
 |
 |
 |
 |
 |
Artigo
| Preliminarily development of a moisture-activated bioresorbable polymeric platform for drug delivery |
|
Renê O. do CoutoI,*; Sven D. SommerfeldII; Koustubh DubeII; Osvaldo de FreitasI; Joachim KohnII
IFaculdade de Ciências Farmacêuticas de Ribeirão Preto, Universidade de São Paulo, Av. do Café - s/n, 14040-903 Ribeirão Preto - SP, Brasil Recebido em 17/12/2014 *e-mail: rocouto@usp.br Bioresorbable polymeric films were prepared by solvent casting using a tyrosine-derived polycarbonate and metronidazole (MDZ) as the model drug at 2.5%, 5% and 10% (w/w). Drug loading did not affect the water uptake, drug release, polymer degradation or erosion profiles. All devices released approximately 85% (w/w) of the drug within a 1.5 h period. This may be attributed to the rapid water uptake of the polymer. An increase in the water uptake correlated with a linear rate increase of the polymer degradation (0.968 < R2 < 0.999). Moreover, MDZ presented a remarkable plasticizing effect for the polymer and drug loading exerted a significant impact on the mechanical properties of the obtained films. The results obtained can be used to further the development of novel biocompatible and biodegradable polymeric platforms for the delivery of metronidazole and other drugs in a broad range of pharmaceutical applications. INTRODUCTION The significant growth of polymer science and technology has provided novel materials for both medical and pharmaceutical fields. Regarding the development of drug delivery systems, the research interests for both academic community and industry have been shifting towards the implementation of biodegradable polymeric systems platform in addition to the conventional approach such as diffusion-controlled and solvent activated systems.1-3 The advantages of degradable polymers are described elsewhere.4,5 Typically, bioresorbable polymers are broken down into (i) hydrolytically (e.g., Poli(α-esters, polyurethanes, poly(alkyl cyanoacrylates) etc.), and (ii) enzymatically (e.g., protein, polysaccharides and poly(amino acids)) degradable.5 As regards the first group aforementioned, special attention has been focused on the rationale development of pseudo poly(amino acids) e.g., tyrosine-derived copolymers, which represent a novel and promising drug delivery platform.6-9In this pursuit, discovery tools such as combinatorial methods, high-throughput experimentation and computational modeling has been applied.10,11 Over the last few decades, these efforts lead to the development of a combinatorial polymer library of tyrosine-derived polycarbonates consisting of desaminotyrosyl-tyrosine alkyl esters (DTR), desaminotyrosyl-tyrosine (DT), and low molecular weight blocks of poly(ethylene glycol) (PEG). This class of polymers is presented by the general master formula: poly(DTR-co-XX%-DT-co-YY%-PEG Mw carbonate) In addition, the notation RXXYY(Mw) is used, where R is the alkyl pendent chain, XX is the mole percent of DT, YY is the mole percent of PEG and Mw is the average molecular weight of PEG.6 Besides their proved long-term biocompatibility,12,13 these polymers present a remarkable structure-activity relationship, once DTR moieties give strength and stability, DT moieties provides degradability and PEG moieties tune water uptake, drug release, and biological and mechanical properties according to the requirements of a specific application.6,14,15 Herein, we report on a systematic investigation of the hydration, degradation and erosion of a resorbable tyrosine-derived polycarbonate E2505(2K) and its impact on the drug release from solvent cast thin films. The effect of drug loading on the mechanical properties of the films was evaluated as well. Metronidazole (MDZ) was used as model drug in this investigation. MDZ is an antibiotic widely used in the development of drug delivery systems for the treatment of several human diseases including periodontal16-27 and vaginal infections.28
MATERIAL Metronidazole (batch SLBG3633V), water, acetonitrile (ACN), d6-dymethyl sulphoxide (DMSO), tetrahydrofuran (THF) and trifluoroacetic acid (TFA) were purchased from (Sigma-Aldrich Co., Steinheim, Germany). Also, phosphate buffer solution (PBS) pH 7.4 (Lonza Inc., Allendale, NJ, USA) and Teflon® filters 0.45 µm x 13mm (Whatman Inc., Florham Park, NJ) were used. All other chemicals were of reagent grade.
EXPERIMENTAL Polymer synthesis and characterization The polymer used was a tyrosine-derived polycarbonate called E2505(2K), which was synthetized via solution polycondensation reactions using the procedure described elsewhere.13 The polymer was placed in the absence of oxygen into sealed aluminum foil bags, and stored under refrigeration (-10 ºC) prior to characterization and further use. The chemical structure was determined by 1H-NMR. A sample of approximately 10 mg was dissolved in 0.75 mL d6-DMSO and analyzed on a Varian VNMRS 400 MHz machine (Varian Inc., Palo Alto, CA, USA). The molecular weight (weight average, Mw) and polydispersity index (PDI) were determined using a Gel Permeation Chromatography (GPC) system consisting of a Waters 717 Plus Autosampler, Waters 2414 Refractive Index Detector, Waters 515 HPLC Pump, serial PLgel columns (5 µm beads; 104 Å and 102 Å pores size; 30 cm long; Polymer Laboratories Inc., subsidiary of Varian Inc., Palo Alto, CA), PLgel 5 µm guard column and Empower® 2 Software (Waters Inc., Milford, MA, USA). The column was operated at room temperature with DMF containing 0.1% TFA as the mobile phase at a flow rate of 1.0 mL min-1. 10 mg sample (n = 3) was dissolved in 1 mL DMF containing 0.1% TFA and filtered with a 0.45 µm Teflon® filter. The injection volume was 20 µL and run time for the analysis was 25 min. Molecular weights were determined relative to polystyrene standards. The total residual solvent content were measured using a Thermogravimetric Analyzer (TGA) Model TGA/SDTA851e with STARe software version 19.10 (Mettler-Toledo Inc., Columbus, OH, USA). Sample of approximately 10 mg (n = 3) were heated from 25 to 300 ºC at a rate of 10 ºC min-1. The glass transition temperature (Tg) was measured using a Differential Scanning Calorimeter (DSC) Model DSC 823e (Mettler-Toledo Inc., Columbus, OH, USA). Approximately 10 mg sample (n = 3) was heated from 25 to 200 ºC at a rate of 10 ºC min-1, and then kept at 200 ºC for 5 min. Then it was cooled down to -40 ºC at the same rate and kept at this temperature for 5 min. Finally, sample was heated from -40 to 200 ºC at a rate of 10 ºC min-1. Fabrication of drug-loaded films Drug was dissolved under low agitation speed in tetrahydrofuran (THF) and then the polymer was added to make a 4% (w/v) solution (final volume of 25 mL) and kept overnight for dispersion. For unloaded sample, the polymer was dissolved in THF to make a 4% (w/v) solution. The dispersions were cast into Teflon® dishes (50 mL, 60 x 20 mm) (VWR International, Philadelphia, PA, USA) in an vacuum oven Isotemp® 280A (Fisher Scientific Inc., USA) at 40 ºC for 72 h to reach a residual solvent content inferior to 1%. Test sample were made by punching 6 mm disks from the casted films and stored in sealed flasks at low moisture and room temperature. Characterization of films Uniformity of mass and thickness Films sample were individually weighed in an analytical balance XS105 (Mettler-Toledo Inc., Columbus, OH, USA). Film thickness was determined using a digital micrometer IP65 (Mytutoyo America Corporation, MA, USA). The experiments were performed in 15 replicates and, in both cases, the results were expressed in average ± standard deviation. Drug loading Test sample were weighted and dissolved in 5 mL ACN with 0.1% TFA using an ultrasonic bath 3510R-MT (Branson Ultrasonics, Danbury, CT, USA) for 40 min. Sample were filtered with 0.45 µm Teflon® filters and analyzed by High Performance Liquid Chromatography (HPLC). The analyses were performed in a LC system comprising a Waters Separations Alliance 2695 Module, Waters 2487 Dual λ Absorbance Ultraviolet (UV) Detector and Empower Pro® Software (Waters Inc., Milford, MA,USA). Chromatographic separation was carried out in a reverse-phase column Synergi Polar RP (250 x 4.6 mm, 4 µm, 80Å) purchased from Phenomenex® (Phenomenex Inc., Torrance, CA, USA). The mobile phase A was water with 0.1% TFA and mobile phase B was ACN with 0.1% TFA. The column was operated at 30 ± 1 ºC at a flow rate of 1.0 mL min-1 using the following gradient of A and B: 0 min, 70% A and 30% B; 8 min, 5% A and 95% B; 9 min, 5% A and 95% B; 10 min, 70% A and 30% B; 12 min, 70% A and 30% B. The injection volume was 20.0 µL and detection was 320 nm. MDZ contents (µg/mg) were determined as compared to a standard curve of MDZ (2.5 - 80 µg mL-1). The experiments were performed in triplicate. Hydration study Samples (n = 3) were individually weighted (W0) and immersed in different vials with 5 mL PBS and stored at 37 ºC. At predetermined times (i.e., 1.5, 3, 6, 12 and 24 h) of incubation, the respective samples were removed from the vials, blotted dry and reweighted (Wt). The percentage increase in mass as a result of water uptake (WU, %) were measured using the following equation 1: 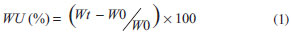 "In vitro" drug release profile Sample (n = 3) were fully immersed in scintillation vials containing 5 mL pre-warmed PBS as the release medium, and placed at 37 ºC into a culture oven Isotemp® 500 Series (Fisher Scientific Inc., USA). At predetermined time intervals (i.e., 0.17, 0.33, 0.75, 1.0, 1.5, 3, 6, 12, 24, 36 and 48 h), 1 mL sample of the releasing medium was withdrawn. The same volume of fresh dissolution medium at the same temperature was added to replace the amount withdrawn after each sampling. MDZ concentration in the samples were determined by HPLC-UV using the method described at the section "Drug loading". The cumulative amounts of MDZ dissolved on the release medium at t time (At, µg) were calculated according to the equation 2: 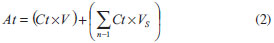 where: Ct = concentration of MDZ in the released medium at t time (µg mL-1); V = total volume of release medium (mL); VS = sample volume (mL). Finally, the percent cumulative amounts of MDZ released were calculated in weight basis as a function of the drug content in each film. "In vitro" polymer degradation and erosion Sample (n = 4) were fully immersed in 5 mL pre-warmed PBS in scintillation vials with one sample per vial and placed into a culture oven Isotemp® 500 Series (Fisher Scientific Inc., USA) at 37 ºC. At predetermined times (i.e., 1.5, 3, 6, 12, 24, 36 and 48 h) both remaining film and eluent solution were analyzed. Degradation study The remaining films were removed from the PBS, blotted dried and dissolved in 1 mL DMF with 0.1% (v/v) TFA. The Mw of the dissolved polymer was determined by GPC as previously described (see polymer synthesis and characterization section). Molecular weights loss was calculated as a ratio of the polymer Mw at time 0. Erosion study 150 µL of 0.1 mol L-1 sodium hydroxide solution were added to the elutant solutions and incubated for 4 hours at room temperature, followed by the addition of 1000 µL of 0.2 mol L-1 hydrochloric acid and incubation for 30 min. at room temperature. The mixture was frozen, then lyophilized in a Labconco FreeZone 2.5 lyophilizer (Labconco Inc., Kansas City, MO, USA) operating at -45 ± 5 ºC and < 0.1 millibar. The lyophilized powder was reconstituted in 1 mL of a solvent mixture containing water: ACN (70:30, v/v), then filtered using a 0.45 µm Teflon® filter. The test sample mixtures were analyzed by HPLC using the method described above (see section Drug loading). Herein, detection was 220 nm and DT contents (µg mL-1) were determined as compared to a DT standard curve (2 - 150 µg mL-1). Results were expressed as ratio of initial polymer mass loss. Thermal analysis Changes on the polymer Tg were analyzed by DSC according the method described earlier (see Polymer synthesis and characterization). Experiments were performed at least in triplicate. Mechanical properties Films were tested using a Sintech 5/D stress strain tester (MTS®, Shakopee, MN, USA) according to ASTM standard D882-0229 at room temperature. Sample width (±5 mm) and thickness (±0.25 mm) were averaged from three measurements prior to analysis. The crosshead speed was 10 mm min-1, and the cell charge was 100 N. The yield point was determined based on the zero slope criterion. The reported values of tensile modulus, tensile strength, strain at yield and strain at break were derived from the stress-strain curves and averaged from five separate runs.
DATA ANALYSIS Comparison of averages of two samples were performed by Student's t-test. For three or more sample, averages were compared by one-way Analysis of Variance (ANOVA), followed by Tukey's test using the software GraphPad Instat 5.1 (GraphPad Software Inc., CA, USA). It was considered as significant values of p lower or equals to 5% (p < 0.05). Analysis of linear regression were performed using the software Minitab 16 (Minitab Inc., USA).
RESULTS AND DISCUSSION Polymer synthesis and characterization The structure of E2505(2K) and 1H NMR spectrum of the polymer are shown in Figure 1. The chemical shifts assignments were addressed for their respective chemical group on the structure of the polymer:
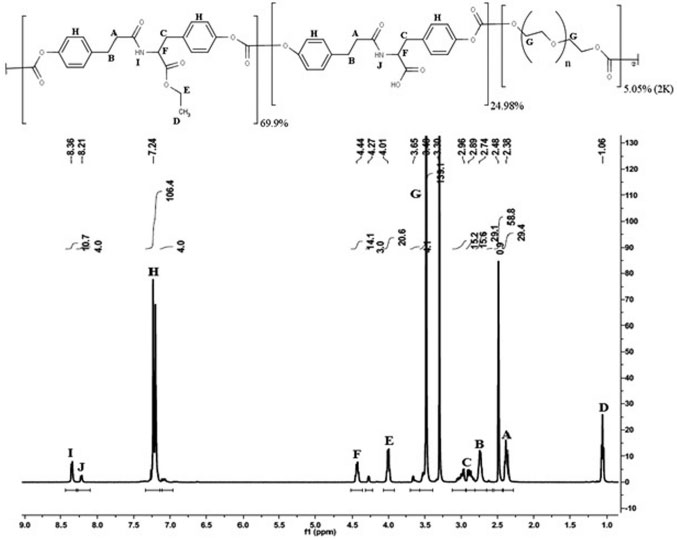 Figure 1. Chemical structure and 1H NMR spectrum for E2505(2K)
A and B protons correspond to the CH2 linked to amide and aromatic groups from the desaminotyrosine respectively. C protons correspond to the CH2 linked to the aromatic ring arising from tyrosine. D and E protons correspond, respectively, to the methyl and ethyl groups of the ethyl-ester from the DTE moieties. F proton corresponds to the α-position in tyrosine. G proton corresponds to the CH2 groups arising from the PEG moieties. H proton corresponds to the aromatic groups in both DTE and DT moieties. I and J proton correspond to the amide groups arising from DTE and DT moieties, respectively. Additionally, the molar content of each monomeric unit was calculated based on the integration of amide protons (I and J) at ~8.3 and ~8.2 ppm, aromatic protons (H) at ~7.2 ppm and PEG methylene protons (G) at ~3.6, ~3.4 and ~3.3 ppm (1H NMR spectrum in Figure 1). Hence, the experimental composition was poly(DTE-co-24.98%DT-co-5.05%PEG2K carbonate), which is close to the theoretical one. Besides the analysis of polymer composition, along the resorbable polymer synthesis the evaluation of several other chemical and physical properties are required. Therefore, the glass transition temperature (Tg), average molecular weight (Mw), polidispersivity index (PDI) and residual solvent content were evaluated in this study. The polymer presented Tg 49 ± 1 ºC, Mw 366 ± 10 KDa, PDI 1.5 ± 0.1 and 0.85 ± 0.12% (w/w) residual solvent. In comparison, E2502(2K) presents Mw 268 KDa, PDI 1.4 and Tg 83 ºC, while E5003.5(2K) presents Mw 239 KDa, PDI 1.4 and Tg 75 ºC.9 Characterization of MDZ loaded films The obtained films presented a light yellow homogeneous color, without characteristic of drug crystallization or bubbles. Moreover, the films showed excellent handling properties due to suitable flexibility. Table 1 presents the results obtained on the characterization of thickness and uniformities of mass and content of the developed resorbable films.
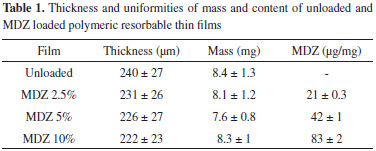
The thickness and mass were distributed around 230 µm and 8 mg, respectively. No significant differences were observed for the thickness and mass of several films. It was expected once the total solid content of the dispersions was kept constant for all formulations. Also, Mw loss ranging from 8.6% up to 17.5% was observed for the dried films as compared to the pure polymer. The Mw loss during the fabrication of the films may be attributed to the residual water content within the solvent used (around 0.1%). Moreover, taking into account the amount of PEG 2000 in the polymer's composition, it also may have drag in some water from the atmosphere along its dispersion procedure (which toke at least 12 h), or even during test sample preparation. Despite the great variation on film thickness and mass, the MDZ content proved to be uniform, with values of relative standard deviation lower than 3%. Furthermore, the MDZ contents obtained are close to the theoretical expected and varied within the limits recommended by the literature i.e., 85 - 115%.30 The water uptake profiles of films are presented in Figure 2. In contrast to the drug release, polymer degradation and erosion, the evaluation of the hydration process was performed within a 24 h interval. Afterwards, the films became weak and evaluation of water uptake beyond this time period was not possible.
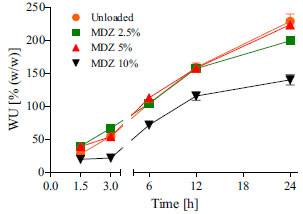 Figure 2. Water uptake profiles for unloaded and several MDZ loaded polymeric resorbable films incubated in PBS at 37 ºC. Mean values of n = 3 samples of n = 3 films ± SD
Monitoring the water uptake of films revealed that unloaded and loaded films all absorbed water in an amount approximately equal to 40% (w/w) of their initial dry weight within a 1.5 h period. Based on these findings, is it possible to assert that the presence of MDZ did not exerted any effect on the hydrophilicity of E2505(2K). Water uptake is a crucial parameter in the characterization of moisture-activated biodegradable polymers.31-33 Several studies have been reporting its effects on drug release performance,7,9,34-36 degradation,9,37,38 erosion,9,31,38 swelling,9,37-39 mechanical properties,40 and biological behavior.13,37,41 There are several methods available to quantify and understand the water uptake phenomena. Gravimetric9,35 and thermogravimetric analysis (TGA),7,33,37 as well 3H-radiolabeled water (3H2O)32,33 and multiscale analysis (e.g., Confocal Raman Spectroscopy, Small-angle scattering, Confocal Raman imaging and cryo-scanning transmission electron microscope)31,32 are the most commonly used. Each method presents intrinsic advantages and disadvantages, that may be evaluated in regard the cost / benefit ratio by the formulator along the characterization of the biodegradable device. Herein, we have chosen to use a rapid gravimetric method, once the other methods available at the laboratory were more expensive and required long time as well great experience to be performed.33 As shown in Figure 2, films presented water uptake ranging from 150% up to around 230% over a period of 24 h, which means that they increased their initial mass up to 2.3 folds. Interestingly, the specimen dimensions increased dramatically, but did not form a thick hydrated gel layer (hydration front) as can be observed for conventional pharmaceutical polymers such as acrylic and cellulosic derivate.42 The high water uptake may be attributed to the amphiphilic character of the polymer, which contains 5% (molar) of PEG in the composition. In comparison, it was reported that the homopolymer, poly(DTE carbonate), absorbs only about 1-2% of water over months.43 Moreover, previous studies compared the properties of several Tyrosine-PEG-derived poly(ether carbonate)s, showing that increased PEG content increased the rate of water uptake as well as the equilibrium water content of the polymer.37 The in vitro MDZ release profiles obtained for the several loaded films are presented in Figure 3.
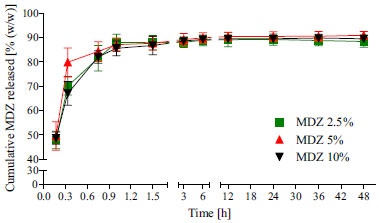 Figure 3. Drug release profiles for MDZ loaded polymeric resorbable films incubated in PBS at 37 ºC. Mean values of n = 3 samples of n = 3 films ± SD
All devices released approximately 85% (w/w) of the drug within a 1.5 h period. For this reason, it was not possible to evaluate the drug release kinetics. This burst release profile is likely attributed to rapid initial water uptake (Figure 2), which leads to polymer chain relaxation and, consequently, increases the driving force for solubilization and diffusion of drug molecules from polymer domains. Similar burst release profile was reported by Lan et al.27 on the development of alginate rings for dental implants. In this case, 33 days sustained release was obtained by using MDZ loaded poly-ε-caprolactone/alginate composites. The results presented in Figure 3 are useful for understanding the lack of effect of drug loading on the polymer hydration. As previously discussed, in the first 1.5 h of water uptake study, no statically significant difference were observed between the amount of water absorbed for the loaded and unloaded films (Figure 2). Taking into account that at this same time span the loaded films all released around 85% of MDZ, the different water uptake profile observed for the film containing MDZ at 10% (w/w) may be attributed essentially to an experimental error, once the drug was almost totally washed out from the films. The in vitro degradation (reduction in polymer Mw of solid phase) and erosion (physical loss of mass and dissolution of films) of the unloaded and MDZ loaded tyrosine-derived polycarbonate films were profiled in aqueous conditions (PBS, 37 ºC) for 48 h and are presented in Figures 4(a) and 4(b), respectively.
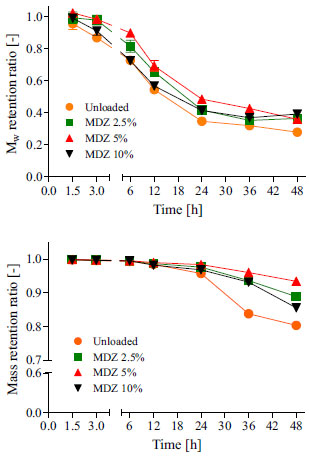 Figure 4. Degradation (a) and erosion (b) profiles for unloaded and MDZ loaded polymeric resorbable films incubated in PBS at 37 ºC. Mean values of n = 4 samples of n = 4 films ± SD
We did not observe any significant differences between the Mw and mass retention ratios for unloaded and MDZ loaded resorbable films over the 48 h study period. Moreover, most of drug was washed out from the films within the 1.5 h of experiment (Figure 3). In conclusion, the hydrolytic degradation of the polymer backbone and erosion of polymeric matrix were not affected by drug loading on the films. Previously, it was reported by Macry et al.9 that the drug loading did not affect the degradation profile from a polymer with close chemical composition i.e., E2502(2K) in fast bioerodible electrospun fiber mats for topical delivery of a hydrophilic peptide. As shown in Figure 4a, E2505(2K) loses around 70% of its initial Mw (degradation) within 48 h. In contrast, mass loss was slower. Only 10-15% (w/w) of the material was eroded (Figure 4b). Noteworthy, the degradation and the erosion are distinctive chemical and physical phenomena. While degradation occurs by covalent bond cleavage, the polymer erosion occurs by dissolution of degradation products after chain scission (oligomers, dimers, monomers) in non-crosslinked systems.5,38,44 Therefore, these two phenomena may be interdependent or may even occur simultaneously. Typically, biodegradable polymers can degrade and erode as: i) bulk process, where no significant alterations occurs in the physical size of the polymer carrier until it is always fully degraded or eroded, but the fraction of polymer remaining on the carrier decreases over time; and ii) surface process, where the polymer matrix is progressively removed from the surface, but the polymer volume fraction remains fairly unchanged.5 According to Burkersroda et al.,38 all degradable polymer can undergo both processes. Beside the diffusivity of water inside the matrix, the dominant process depends on the degradation rate of the polymer's functional group, and the matrix dimension. Upon visual inspection, films were found to be completely eroded and dissolved in the aqueous medium within 3 to 4 days. However, after 2 days it was not possible to evaluate the Mw retained by the remaining device anymore, once film became very weak. These results are in agreement with both Magno et al.,13 who reported that E2502(2K) scaffolds eroded within 3 to 7 days and Macri et al.,9 who showed that E2502(2K) electrospun fiber mats eroded in 4 days. Interestingly, the unloaded films had the tendency to become opaque and eroded into larger particulates before completely dissolving. The loaded films however, turned into clear gel-like material prior to complete dissolution. In order to evaluate the effect of hydration on the degradation of E2505(2K) films, we have correlated the data obtained from the water uptake and Mw retention ratio over a 24 h study period. The obtained linear regression plots, as well their respective fit curve equations and determination coefficients (R2) are presented in Figure 5.
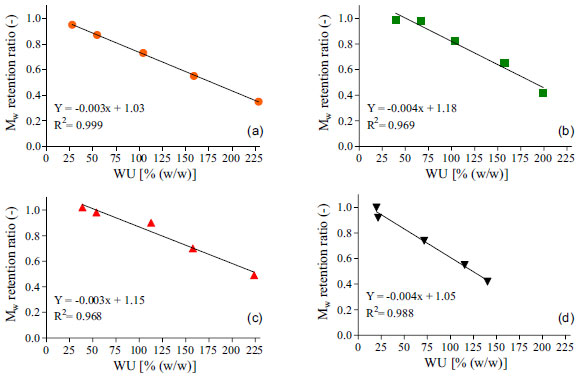 Figure 5. Linear regression plots between Water Uptake (WU) and Mw retention ratios for unloaded (a) and loaded resorbable polymeric films containing 2.5% (b), 5% (c) and 10% (d) of metronidazole. Plotted data are the average values obtained over 24 h period of experiments
Results from Figure 5 all show a significant negative correlation between the evaluated factors (0.968 < R2 < 0.999), which means that increasing the water uptake decreased the Mw retention ratio from the polymer. These findings corroborate with other results reported earlier9,31,37,38 and sustain the hypothesis that water uptake exerts a key role on the degradation of resorbable polymers. For degradation to occur by hydrolysis, polymer must interact with water by mean of charge interactions or hydrogen bonding mechanisms and absorb the surrounding aqueous solvent. Obviously, the presence of hydrolytically labile bonds on the polymers backbone or crosslinker is mandatory to enable this mechanism.5 Comparing Figure 3 with Figures 4a and 4b, it is clear that nor the degradation neither the erosion of the polymeric matrix showed significant correlation with drug release profiles. This was expected due to bulk erosion properties of the polymer. Typically, for drug delivery purpose, bulk-eroding polymers may give lower release rates, as they have no constant erosion velocity. On the other hand, a surface-eroding polymer can provide constant and easily controllable drug release rates. In that case, erosion proceeds at constant velocity at any time over this phenomena duration.5 Although being very useful for pharmaceutical applications, achieving a surface erosion characteristic for a bioerodible polymer displays a difficult task. In general, it requires adjusts on the material's dimensions and shape, as well changes in its chemical properties. Accordingly, most polymers present bulk erosion characteristics, since surface erosion can occurs slower than the penetration of water and degradation of the interior of the materials.44 The results for the thermal and mechanical properties for the obtained resorbable films are presented on Table 2.
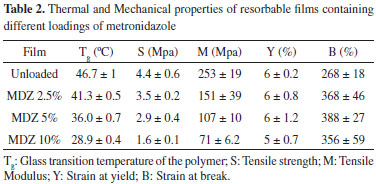
The Tg of the polymer ranged from 28.9 up to 46.7 ºC, in a way that increasing the MDZ loading decreased the Tg. This trend can also be seen graphically in Figure 6, where a zoom in the glass transition region from the second heating DSC curve is presented.
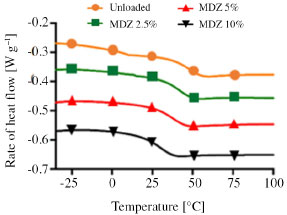 Figure 6. Second heating DSC curves showing the changes in the Tg as a function of MDZ loading on films
The Tg, is the temperature at which the amorphous phase of the polymer is converted between rubbery and glassy states. At the Tg an amorphous polymer softens, undergoing a transition from a glassy state to a rubbery state because of increased segmental mobility.35 Typically, decreasing the Tg of polymers can improve its film forming properties and the appearance of the obtained films. Moreover, the decrease on the Tg is noticed to exert crucial impact in the physical-mechanical properties of polymers. For instance, it prevents film cracking and improves film flexibility and processability.45-47 The mechanism by which it occurs is already very well understood and can be explained by the reduction (weakening) on the interactions of the polymer chains. This allow polymer chains to more easily slide across one another. Substances that act decreasing the Tg of a polymer are called plasticizers.48 The visual difference on the erosion behaviors reported earlier for unloaded and loaded films may be attributed to the plasticizing effect that MDZ exerted over the polymer. There are just a few works in the literature reporting the plasticizing effect of drugs so far.45,47,49 Furthermore, despite its importance on the development of drug delivery systems, to date no study has attempted the effect of MDZ on the Tg of polymers. This ratifies the unprecedented nature of our work. The same trend was also observed for both tensile strength (S) and Tensile or Young's Modulus (M), which varied from 1.6 MPa up to 4.4 MPa and 71 MPa up to 253 MPa, respectively. The S of a film is defined as the resistance of the material to a force tending to tear it apart. On the other hand, the M is a fundamental measurement of the stiffness of the material i.e., how it deforms in an elastic region. In both cases, the higher are their values, the more resistant the material is to being stretched.50 Elgindy and Samy51 evaluated the mechanical properties and drug release of cross-linked Eudragit films containing metronidazole. Using different amounts of succinic or citric acid (3.5 and 7% w/w) as cohesion promoters and triacetin (10. 25 and 45% w/w) as plasticizing agent, values of S and M ranging from 0.98 N cm-2 up to 30.98 N cm-2 and 0.28 N cm-2 up to 10.13 N cm-2, respectively, were obtained. Regarding the parameter of strain at yield (Y), no significant differences were observed between the values obtained, which varied around 5.5%. This parameter represents the stress at which a material begins to deform plastically. Knowledge of the yield point is important when designing a polymeric device since it generally represents an upper limit to the load that can be applied.50 Additionally, independently of the MDZ charge on the films, loaded films presented greater values of strain at break (B) as compared to the unloaded films. The B is the measurement of the maximum deformation the film can undergo before tearing it apart.51 In the light of the plasticizing effect of MDZ in the obtained films, our results are in agreement with others published elsewhere, which reported that the strain at break increases with the increase of plasticizer in a certain formulation.52 For better understanding how the drug loading, glass transition temperature, Young's Modulus and Tensile Strength correlate one with each other, a matrix correlation was prepared and is shown in Table 3.
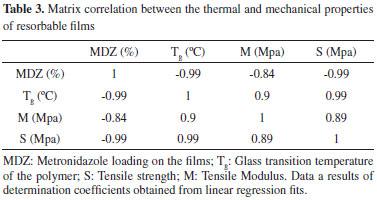
Based on the obtained determination coefficients it is possible to assert that all parameter evaluated have a significant interdependence with each other. Increasing drug loading decreases Tg of the polymer as well M and S of the films. On the other hand, increasing the Tg of the polymer increases both M and S of the films. Finally, the Tensile Modulus and the Tensile strength showed to be positively correlated, which means that increasing one factor the other also increases and vice versa. For the first time the effects of metronidazole loading on the physical-mechanical properties of a polymer are reported. A knowledge of the nature of drug - polymer interaction and their impacts on properties of bioresorbable films would be extremely useful for effective formulation design and its end usage properties.
CONCLUSIONS The obtained results suggest possible pharmaceutical applications for metronidazole loaded resorbable films from tysorine-derived polycarbonates. Due to fast hydration process, the film would most likely be useful in applications where rapid drug release is desired or for prolonged release from a poorly hydrated region such as for wound dressing or to be applied on the short term periodontal pocket treatment. Aiming to enable their application for sustained drug release purpose, the formulations must be engineered in order to control the water uptake. For that, the hydrophobicity of both drug and polymer must be adjusted. This can be achieved by i) using metronidazole benzoate or another hydrophobic drug or macromolecule as model; ii) using a tyrosine-derived copolymer comprising a smaller amount of PEG and/or a smaller average molecular weight of PEG in its composition e.g., E2005(1K); and/or iii) blending this polymer with others biodegradable polymers of higher degree of hydrophobicity (e.g., poly-caprolactone, poly(lactic acid), poly(lactic-co-glycolic acid), chitosan etc.).
ACKNOWLEDGMENTS Grants number 2012/06167-1 and 2013/12174-3 and project award number 2012/07251-6 from the State of São Paulo Sponsorship Agency (FAPESP) supported this work. The authors greatly thanks Barry Caninghan for help with polymer synthesis.
REFERENCES 1. Liechty, W.; Kryscio, D.; Slaughter, B.; Peppas, N.; Annu Rev. Chem. Biomol. Eng. 2010, 1, 149. DOI: http://dx.doi.org/10.1146/annurev-chembioeng-073009-100847 PMID: 22432577 2. Fu, Y.; Kao, W. J.; Expert Opin. Drug Delivery 2010, 7, 429. DOI: http://dx.doi.org/10.1517/17425241003602259 3. Mansour, H. M.; Sohn, M.; Al-Ghananeem, A.; Deluca, P. P.; Int. J. Mol. Sci. 2010, 11, 3298. DOI: http://dx.doi.org/10.3390/ijms11093298 PMID: 20957095 4. Wood, D. A.; Int. J. Pharm. 1980, 7. DOI: http://dx.doi.org/10.1016/0378-5173(80)90094-0 5. Nair, L. S.; Laurencin, C. T.; Prog. Polym. Sci. 2007, 32, 762. DOI: http://dx.doi.org/10.1016/j.progpolymsci.2007.05.017 6. Bourke, S.; Kohn, J.; Adv. Drug Deliv. Rev. 2003, 55, 447. DOI: http://dx.doi.org/10.1016/S0169-409X(03)00038-3 PMID: 12706045 7. Schachter, D. M.; Kohn, J.; J. Controlled Release 2002, 78, 143. DOI: http://dx.doi.org/10.1016/S0168-3659(01)00487-4 8. Lewitus, D.; Smith, K. L.; Shain, W.; Kohn, J.; Acta Biomater. 2011, 7, 2483. DOI: http://dx.doi.org/10.1016/j.actbio.2011.02.027 PMID: 21345383 9. Macri, L. K.; Sheihet, L.; Singer, A. J.; Kohn, J.; Clark, R. A. F.; J. Controlled Release 2012, 161, 813. DOI: http://dx.doi.org/10.1016/j.jconrel.2012.04.035 10. Lewitus, D. Y.; Rios, F.; Rojas, R.; Kohn, J.; J. Mater. Sci. Mater. Med. 2013, 24, 2529. DOI: http://dx.doi.org/10.1007/s10856-013-5008-0 PMID: 23888354 11. Kohn, J.; Welsh, W. J.; Knight, D.; Biomaterials 2007, 28, 4171. DOI: http://dx.doi.org/10.1016/j.biomaterials.2007.06.022 PMID: 17644176 12. Tangpasuthadol, V.; Pendharkar, S. M.; Peterson, R. C.; Kohn, J.; Biomaterials 2000, 21, 2379. DOI: http://dx.doi.org/10.1016/S0142-9612(00)00105-8 PMID: 11055285 13. Magno, M. H. R.; Kim, J.;Srinivasan, A.; McBride, S.; Bolikal, D.; Darr, A.; Hollinger, J. O.; Kohn, J.; J. Mater. Chem. 2010, 20, 8885. DOI: http://dx.doi.org/10.1039/c0jm00868k 14. Pulapura, S.; Li, C.; Kohn, J.; Biomaterials 1990, 11, 666. DOI: http://dx.doi.org/10.1016/0142-9612(90)90025-L PMID: 2090301 15. James, K.; Levene, H.; Parsons, J. R.; Kohn, J.; Biomaterials 1999, 20, 2203. DOI: http://dx.doi.org/10.1016/S0142-9612(99)00151-9 PMID: 10614927 16. Jones, D. S.; Woolfson, A. D.; Brown, A. F.; Neill, M. J. O.; J. Controlled Release 1997, 49, 71. DOI: http://dx.doi.org/10.1016/S0168-3659(97)00060-6 17. Perioli, L.; Ambrogi, V.; Rubini, D.; J. Controlled Release 2004, 95, 521. DOI: http://dx.doi.org/10.1016/j.jconrel.2003.12.018 18. Golomb, G.; Friedman, M.; Soskolne, A.; Stabholz, A.; Sela, M. N.; J. Dent. Res. 1984, 63, 1149. DOI: http://dx.doi.org/10.1177/00220345840630091101 PMID: 6589279 19. Reise, M.; Wyrwa, R.; Müller, U.; Zylinski, M.; Völpel, A.; Schnabelrauch, M.; Berg, A.; Jandt, K. D.; Watts, D. C.; Sigusch, B. W.; Dent. Mater. 2012, 28, 179. DOI: http://dx.doi.org/10.1016/j.dental.2011.12.006 PMID: 22226009 20. Miani, P. K.; do Nascimento, C.; Sato, S.;Filho, A. V; da Fonseca, M. J. V; Pedrazzi, V.; Eur. J. Clin. Microbiol. Infect. Dis. 2012, 31, 1611. DOI: http://dx.doi.org/10.1007/s10096-011-1484-7 PMID: 22138847 21. Shifrovitch, Y.; Binderman, I.; Bahar, H.; Berdicevsky, I.; Zilberman, M.; J. Periodontol. 2009, 80, 330. DOI: http://dx.doi.org/10.1902/jop.2009.080216 PMID: 19186975 22. El-Kamel, A. H.; Ashri, L. Y.; Alsarra, I. A.; AAPS PharmSciTech 2007, 8, E75. DOI: http://dx.doi.org/10.1208/pt0804091 PMID: 17915825 23. Urb, E.; Er, I.; Szab, P.; Cs, E.; Feh, A.; Int. J. Pharm. 2008, 358, 23. DOI: http://dx.doi.org/10.1016/j.ijpharm.2008.02.025 24. Sato, S.; Fonseca, M.; Ciampo, J.; Braz. Oral Res. 2008, 22, 145. PMID: 18622484 25. Prabhushankar, G. L.; Sathesh, P. R. Int. J. PharmTech Res. 2010, 2, 2188. 26. Magnusson, I.; J. Clin. Periodontol. 1998, 959. DOI: http://dx.doi.org/10.1111/j.1600-051X.1998.tb02398.x PMID: 9839853 27. Lan, S. F.; Kehinde, T.; Zhang, X.; Khajotia, S.; Schmidtke, D. W.; Starly, B.; Dent. Mater. 2013, 29, 656. DOI: http://dx.doi.org/10.1016/j.dental.2013.03.014 PMID: 23602170 28. Perioli, L.; Ambrogi, V.; Pagano, C.; Scuota, S.; Rossi, C.; Int. J. Pharm. 2009, 377, 120. DOI: http://dx.doi.org/10.1016/j.ijpharm.2009.05.016 PMID: 19454304 29. American Society for Testing and Materials (ASTM) Standard Test Method for Tensile Properties of Thin Plastic Sheeting; 2002; Vol. 14, pp. 1 - 10. 30. Dixit, R. P.; Puthli, S. P. ; J. Controlled Release 2009, 139, 94. DOI: http://dx.doi.org/10.1016/j.jconrel.2009.06.014 31. Sousa, A.; Schut, J.; Kohn, J.; Libera, M.; Macromolecules 2006, 39, 7306. DOI: http://dx.doi.org/10.1021/ma061286g 32. Valenzuela, L. M.; Zhang, G.; Flach, C.; Murthy, S.; Mendelsohn, R.; Michniak-Kohn, B.; Kohn, J.; Polym. Degrad. Stab. 2012, 97, 410. DOI: http://dx.doi.org/10.1016/j.polymdegradstab.2011.12.001 PMID: 22368310 33. Valenzuela, L. M.; Michniak, B.; Kohn, J.; J. Appl. Polym. Sci. 2011, 121, 1311. DOI: http://dx.doi.org/10.1002/app.33485 34. Soares, J. S.; Zunino, P.; Biomaterials 2010, 31, 3032. DOI: http://dx.doi.org/10.1016/j.biomaterials.2010.01.008 PMID: 20129660 35. Hunt, J. A.; Joshi, H. N.; Stella, V. J.; Topp, E. M.; J. Controlled Release 1990, 12, 159. DOI: http://dx.doi.org/10.1016/0168-3659(90)90092-8 36. Fulzele, S. V; Satturwar, P. M.; Dorle, A. K.; Int. J. Pharm. 2002, 249, 175. DOI: http://dx.doi.org/10.1016/S0378-5173(02)00529-X PMID: 12433446 37. Yu, C.; Kohn, J.; Biomaterials 1999, 20, 253. DOI: http://dx.doi.org/10.1016/S0142-9612(98)00169-0 PMID: 10030602 38. Von Burkersroda, F.; Schedl, L.; Göpferich, A.; Biomaterials 2002, 23, 4221. DOI: http://dx.doi.org/10.1016/S0142-9612(02)00170-9 PMID: 12194525 39. Graham, N. B.; Zulfiqar, M.; Polymer 1989, 30, 2130. DOI: http://dx.doi.org/10.1016/0032-3861(89)90305-4 40. Blasi, P.; D'Souza, S. S.; Selmin, F.; DeLuca, P. P.; J. Controlled Release 2005, 108, 1. DOI: http://dx.doi.org/10.1016/j.jconrel.2005.07.009 41. Tanaka, M.; Mochizuki, A.; J. Biomed. Mater. 2004, 68, 684. DOI: http://dx.doi.org/10.1002/jbm.a.20088 42. Lopes, C.; Lobo, J.; Costa, P.; Braz. J. Pharm. Sci. 2005, 41, 143. 43. Ertel, S. I.; Kohn, J.; J. Biomed. Mater. Res. 1994, 28, 919. DOI: http://dx.doi.org/10.1002/jbm.820280811 PMID: 7983090 44. Tamada, J. a; Langer, R.; Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 552. DOI: http://dx.doi.org/10.1073/pnas.90.2.552 PMID: 8421690 45. Nair, R.; Nyamweya, N.; Int. J. Pharm. Sci. Nanotechnol. 2001, 225, 83. DOI: http://dx.doi.org/10.1016/S0378-5173(01)00767-0 46. Rabek, C. L.; Van Stelle, R.; Dziubla, T. D.; Puleo, D. A.; J. Biomater. Appl. 2014, 28, 779. DOI: http://dx.doi.org/10.1177/0885328213480979 PMID: 23520360 47. Repka, M. A.; Gerding, T. G.; Repka, S. L.; McGinity, J. W.; Drug Dev. Ind. Pharm. 1999, 25, 625. DOI: http://dx.doi.org/10.1081/DDC-100102218 PMID: 10219532 48. Güngör, S.; Erdal, M.; Özsoy, Y. In Recent Advances in Plasticizers; Luqman, M., ed.; InTech: Rijeka, 2012; pp. 91-112. 49. Kalogeras, I. M.; Eur. J. Pharm. Sci. 2011, 42, 470. DOI: http://dx.doi.org/10.1016/j.ejps.2011.02.003 PMID: 21324354 50. Morales, J. O.; Mcconville, J. T.; Eur. J. Pharm. Biopharm. 2011, 77, 187. DOI: http://dx.doi.org/10.1016/j.ejpb.2010.11.023 PMID: 21130875 51. Elgindy, N.; Samy, W.; Int. J. Pharm. 2009, 376, 1. DOI: http://dx.doi.org/10.1016/j.ijpharm.2009.03.026 PMID: 19450940 52. Felton, L. A.; O'Donnell, P. B.; McGinity, J. W. In Aqueous Polymeric Coatings for Pharmaceutical Dosage Forms; McGinity, J. W.; Felton, L. A., eds.; Informa Healthcare: Londres, 2008; pp. 105-128. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access