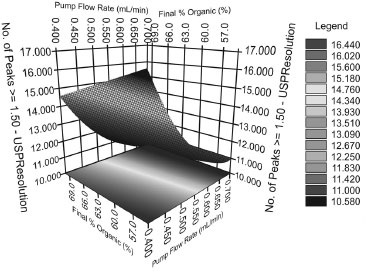 Figure 1. Response surface: flow rate/final concentration of organic modifier (MeOH) versus number of peaks with R > 1.5
After the optimized chromatographic condition was achieved, these parameters were applied in the UHPLC-MS analyses to search in the samples of the three selected markers: theobromine, chlorogenic acid and caffeine. These metabolites are generally related to I. paraguariensis and are found in high concentrations. Furthermore, theobromine and caffeine are responsible for the stimulating effect of the plant and chlorogenic acid is an important antioxidizing agent.30,31 The UHPLC-MS analyses for theobromine and caffeine were carried out in the positive ion mode because these compounds have basic groups and it is relatively easy for these compounds to be protonated. The analyses in the positive mode showed peaks at m/z 181.1 [M+H]+ for theobromine and m/z 195.0 [M+H]+ for caffeine. On the other hand, for the chlorogenic acid (which is easier to deprotonate and analyze using the negative ion mode) a peak at m/z 353.0 [M-H]- was verified (Figure 2) The results that were obtained were attributed to the ions of the three markers, which are normally found in I. paraguariensis extracts.12 The main fragments (daughter ions) observed in the positive mode were m/z 96 [M+H]+ and m/z 138 [M+H]+, which were attributed to theobromine and caffeine molecules, respectively. For the chlorogenic acid molecular analysis in the negative mode, the daughter ion fragment was m/z 165 [M-H]-. These experiments confirmed the presence of these three markers and the results described in the literature.12,14
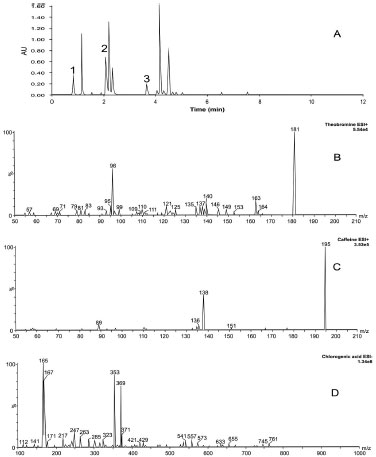 Figure 2. (A) UHPLC-MS, 1 - tr: 0.83; 2 - tr: 2.18, 3 - tr: 3.87. Chromatogram of total ions. B - m/z: 181.1 [M+H]+ (positive mode), C - m/z: 195.0 [M+H]+ (positive mode), D - m/z: 353.0 [M-H]- (negative mode). Mobile phase: methanol (B) in aqueous solution of 0.1% formic acid (v/v, pH=2.17, A) - (gradient: 5% of B for 1.55 min; 5.0-70.0% of B for 12.05 min; maintained for 1.0 min at 70.0%; 70.0-90.0% of B for 1 min), flow rate: 0.4 mL min-1
The same optimized chromatographic conditions obtained through the processing of this data using UHPLC were simulated for the HPLC platform (Table 1) using Selectivity Chart and Acquity Columns Calculator sofware (Waters Corporation, Milford, MA).
The HPLC chromatogram of yerba-mate using the developed method was obtained and the peaks of the chemical markers were well resolved. The next stage of the study was to evaluate and try to quantify the chlorogenic acid, caffeine and theobromine in the reference extract, and in the commercial samples. Therefore, the validation of the method was conducted according to the parameters of ANVISA using HPLC-DAD equipment.28 The linearity of the method was confirmed by the linear regression from the calibration curve data for the substances.The selectivity of the caffeine, theobromine and chlorogenic acid were examined by comparing the diode array spectra and the retention factors of these substances in the analytical runs, and in the injections of the isolated standards. The parameters of regression equation, linearity, precision, limits of detection and quantification, (LOD and LOQ, respectively) and recovery are shown in Table 2.
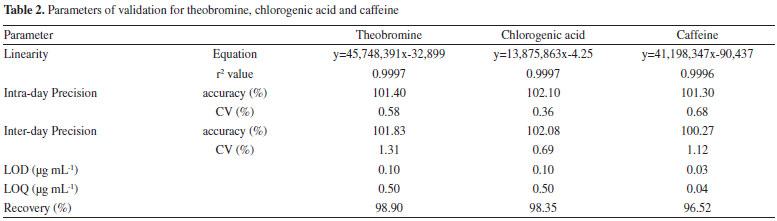
The residual analyses of the calibration curves were evaluated and no problems were found such as lack of fit or deviations of linearity. The three adjusted r2 values were greater than 0.9990. The matrix inteference was evaluated by calculating a second calibration curve using the standard addition method. This curve also showed as linear (y = 45,602,318x + 390,026 and r2 = 0.9999 for theobromine; y = 14,029,018x + 720,117 and r2 = 0.9999 for chlorogenic acid; and y = 41,839,984x + 2,016,217 and r2 = 0.9988 for caffeine). The parallelism and agreement between these two curves was observed, which made it possible to infer that the matrix did not interfere in the analyses of the phytochemical markers.32 Given that the validated HPLC-DAD methods were suitable for the quantitative analyses of chlorogenic acid, caffeine and theobromine, the method was applied to analyze the samples used in this study and the results of the quantification of the marker samples are presented in Table 3.
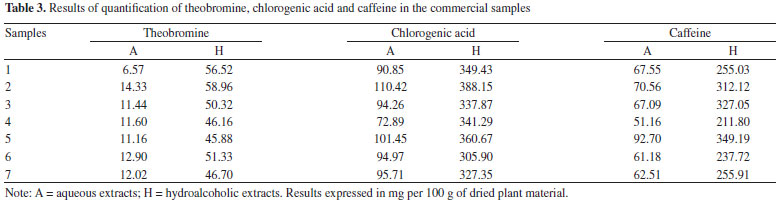
The differences in the quantification of the markers between the hydroalcoholic and the aqueous extracts were statistically significant for the three markers (p < 0.05, unpaired Student's t-test). These differences were related to the solubility of the substances in the extraction solvents. It is known that xanthines are poorly soluble in water.33 Other studies have quantified the phenolics and xanthines in yerba-mate: Borré et al.34 detected 0.86 g of caffeine and 0.15 g of theobromine per 100 g of leaves (dry material), with a caffeine/theobromine ratio of 5.7. The level of chlorogenic acid was quantified as 0.14 g per 100 g dry plant material (leaves) by the same authors. In another study, Gnoatto et al.35 concluded that the extraction method affected the content of xanthines in I. paraguariensis, and they found a caffeine/theobromine ratio that ranged from 1.4 to 14.0. Similarly, in our study the caffeine/theobromine ratio varied from 4.8 to 10.3. The quantitative determination of some components is not sufficient to determine the identity of a species because it does not evaluate the totality of chemical data from the chromatographic analyses. Thus, analyses using multivariate fingerprinting may increase the effectiveness of the quality control approach for plant materials (phytocomplex). Multivariate analysis is also important in highlighting and identifying samples based on chemical differences (in this case the chromatographic profile). Van der Kooy et al.36 applied nuclear magnetic resonance (NMR) and mass spectrometry (MS) coupled to multivariate analysis in the quality control of herbal materials and phytopharmaceuticals, and it seems that their study provided promising future perspectives and development for these techniques. Martins, Pereira-Filho and Cass25 analyzed the chromatographic profile of six Phyllanthus species, and by using chemometric models they classified and authenticated 25 commercial samples. Kim et al.37 used NMR and multivariate analysis to classify eleven South American Ilex species and clearly showed that metabolomic discrimination helped in the chemotaxonomic classification of the samples. The chromatographic fingerprints obtained by HPLC at 280 nm for different extracts of yerba-mate revealed a great similarity between the samples (Figure 3) but did not provide conclusive information. Using visual comparison of the chromatograms, it was inferred that all the evaluated samples seemed to be yerba-mate and that the variations in the intensity of the peaks presented differences that reinforced the quantitative analyses. Thus, PCA was applied in order to evaluate and to corroborate the data.
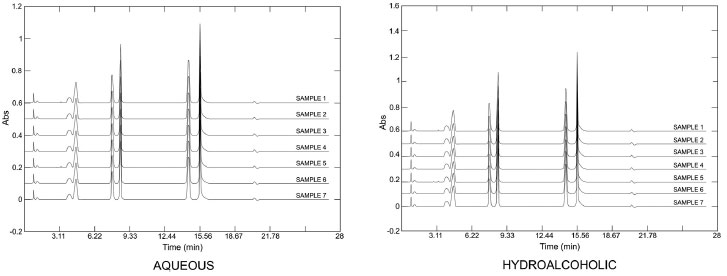 Figure 3. Fingerprint chromatograms of aqueous and hydroalcoholic extracts of Ilex paraguariensis
Principal component 1 (PC1) explained up to 98.56% of total variance for the HPLC data of the hydroalcoholic extracts PC2 explained 0.82% and PC3 explained 0.41%. Likewise, PC1 explained 99.05% of total variance for the HPLC data of the aqueous extracts, PC2 explained 0.57% and PC3 explained 0.21%. Using PCA it was possible to separate the I. paraguariensis extracts into two groups; the first group, in the positive quadrant, included the aqueous extracts, and the negative quadrant included the hydroalcoholic extracts (Figure 4). The difference between the hydroalcoholic and aqueous groups was due to the chemical affinity of many of the constituents, which vary according to the polarity of the substances. This causes qualitative and quantitative changes in the samples, which leads to a distinct classification.38 These results showed the efficiency of the method in grouping the samples according to the similarities and diferences between them.
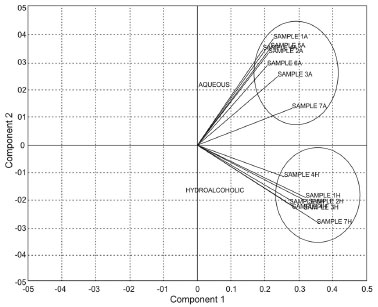 Figure 4. PCA analyses of the hydroalcoholic (H) and aqueous (A) extracts of Ilex paraguariensis
It was possible to identify some reasons for these differences. Extract preparation is an important factor in the variation in plant composition, as presented above. Due to thermic exposure, depending on the time and temperature, degradation of the chemical constituents may occur in industrial process; inefficient stabilization may result in enzymatic reactions that also change this composition.38 Furthermore, edaphic factors influence the synthesis of secondary metabolities and localization may be an explication for this classification.35
CONCLUSION Yerba-mate is a very common species in South America and the industries that process this plant need to be concerned about efficient quality control of this raw material. Chromatographic methods have been shown to be fast and efficient tools for the qualitative and quantitative evaluation of plant materials. In an attempt to optimize chromatographic conditions, the 'Design Space' approach was applied because it makes it possible to improve the analysis of the critical factors that influence the separation. Consequently, in the first part of this study QbD was applied to the UHPLC platform (performed in the Luis Barssoti Application Laboratory in Waters (Sao Paulo)) to obtain, in a short period of time, an efficient method to separate and identify the three main phytomarkers of I. paraguariensis in the same run. Although the UHPLC system provides analyses with a rapid outcome, the reality is that Brazilian phytopharmaceutical industries use HPLC platforms for quality control on a routine basis. Taking that into consideration, the developed conditions were transferred to HPLC-DAD equipment. The validation was performed and the method was applied to allow the identification and quantification of the differences between the samples commercialized as yerba-mate raw material. The use of multivariate analysis aided the chromatographic experiments because it permited the evaluation of several factors simultaneously, which was very important because the identity of the species cannot be confirmed simply by the quantification of the main phytochemical markers. The use of HPLC-DAD for the quantification of the main phytochemical markers, together with the application of multivariate analysis fingerprinting, can contribute to a routine quality control evaluation of raw materials, enabling the authentication of I. paraguariensis samples
SUPPLEMENTARY MATERIAL A table describing the factors evaluated in the 'Design Space' screening stage and factors evaluated in the 'Design Space' optimization stage can be found in pdf format at http://quimicanova.sbq.org.br/, with free access.
ACKNOWLEDGEMENTS The authors would like to thank CAPES and the Fundaçao Araucária for financial support, and also the Luis Barssoti Application Laboratory (Waters, Sao Paulo) for the analyses.
REFERENCES 1. Cansian, R. S.; Teses de Doutorado, Universidade Federal de Sao Carlos, Brasil, 2003. 2. Meinhart, A. D.; Bizzotto, C. S.; Ballus, C. A.; Rybka, A. C. P.; Sobrinho, M. R.; Cerro-Quintana, R. S.; Teixeira-Filho, J.; Godoy, H. R. T.; J. Agric. Food Chem. 2010, 58, 2188. DOI: http://dx.doi.org/10.1021/jf903781w PMID: 20058928 3. Pereira, M. F.; Ciampi, A. Y.; Inglis, P. W.; Souza, V. A.; Azevedo, V. C. R.; Applications in Plant Sciences 2013, 1, 1. 4. Parana, Secretaria Estadual de Agricultura e Abastecimento. In: Yerbamate, o ouro verde do Paraná. Available at http://www.gazetadopovo.com.br/vida-e-cidadania/especiais/erva-mate/index.jpp, accessed May, 2014. 5. Matsumoto, R. L. T.; Bastos, D. H. M.; Mendonça, S.; Nunes, V. S.; Bartchewsky, W.; Ribeiro Jr., M. L.; Carvalho, P. O.; J. Agric. Food Chem. 2009, 57, 1775. DOI: http://dx.doi.org/10.1021/jf803096g PMID: 19219987 6. Wnuk, M.; Lewinska, A.; Oklejewicz, B.; Bugno, M.; Slota, E.; Bartosz, G.; Mutat. Res. 2009, 679, 18. DOI: http://dx.doi.org/10.1016/j.mrgentox.2009.07.017 PMID: 19733686 7. Arçari, D. P.; Bartchewsky, W.; dos Santos, T. W.; Oliveira, K. A.; de Oliveira, C. C.; Gotardo, E. M.; Pedrazzoli, J.; Gambero, A.; Ferraz, L. F.; Carvalho, P. O.; Ribeiro, M. L.; Mol Endocrinol 2011, 335, 110. DOI: http://dx.doi.org/10.1016/j.mce.2011.01.003 8. Burris, K. P.; Davidson, P. M.; Stewart, C. N.; Harte, F. M.; J. Food Sci. 2011, 76, 456. DOI: http://dx.doi.org/10.1111/j.1750-3841.2011.02255.x 9. Boaventura, B. C.; Di Pietro, P. F.; Stefanuto, A.; Klein, G. A.; de Morais, E. C.; de Andrade, F.; Wazlawik, E.; da Silva, E. L.; Nutrition 2012, 28, 657. DOI: http://dx.doi.org/10.1016/j.nut.2011.10.017 PMID: 22578980 10. Kim, J. H.; Kang, Y. R.; Lee, H. Y.; Moon, D. I.; Seo, M. Y.; Park, S. H.; Choi, K. H.; Kim, C. R.; Kim, S. H.; Oh, J. H.; Cho, S. W.; Kim, S. Y.; Kim, M. G.; Chae, S. W.; Kim, O.; Oh, H. G.; Lab. Anim. Res. 2012, 28, 23. DOI: http://dx.doi.org/10.5625/lar.2012.28.2.109 11. Pharmacopoeia dos Estados Unidos do Brasil, 1st ed., Nacional: Sao Paulo, 1926. 12. Dugo, P.; Cacciola, F.; Donato, P.; Jacques, R. A.; Caramao, E. B.; Mondello, L.; J. Chromatogr. A 2009, 1216, 7213. DOI: http://dx.doi.org/10.1016/j.chroma.2009.08.030 PMID: 19726042 13. Dartora, N.; Souza, L. M.; Santana-Filho, A. P.; Iacomini, M.; Valduga, A. T.; Gorin, P. A. J.; Sassaki, G. L.; Food Chem. 2011, 129, 1453. DOI: http://dx.doi.org/10.1016/j.foodchem.2011.05.112 14. Bravo, L.; Goya, L.; Lecumberry, E.; Food Res. Int. 2007, 40, 393. DOI: http://dx.doi.org/10.1016/j.foodres.2006.10.016 15. Peixoto, M. P. G.; Kaiser, S.; Verza, S. G.; Resende, P. E.; Treter, J.; Pavei, C.; Borré, G. L.; Ortega, G. G.; Phytochem. Anal. 2012, 23, 415. DOI: http://dx.doi.org/10.1002/pca.1374 PMID: 22105927 16. Reif, K.; Sievers, H.; Steffen, J. P.; HerbalGram 2004, 63, 38. 17. Alaerts, G.; Matthijs, N.; Smeyers-Verbeke, J.; Vander-Heyden, Y.; J. Chromatogr. A 2007, 1172, 1. DOI: http://dx.doi.org/10.1016/j.chroma.2007.07.080 PMID: 17942105 18. Xu, C.; Liang, Y.; Chau, F.; Heyden, Y. V.; J. Chromatogr. A 2006, 1134, 253. DOI: http://dx.doi.org/10.1016/j.chroma.2006.08.097 PMID: 16962124 19. Gan, F.; Ye, R.; J. Chromatogr. A 2006, 1104, 100. DOI: http://dx.doi.org/10.1016/j.chroma.2005.11.099 PMID: 16360164 20. Snyder, L. R.; Dolan, J. W.; J. Chromatogr. A 1996, 721, 3. DOI: http://dx.doi.org/10.1016/0021-9673(95)00771-7 21. Swartz, M. E.; Krull, I. S.; LCGC North Am. 2008, 11, 1. 22. Molnár, I.; Rieger, H. J.; Monks, K. E.; J. Chromatogr. A 2010, 1217, 3192. 23. Bianchini, R. M.; Castellano, P. M.; Kaufman, T. S.; Anal. Chim. Acta 2009, 654, 141. DOI: http://dx.doi.org/10.1016/j.aca.2009.09.022 PMID: 19854345 24. Liang, Y. Z.; Xie, P.; Chan, K.; J. Chromatogr. B 2004, 812, 53. DOI: http://dx.doi.org/10.1016/S1570-0232(04)00676-2 25. Martins, L. R. R.; Pereira-Filho, E. R.; Cass, Q. B.; Anal. Bioanal. Chem. 2011, 400, 469. DOI: http://dx.doi.org/10.1007/s00216-011-4749-1 PMID: 21336795 26. Skov, T.; Van den Berg, F.; Tomasi, G.; Bro, R.; J. Chemometr. 2006, 20, 484. DOI: http://dx.doi.org/10.1002/cem.1031 27. Zielinski, A. A. F.; Haminiuk, C. W. I.; Nunes, C. A.; Schnitzler, E.; Van Ruth, S. M.; Granato, D.; Compr. Rev. Food Sci. Food Saf. 2014, 13, 300. DOI: http://dx.doi.org/10.1111/1541-4337.12060 28. Brasil, Agência Nacional de Vigilância Sanitária. Resoluçao nº 899/2003. Diário Oficial da Uniao, 2003. 29. Chen, S.; Kord, A.; J. Chromatogr. A 2009, 1216, 6204. DOI: http://dx.doi.org/10.1016/j.chroma.2009.06.084 PMID: 19616783 30. Bastos, D. H. M.; Saldanha, L. A.; Catharino, R. R.; Sawaya, A. C. H. F.; Cunha, I. B. S.; Carvalho, P. O.; Eberlin, M. N.; Molecules 2007, 12, 423. DOI: http://dx.doi.org/10.3390/12030423 PMID: 17851401 31. Bracesco, N.; Sanchez, A. G.; Contreras, V.; Menini, T.; Gugliucci, A.; J. Ethnopharmacol. 2011, 136, 378. DOI: http://dx.doi.org/10.1016/j.jep.2010.06.032 PMID: 20599603 32. Beltrame, F. L.; Filho, E. R.; Barros, F. A. P.; Cortez, D. A. G.; Cass, Q. B.; J. Chromatogr. A 2006, 1119, 257. DOI: http://dx.doi.org/10.1016/j.chroma.2005.10.050 PMID: 16360665 33. Roth, H. J.; Eger, K.; Trochütz, R.; Pharmaceutical Chemistry, 1st ed., Elis Horwood: New York, 1991, p. 589-601. 34. Borré, G. L.; Kaiser, S.; Pavei, C.; Silva, F. A.; Bassani, V. L.; Ortega, G. G.; J. Liq. Chromatogr. Relat. Technol. 2010, 33, 362. DOI: http://dx.doi.org/10.1080/10826070903526055 35. Gnoatto, S. C. B., Bassani, B. L., Coelho, G. C., Schenkel, E. P.; Quim. Nova 2007, 30, 304. DOI: http://dx.doi.org/10.1590/S0100-40422007000200012 36. Van der Kooy, F.; Maltese, F.; Choi, Y. H., Kim, H. K.; Verpoorte, R.; Planta Med. 2009, 75, 763. DOI: http://dx.doi.org/10.1055/s-0029-1185450 PMID: 19288400 37. Kim, H.; Khan, S.; Wilson, E. G.; Kricun, S. D. P.; Meissner, A.; Goraler, S.; Deelder, A. M.; Choi, Y. H.; Verpoorte, R.; Phytochemistry 2010, 71, 773. DOI: http://dx.doi.org/10.1016/j.phytochem.2010.02.001 PMID: 20199787 38. Isolabella, S.; Cogoi, L.; López, P.; Anesini, C.; Ferraro, G.; Filip, R.; Food Chem. 2010, 122, 695. DOI: http://dx.doi.org/10.1016/j.foodchem.2010.03.039 |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access