
 |
 |
 |
 |
 |
Revisão
|
|
| Recentes aplicações em síntese orgânica de catálise foto redox mediada por luz visível Recent applications in organic synthesis of visible light photoredox catalysis |
|
Angelina M. de Almeida; Mauro V. de Almeida; Giovanni W. Amarante*
Departamento de Química, Universidade Federal de Juiz de Fora, 36036-900 Juiz de Fora - MG, Brasil Recebido em 22/04/2015 *e-mail: giovanni.amarante@ufjf.edu.br In the past few years, photoredox catalysis has become a powerful tool in the field of organic synthesis. Using this efficient method, it is possible to excite organic compounds from visible light and attain alternative mechanistic pathways for the formation of chemical bonds, a result which is not obtainable by classical methods. The rapid growth of work in the area of photoredox catalysis is due to its low cost, broad chemical utility protocols, and, especially, its relevancy from the green and sustainable chemistry viewpoints. Thus, this study proposes a brief theoretical discussion of and highlights recent advances in visible-light-induced photoredox catalysis through the analysis of catalytic cycles and intermediates. INTRODUÇÃO A fotoquímica é a ciência que investiga a interação entre a radiação na região do visível e ultravioleta sobre íons e moléculas. Os fenômenos fotoquímicos são de extrema importância para a vida através do processo de fotossíntese e na área industrial podem ser aplicados na catálise, degradação de poluentes, etc.1 Há mais de cem anos, a luz solar já era reconhecida como fonte energética limpa, de baixo custo, abundante e renovável para química orgânica.2 Entretanto, o principal fator limitante para o uso desta fonte de energia em síntese foi a inabilidade de absorção de luz visível pela maioria dos compostos orgânicos.3 Uma alternativa elegante encontrada para solucionar essa questão foi o desenvolvimento de fotocatalisadores hábeis a absorver radiação na região do visível e formar estados excitados altamente reativos. Sob esses aspectos, a catálise foto redox mediada por luz visível surge como alternativa no campo da catálise para ativação de moléculas orgânicas através da transferência de elétrons efetuada pelos fotocatalisadores.4 A maior parte dos fotocatalisadores empregados em síntese orgânica são metálicos. Dentre eles, merece destaque o emprego de complexos polipiridínicos de rutênio e irídio em catálise foto redox, como por exemplo, o complexo de tris(2,2'-bipiridina) rutênio(II), ou Ru(bpy)3+2 (Figura 1).
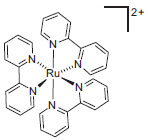 Figura 1. Estrutura geométrica do íon complexo [Ru(bpy)3]+2
O emprego destes complexos metálicos na área de inorgânica e de materiais é amplamente estudado e encontram-se aplicados à formação de H2 e O2 a partir da água,5 redução de CO2 em CH4,6 células solares sensíveis a corantes7 e reações de polimerização.8 Em contrapartida, o uso dos complexos em síntese orgânica era observado apenas pontualmente até pouco tempo atrás. Um dos primeiros trabalhos nesta área foi em 1984. Cano-Yelo e Deronzier desenvolveram a síntese do ácido fenatreno-9-carboxílico (2) através do uso da reação de Pschorr catalisada por Ru(bpy)3+2 e mediada por luz visível (Esquema 1).9
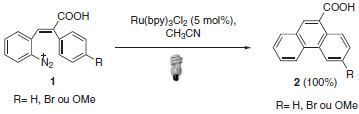 Esquema 1. Catálise foto redox aplicada à Reação de Pschorr
Em 2008, os grupos de pesquisas de Yoon e MacMillan descreveram o uso de [Ru(bpy)3]2+ como catalisador foto redox em reações de cicloadição [2+2]10 e α-alquilação de aldeídos,11 respectivamente. Em 2009, Stephenson e colaboradores divulgaram a desalogenação foto redox de haletos de alquila.12 A partir dessas contribuições, a busca de aplicações de catálise foto redox tornou-se sinônimo de possibilidade de obtenção de metodologias inovadoras na área de síntese orgânica. Como ilustra a Figura 2, o aumento no interesse da comunidade acadêmica pode ser comprovado pelo número de publicações desde 2008 e um contínuo crescimento nessa área de pesquisa.
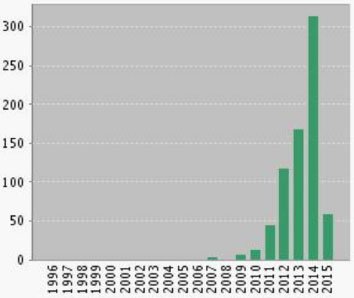 Figura 2. Número de publicações sobre "catálise foto redox" nos últimos 20 anos (Web of Science 13/03/15)
PROPRIEDADES FOTOQUÍMICAS DE [Ru(bpy)3]+2 Os complexos metálicos devem apresentar algumas características fundamentais para que possam ser empregados como catalisadores foto redox. Em primeiro lugar, é necessário que o complexo tenha absorção máxima na região do visível (aproximadamente 450 nm) para que possa ser excitado através de luz visível. A formação de um estado excitado estável, com tempo de vida moderado (τ = 1100 ns) é fundamental para que possa haver interações químicas a partir do mesmo. O estado excitado deve apresentar um eficiente caráter redutor e oxidante, uma vez que deve atuar como mediador em reações de transferência de elétrons com moléculas orgânicas.13 O estado excitado formado a partir da excitação dos complexos de metais de transição coordenados a ligantes insaturados (equação 1) apresenta características intrínsecas devido à reorganização eletrônica na molécula. A estabilidade moderada induzida no estado excitado é responsável por interações bimoleculares, que podem interagir na transferência de energia (equação 2) ou elétrons (equação 3) com moléculas não excitadas. Existem outras formas de interação bimoleculares e diferentes modos de desativação do estado excitado, como por exemplo, emissão de luz, mas que não será detalhado no escopo deste trabalho.14  A explicação formal para este processo encontra-se em termos eletrônicos de orbitais ocupados e não ocupados no complexo metálico. Os complexos metálicos de Ru+2 apresentam sistema d6 e ligantes polipiridínicos com orbitais aceptores π*. No processo de excitação, a absorção de um fóton na região do visível ocasiona a transferência de um elétron do orbital dπ do metal para o orbital π* do ligante polipiridínico (Esquema 2).15
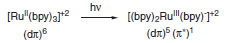 Esquema 2. Processo de absorção de luz para formação do estado excitado
O processo de formação do estado excitado do complexo de Ru é denominado como processo de Transferência de Carga Metal-Ligante (TCML) e é responsável pela oxidação do metal e redução do ligante. O estado excitado tripleto de mais baixa energia, *[Ru(bpy)3]+2, pode atuar com oxidante ou redutor, ativando moléculas orgânicas externas (Esquema 3).16
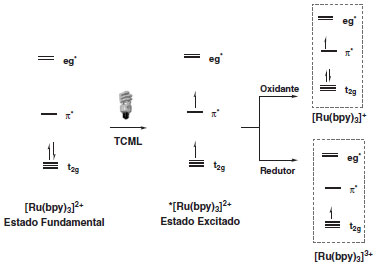 Esquema 3. Representação simplificada do orbital molecular do complexo [Ru(bpy)3]+2
As reações químicas de transferência de elétrons entre o estado excitado e moléculas doadoras (D) ou aceptoras (A) podem ser feitas por intermediação de dois ciclos distintos: ciclo de supressão redutiva ou ciclo de supressão oxidativa (Esquema 4).
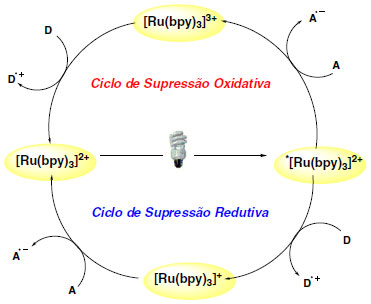 Esquema 4. Ciclo foto redox para o complexo [Ru(bpy)3]+2
No mecanismo de supressão oxidativa há transferência de um elétron proveniente do orbital π* para uma molécula aceptora (A) de elétrons. Em seguida, a molécula doadora de elétrons (D) transfere um elétron para o orbital πd do metal para finalizar o ciclo e regenerar o catalisador. No ciclo de supressão redutiva, *[Ru(bpy)3]+2 atua como agente oxidante recebendo elétrons da espécie D com a formação do intermediário de RuI, que, por sua vez, exercerá função de agente redutor doando elétrons para o substrato aceptor A. De maneira geral, os termos "supressão oxidativa" e "supressão redutiva" são usados para indicar o caminho reacional onde o fotocatalisador em seu estado excitado é oxidado e reduzido, respectivamente.4 Para melhor compreensão do processo foto redox é necessário levar em consideração o potencial de todas as espécies químicas envolvidas no ciclo catalítico. O potencial de redução para a espécie metálica no estado excitado (E1/2*II/I = +0,77V vs SCE) comprova que o fotocatalisador excitado é melhor oxidante que o estado fundamental (E1/2II/I = -1,33V vs SCE).17 Os diferentes centros metálicos e estrutura dos ligantes também podem fornecer fotocatalisadores com diferentes potenciais redox resultando em uma ampla possibilidade de condições catalíticas.18 A presença de grupos substituintes doadores de elétrons em ligantes heteroaromáticos dão origem a espécies mais redutoras, enquanto a presença de substituintes retiradores de elétrons permitem a formação de catalisadores mais oxidantes.19 O uso de reagentes de caráter doador (aminas terciárias) ou aceptor (Metil viologênio: MV+2, poli-halometano, dicianobenzeno, sal de arildiazônio, etc) é uma estratégia clássica para promover reações foto redox, entretanto o emprego quantitativo dessas substâncias como fonte e reserva de elétrons não é bem vista do ponto de vista da química verde. Existem várias possibilidades didáticas para classificar as reações foto redox e destacar as possíveis aplicações e usos como uma nova ferramenta para a síntese orgânica.19,20 As seções seguintes irão abordar a catálise foto redox mediada por luz visível de acordo com o ciclo de supressão: supressão oxidativa; supressão redutiva; e reações redox neutras. Durante a discussão dos exemplos será evidenciado o uso da catálise foto redox em recentes aplicações em síntese orgânica através da descrição mecanística do ciclo catalítico, identificação de importantes intermediários radicalares formados e ainda identificar a diversidade de produtos que podem ser obtidos através desta metodologia.
REAÇÕES FOTO REDOX POR SUPRESSÃO OXIDATIVA Os primeiros exemplos abordados para demostrar as aplicações da catálise foto redox na síntese orgânica serão as reações que apresentam ciclo catalítico por supressão oxidativa. Em geral, essa classe de reações apresenta em comum a necessidade de quantidade estequiométrica de uma espécie aceptora de elétrons. Em 2010, Yoon e colaboradores desenvolveram uma metodologia de foto cicloadição [2+2] para a síntese de anéis ciclobutano (Esquema 5).21 Apenas 1.0 mol% do fotocatalisador foi empregado e produtos bicíclicos com estereoquímica relativa cis puderam ser acessados em rendimentos que variaram de 54%-92%.
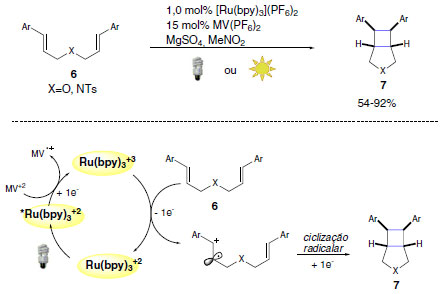 Esquema 5. Metodologia desenvolvida por Yoon e colaboradores para foto cicloadição [2+2]
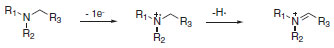
O produto ciclizado pode ser obtido empregando-se bis-alcenos ricos em elétrons (6), na presença de luz visível, fotocatalisador [Ru(bpy)3]+2 e metil viologênio (MV+2) como aceptor de elétrons. A oxidação de aminas é um item a ser destacado dentro da catálise foto redox oxidativa. Isto se deve ao caráter doador de elétrons desta classe, o que possibilita a oxidação das mesmas por doação de um elétron e formação do intermediário reativo cátion radicalar. Como indicado no Esquema 6, os intermediários formados (íon imínio) são potentes eletrófilos e úteis para a formação de novas ligações em posição α das aminas.
 Esquema 6. Formação dos intermediários reativos na oxidação de aminas
A reação de α-cianação demostrada por Koenigs e colaboradores fornece acesso a aminonitrilas através de ativação da ligação C-H.22,23 Para esta transformação, cianeto de potássio foi utilizado como provedor de cianeto e ácido acético atua como co-catalisador, liberando ácido cianídrico em solução. Nestas condições e com apenas 1,0 mol% de fotocatalisador de irídio forneceu o produto de α-cianação com 94% de rendimento. O fotocatalisador de irídio Ir(ppy)2(bpy)PF6, em seu estado excitado, é capaz de oxidar N-feniltetrahidroisoquinolina (8) ao cátion radicalar. Oxigênio molecular participa do ciclo catalítico e apresenta como função a reoxidação do fotocatalisador e formação do ânion superóxido (O2-•). O ânion superóxido, por sua vez, atua interagindo com o hidrogênio do cátion radicalar para a formação do íon imínio (9). Ao mesmo tempo, os autores sugerem também um caminho mecanístico iônico para a formação do íon imínio. Nesse aspecto, o cátion radicalar poderia ser desprotonado na posição α e o radical α-amina (10) poderia ser estabilizado pela sobreposição do orbital não-ligante preenchido do nitrogênio (Esquema 8).23
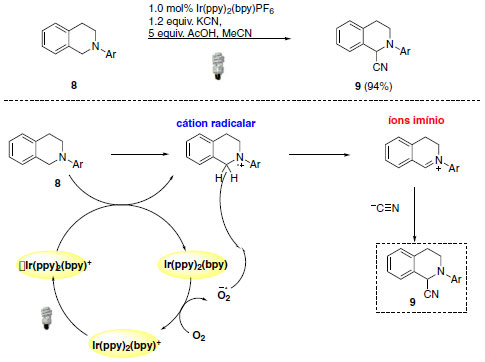 Esquema 7. Metodologia de α-cianação através de ativação da ligação C-H
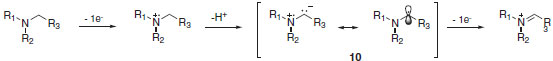 Esquema 8. Caminho mecanístico iônico para a formação do íon imínio
Ainda sob o contexto de α-funcionalização de aminas através da ativação de ligações C(sp3)-H, Vila e colaboradores descreveram o primeiro exemplo de uso simultâneo de oxidação aeróbica mediada por luz visível e catálise metálica.24 Como pode ser observado no Esquema 9, essa metodologia proporcionou a formação de uma nova ligação C-C através da adição do alcino 11. O produto 12 foi obtido com 88% de rendimento.
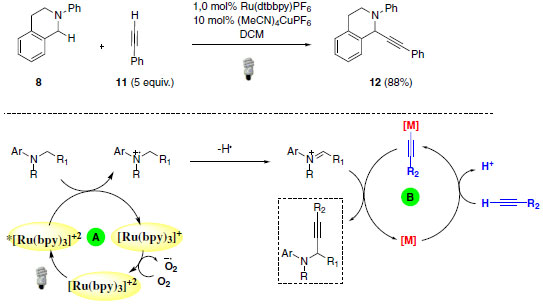 Esquema 9. Catálise dupla: oxidação aeróbica fotocatalítica e catálise metálica
O mecanismo proposto sugere a formação do íon imínio através da abstração de hidrogênio do cátion radicalar pelo superóxido formado na etapa de regeneração do catalisador. O íon imínio, formado no ciclo A, irá participar do segundo ciclo através de reação de adição 1,2 pelo acetileto de cobre formado no ciclo B. A eficiência da catálise dupla empregada neste exemplo se deve, principalmente, à compatibilidade entre as diferentes catálises empregadas. Nesse tipo de metodologia, onde há vários reagentes com funções particulares, todos os componentes devem ser cuidadosamente analisados. Fatores como a fonte de luz, tipo de fotocatalisador e catalisador metálico usado no segundo ciclo é crucial para o sucesso reacional.25 Muitos outros exemplos poderiam ainda ser empregados para enfatizar a ampla aplicação das reações foto redox por supressão oxidativa.26-28 Dentre eles, pode-se destacar a importância de obtenção de produtos tipo Mannich e a α-arilação de compostos α-aminocarbonílicos.29,30 O produto tipo Mannich 14, onde o íon imínio formado pela catálise foto redox é interceptado pelo silil enol éter 13, foi isolado com 96% de rendimento (Esquema 10).31
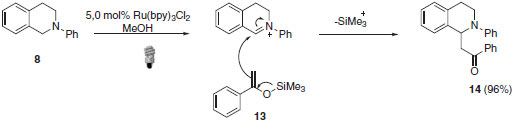 Esquema 10. Obtenção de produtos tipo Mannich: acoplamento entre íon imínio silil enol éter
A reação de α-arilação de compostos α-aminocarbonílicos é conhecida pela formação de subprodutos e baixos rendimentos devido à alta reatividade dos materiais de partida, constituindo um tópico desafiador para os químicos sintéticos. Li e colaboradores desenvolveram uma metodologia branda para obtenção dos produtos da α-arilação de compostos α-aminocarbonílicos (Esquema 11).32 Como resultado, a metodologia apresentou tolerância à presença de grupos funcionais, além de não utilizar bases, ligantes ou peróxidos.
 Esquema 11. α-Arilação de compostos α-amino carbonílicos
REAÇÕES FOTO REDOX POR SUPRESSÃO REDUTIVA Em reações foto redox por supressão redutiva são empregadas substâncias doadoras de elétrons como redutores em quantidade estequiométrica. Por exemplo, Stephenson e colaboradores utilizaram essa classe de catálise foto redox em reações de ciclização radicalar de pirróis e indóis (Esquema 12).33 Compostos cíclicos puderam ser sintetizados, como por exemplo o 19, com rendimento de 60%. Importante ressaltar a alta seletividade em favor da reação envolvendo a posição C-2 do indol.
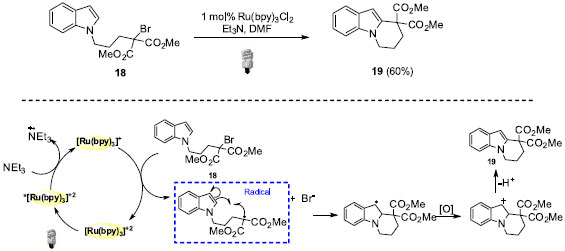 Esquema 12. Reação de ciclização intramolecular de pirróis e indóis
Em termos do mecanismo, o intermediário radicalar é formado via redução da ligação C-Br simultaneamente com a reoxidação da espécie fotocatalítica de rutênio. Essas características associadas à formação do radical benzílico como intermediário são responsáveis pelo sucesso dessa abordagem. Recentemente, Zheng e colaboradores evidenciaram o uso de catálise foto redox para a formação de ligação C-N através da preparação foto catalítica de N-aril indol (Esquema 13).34 Os derivados indólicos de interesse foram obtidos após subsequentes etapas de oxidação e aromatização e com rendimentos de até 88%.
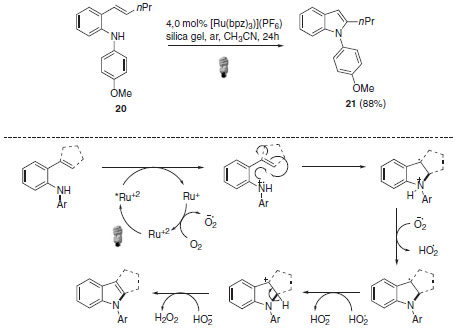 Esquema 13. Preparação foto catalítica de N-aril indol
A formação do cátion radicalar se dá através da redução do estado excitado do fotocatalisador metálico, seguida da formação do anel de cinco membros. As reações de catálise foto redox, além de possibilitar a formação de produtos inovadores, são capazes de empregar seletivamente materiais de partida e dar origem a diferentes compostos a partir deles. Cho e colaboradores foram capazes de aplicar a metodologia foto redox no controle de seletividade em reações de trifluorometilação de alcinos.35 Reações de trifluorometilação são amplamente estudadas devido à importância do grupo CF3 na área farmacêutica e agroquímica.36 Diversas possibilidades podem ser visualizadas para a obtenção seletiva de diferentes produtos através de uma pequena variação no sistema foto catalítico (Esquema 14). As diferentes condições reacionais levaram aos produtos provenientes de reações de iodotrifluormetilação (22),37,38 hidrotrifluorometilação (23)39 e trifluorometilação de alcinos (24).40
 Esquema 14. Controle de seletividade em reações de trifluorometilação de alcinos
Inicialmente, as reações de iodotrifluorometilação e hidrotrifluorometilação foram optimizadas a partir do emprego de diferentes fotocatalisadores e bases.41 A condição ideal para a metodologia de iodotrifluorometilação foi o emprego do fotocatalisador [Ru(phen)3]Cl2 (0,5 mol%), 2 equivalentes de base TMEDA (N,N,N',N'-tetrametiletilenodiamina) em acetonitrila e mediada por luz Led azul por 3 h à temperatura ambiente. Para obtenção dos produtos da hidrotrifluorometilação, as condições ideais foram o emprego do fotocatalisador fac-[Ir(ppy)3] (3,0 mol%) e 10 equivalentes de base DBU com irradiação prolongada da mesma fonte de luz visível (Esquema 15). Como resultado, altas seletividades em favor do isômero E foram alcançadas na reação de iodotrifluorometilação (relação E/Z = 18:1).
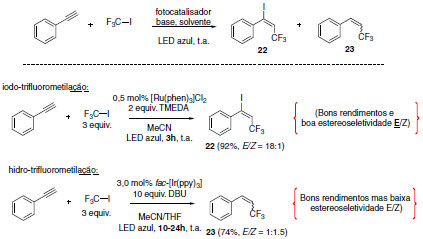 Esquema 15. Condições optimizadas para as metodologias de iodotrifluorometilação e hidrotrifluorometilação
O mecanismo para a hidrotrifluorometilação de alcinos é iniciado pela foto excitação do catalisador seguido da redução do mesmo pela base. O ânion radicalar metálico é a espécie responsável pela redução da ligação CF3-I, com simultânea regeneração do fotocatalisador. A espécie radicalar CF3 é adicionada ao alcino e fornece o radical vinílico 24, que por sua vez abstrai o hidrogênio da base para a formação do produto 23. O produto da reação de iodotrifluorometilação pode ser obtido em competição no ciclo catalítico por abstração de iodeto diretamente do reagente CF3I. O mecanismo sugere a ideia de reação de catálise em cascata através da de-iodinação de 22. Logo, a espécie 22 também pode estar envolvida no mecanismo de hidrotrifluorometilação (Esquema 16).
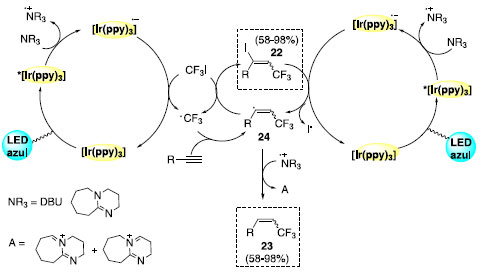 Esquema 16. Mecanismo proposto para as metodologias de iodotrifluorometilação e hidrotrifluorometilação
A metodologia de trifluorometilação de alcinos mostrou-se menos eficiente que as reações de iodotrifluorometilação e hidrotrifluorometilação. O rendimento máximo obtido no escopo da reação foi de 68% e isto se deve à formação de subprodutos bistrifluorometilado (rendimento de aproximadamente 15%) e iodeto de alquinila (em torno de 5%) (Esquema 17).
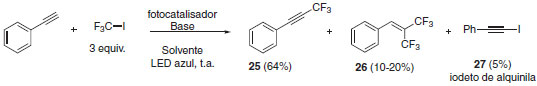 Esquema 17. Produtos e subprodutos na reação de trifluorometilação de alcinos
A metodologia otimizada emprega 2,0 mol% de fac-[Ir(ppy)3], 3,0 equivalentes de base (KOtBu), sob irradiação por 20 h de Led azul. Nestas condições de reação substratos contendo substituintes doadores de elétrons e halogênios puderam ser sintetizados em bons rendimentos (Esquema 18).
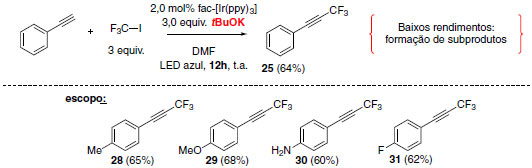 Esquema 18. Condição reacional optimizada na reação de trifluorometilação de alcinos
Uma proposta de mecanismo para esta reação envolve formação de iodeto de alquinila e posterior interação com o radical CF3 formado através de catálise foto redox. O radical alquenil trifluorometilado A, originado na etapa citada anteriormente, poderá abstrair um próton levando à formação do intermediário B com posterior eliminação de HI (Esquema 19).
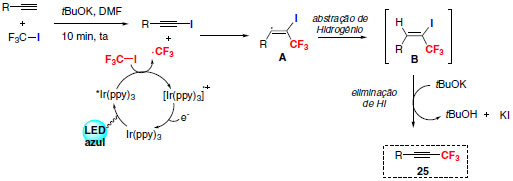 Esquema 19. Mecanismo proposto para a reação de trifluorometilação de alcinos S
REAÇÕES FOTO REDOX NEUTRAS Apesar da diversidade de aplicações em síntese via reações foto redox por supressão redutiva e oxidativa, as aplicações atuais na área de catálise foto redox mediada por luz visível apresentam-se voltadas para caminhos reacionais neutros.42 A utilização de substâncias em quantidades estequiométricas como fonte e reservatório de elétrons é um problema que pode ser contornado por intermédio desta classe de reações.43 Para esta finalidade, os materiais de partida de reações foto redox neutras devem participar simultaneamente dos processos de oxidação e redução em diferentes pontos do ciclo catalítico, dispensando o uso extra de compostos oxidantes e redutores quantitativos.44 Como resultado, os materiais de partida e os produtos obtidos devem apresentar o mesmo número de oxidação.45 Em 2014, um exemplo foi descrito de bifuncionalização de alcenos através de metodologia foto redox neutra. Nesta reação foi possível introduzir os grupos CF3 e Cl à dupla ligação com formação de apenas dióxido de enxofre como produto secundário.46 Os produtos bifuncionalizados puderam ser sintetizados em rendimentos de até 83% (Esquema 20).
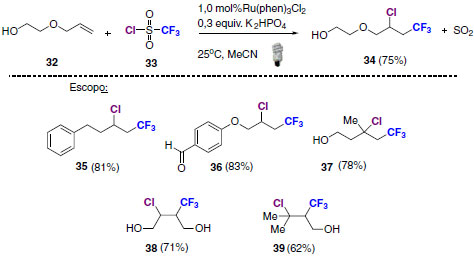 Esquema 20. Reação de bifuncionalização de alcenos
O ciclo catalítico proposto na reação de bifuncionalização de alcenos aborda a excitação do catalisador metálico para a formação do estado excitado *[Ru(phen)3]+2. O estado excitado irá atuar com agente redutor e a espécie radicalar de cloreto de trifluorometanosulfonila dá origem à espécie radicalar de CF3, dióxido de enxofre e íon cloreto. O radical CF3 será inserido na dupla ligação do alceno com formação do intermediário radicalar (A), que por sua vez tem função de regenerar o fotocatalisador através da doação de um elétron. O cátion (A') formado nesse processo irá interagir com o íon cloreto presente no meio reacional para a bifuncionalização dos alcenos (Esquema 21).
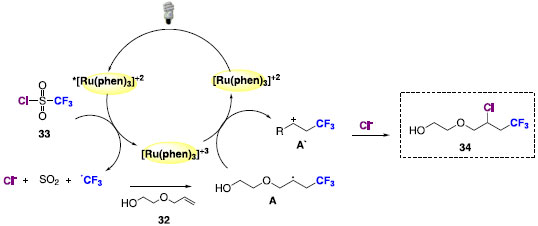 Esquema 21. Ciclo catalítico para a reação de bifuncionalização de alcenos
Uma interessante aplicação para a metodologia mostrada anteriormente é a funcionalização de duplas ligações em compostos biologicamente ativos. A Rotenona, uma substância com caráter pesticida e inseticida, foi utilizada com êxito como material de partida para obtenção de seu análogo cloro-trifluorometilado (40) com rendimento de 92% (Esquema 22).
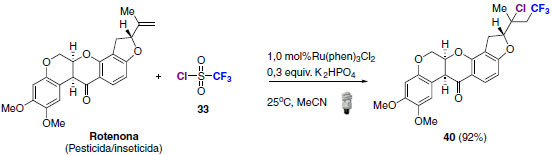 Esquema 22. Aplicação da metodologia de bifuncionalização de alcenos para a síntese de análogos de substâncias biologicamente ativas
A catálise foto redox assimétrica é outro ponto de destaque a ser discutido. As propriedades distintas de estereoisômeros no contexto farmacêutico e biológico resultam na necessidade de desenvolvimento de sínteses estereosseletivas e torna este tópico um desafio à parte na área da síntese orgânica.47 A elaboração de complexos metálicos enantiomericamente puros e que apresentem atividade foto redox pode ser uma alterativa no desenvolvimento de reações foto redox estereosseletivas mediadas por luz visível. Nesse contexto, o grupo de pesquisa de Meggers tem desenvolvido complexos de irídio e rutênio enantiomericamente puros para serem aplicados como catalisadores quirais em reações foto redox (Figura 3).48
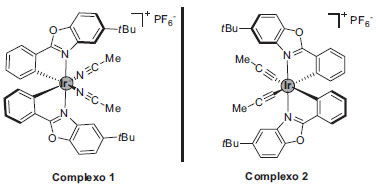 Figura 3. Complexos metálicos enantiomericamente puros com atividade foto catalítica desenvolvidos por Meggers e colaboradores
Uma aplicação eficaz desses complexos está relacionada com a síntese de 2-acilimidazol α-alquilado. A metodologia desenvolvida na formação de novas ligações C-C exibe elevados rendimentos e excesso enantiomérico (Esquema 23).49 Os complexos metálicos mencionados anteriormente desempenharam diferentes funções no mecanismo da reação, tais como, foto catalisador, ácido de Lewis e também como ativador do substrato 2-acilimidazol.
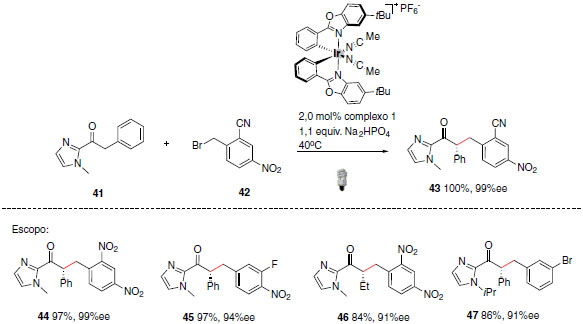 Esquema 23. Aplicação dos complexos metálicos desenvolvidos por Meggers e colaboradores na síntese de 2-acilimidazol α-alquilado
A interação entre catálise foto redox mediada por luz visível e organocatálise50 é uma discussão que merece destaque devido à possibilidade de sínteses inovadoras. Dentre as inúmeras contribuições do grupo de pesquisa de D. W. C. MacMillan nesta área, a α-benzilação enantioseletiva de aldeídos pode ilustrar a importância dessa metodologia (Esquema 24).11,51,52 Altos rendimentos e enantiosseletividades foram alcançadas para essa transformação, utilizando uma imidazolidinona como organocatalisador.
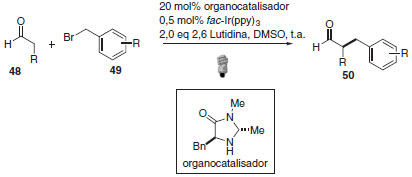 Esquema 24. α-Benzilação enantioseletiva de aldeídos: correlação entre catálise foto redox e organocatálise
No mecanismo da organocatálise foto redox, o complexo de irídio em seu estado excitado apresenta caráter altamente redutor e pode transferir um elétron para o brometo de benzila com formação do radical eletrofílico benzílico e íon brometo. Simultaneamente, há condensação do organocatalisador (imidazolidinona) com o aldeído 48 com resultante formação da enamina quiral 51. A formação da ligação C-C e do radical α-amino 52 se deve à adição do radical eletrofílico à enamina. A interseção dos ciclos catalíticos é responsável pela oxidação de 52 para regenerar o foto catalisador e formação do íon imínio 53. Através da hidrólise do imínio 53 o organocatalisador é regenerado e fornece os produtos α-benzilados (Esquema 25).
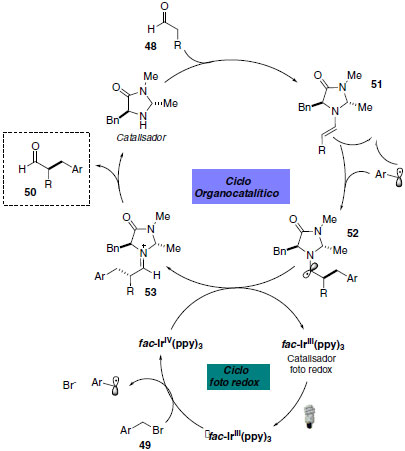 Esquema 25. Mecanismo da reação de α-benzilação enantioseletiva de aldeídos
Outra contribuição recente de organocatálise foto redox realizada por MacMillan e colaboradores destaca a importância da reação de arilação de ligações alílica C(sp3)-H (Esquema 26).53 Importante ressaltar que a ativação direta de ligações C(sp3)-H para a formação de novas ligações C-C a partir de metodologias brandas ainda não é uma ferramenta trivial. Altos rendimentos foram alcançados para formação de produtos interessantes do ponto de vista sintético, como por exemplo, arilação de hidrocarboneto cíclico de oito membros, fornecendo o produto desejado com rendimento de 86%.
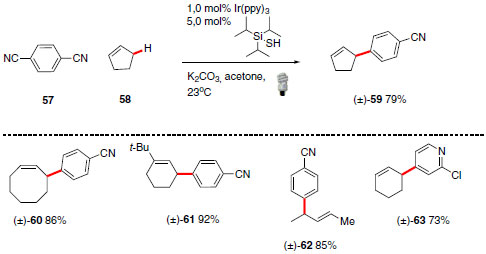 Esquema 26. Reação de arilação de ligações alílica C(sp3)-H desenvolvida por MacMillan e colaboradores
O fotocatalisador metálico, após ser excitado com luz visível, pode transferir um elétron para arenos deficientes em elétrons para formação do ânion radicalar areno 64. A espécie de IrIV só atua na oxidação do organocatalisador tiol em presença de base. Isto se deve à desprotonação do tiol (pKa = 7,91)54 pela base para formação do íon tiolato, que por sua vez, apresenta menor potencial de redução e pode ser prontamente oxidado pelo foto catalisador. Em seguida, o radical 66 formado atua abstraindo um próton do alceno fornecendo o radical alílico 65 e regenerando o organocatalisador. A nova ligação C-C tem origem a partir do acoplamento intermolecular entre os radicais 64 e 65, seguido de aromatização via eliminação de cianeto (Esquema 27).
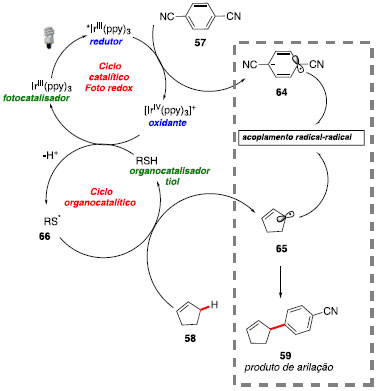 Esquema 27. Mecanismo da reaçao de arilaçao de ligaçoes alílica C(sp3)-H
Dentro desta abordagem de catálise dupla e ativação de ligação C-H foram ainda descritas a arilação direta de éteres benzílicos55 e a metodologia de acoplamento de éteres benzílicos com aldiminas para fornecer β-aminoéteres (Esquema 28).56 Em ambos os casos o mecanismo envolve o acoplamento direto das espécies radicalares geradas cataliticamente nos ciclos envolvidos.
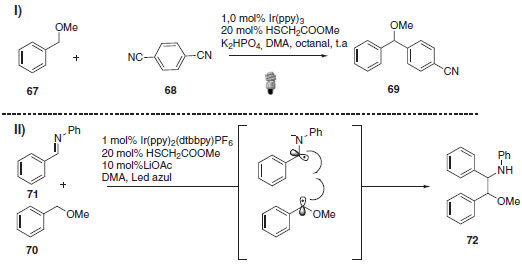 Esquema 28. I) Arilação de éteres benzílicos; II) Acoplamento de éteres benzílicos com aldiminas para síntese de β-aminoéteres
Além da combinação da catálise foto redox e organocatálise, a interação entre catálise foto redox e catálise metálica57 também pode representar uma nova rota de síntese não convencional. Há vários exemplos na literatura que empregam, dentre outros metais, ouro,57 cobre,58 níquel,59 paládio.60 Os complexos de metais de transição cobre, níquel e ouro também podem apresentar função fotocatalítica, substituindo os tradicionais complexos de rutênio e irídio empregados em catálise foto redox.61-64 A reação de alquilação foto induzida de amidas catalisada por cobre desenvolvida por Fu e colaboradores ilustra a aplicação desta classe de foto catalisadores.65 Nessa metodologia, diferentes haletos secundários cíclicos e acíclicos foram empregados de forma eficaz, além da alta tolerância funcional exibida pela reação. O escopo pode ser aplicado a amidas primária alifáticas, incluindo exemplos de grupos impedidos do ponto de vista estéreo e ainda alquilação de amidas aromáticas e α.β-insaturadas (Esquema 29).
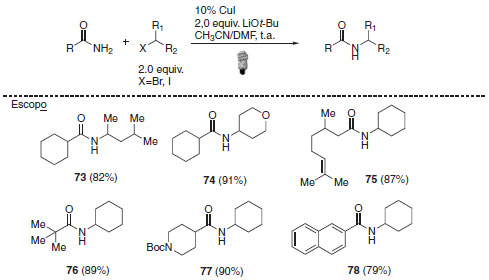 Esquema 29. Reação de alquilação foto induzida de amidas catalisada por cobre
Um dos prováveis caminhos mecanísticos proposto pelos autores postula que o complexo de cobre(I) [LnCu(Nu)] formado apresenta papel de destaque no ciclo catalítico sofrendo excitação na presença de luz visível e possibilitando a transferência de elétrons. O estado excitado de Cu(I) seria suficientemente redutor para transferir um elétron para o aduto eletrofílico com formação de radical alquila. A interação entre a espécie foto catalítica oxidada e o radical alquila dá origem ao produto de interesse e o nucleófilo presente no meio reacional atua na regeneração da espécie foto sensível para reiniciar o ciclo catalítico (Esquema 30).
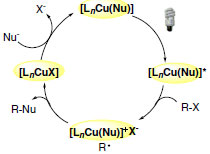 Esquema 30. Mecanismo proposto para a reação de alquilação foto induzida de amidas catalisada por cobre
Além dos catalisadores metálicos, alguns corantes orgânicos podem interagir com a luz visível para a formação de um estado excitado que pode interagir com outros substratos orgânicos em reações de catálise foto redox. O uso de corantes como foto catalisadores apresenta algumas vantagens quando comparado ao uso dos usuais complexos de rutênio e irídio. De maneira geral, os corantes orgânicos apresentam baixo custo e são menos tóxicos. Logo, essas substâncias são mais fáceis de serem manuseadas e agridem menos o meio ambiente quando comparadas aos catalisadores metálicos.63 Um exemplo recente da aplicação de corantes orgânicos como foto catalisadores desenvolvidos por Xiao e colaboradores aborda a alcoxicarbonilação radicalar de sais de arildiazônio mediada por luz visível.66 As condições reacionais otimizadas empregadas foram 0,5 mol% de fluoresceína, 80 atm de monóxido de carbono e irradiação de luz Led azul de 16 W em metanol (Esquema 31).
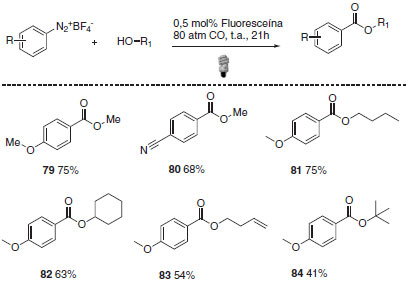 Esquema 31. Reação de alcoxicarbonilação radicalar de sais de arildiazônio
Foram empregados grupos substituintes doadores e retiradores no anel aromático dos sais de arildiazônio e diferentes álcoois alifáticos para comprovar a eficiência da metodologia desenvolvida. O mecanismo proposto mostra a redução do substrato arildiazônio pelo catalisador foto excitado com formação da espécie fenil radicalar A (Esquema 32). Posterior interação com monóxido de carbono origina o radical B, que por sua vez atua como agente redutor para regenerar o foto catalisador e fornecer a espécie C. Os ésteres sintetizados por alcoxicarbonilação são provenientes da reação de C com o álcool (metanol).
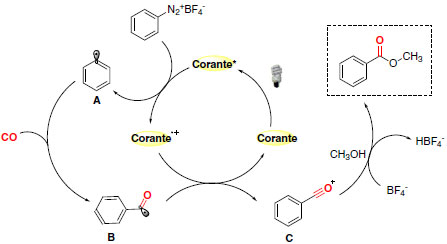 Esquema 32. Mecanismo da reação de alcoxicarbonilação radicalar de sais de arildiazônio
CONCLUSÃO E PERSPECTIVAS Esta breve revisão ilustra claramente a importância que a catálise foto redox adquiriu nos últimos anos na área de síntese orgânica. O grande número de trabalhos implica no desenvolvimento de novas metodologias e sínteses inovadoras devido ao modo único de ativação foto redox, além da possibilidade de empregar diferentes conceitos e ciclos catalíticos. O ponto de vista ambiental também merece destaque, uma vez que a catálise foto redox utiliza fonte de luz limpa e renovável (luz visível) como energia de ativação dos foto catalisadores, além de apresentar um custo operacional baixo e possibilitar o uso de corantes orgânicos como catalisadores em suas reações. Para elaboração de um sistema catalítico efetivo é necessária a escolha adequada do fotocatalisador, substratos e precursores radicalares. As transformações redox neutras, ou seja, sem o uso de oxidantes e redutores em excesso são metodologias favoráveis no âmbito da química verde. Mesmo com o avanço nesta área de pesquisa, existem algumas perspectivas que merecem especial atenção e necessitam estudos mais aprofundados. O desenvolvimento de intermediários que possam ser reativos, porém isoláveis, é de extrema importância para o entendimento e propostas de mecanismos reacionais. A abordagem de uma maior variedade de substratos no escopo também deve ser questionada. Podemos ainda enfatizar a aplicação da catálise foto redox mediada por luz visível em escala industrial. Esse é um grande desafio, mas que pode ter potencial resposta em metodologias de química em fluxo.67,68 Pelo exposto, a catálise foto redox mediada por luz visível tem muito a contribuir para a síntese orgânica e ainda necessita de amadurecimento e avanços para facilitar a sua aplicabilidade.
AGRADECIMENTOS Os autores agradecem ao CNPq, à CAPES, à FAPEMIG, à Rede Mineira de Química e à UFJF pelo suporte financeiro.
REFERÊNCIAS 1. Ferraudi, G. J.; Elements of Inorganic Photochemistry, 1th ed., Willey: New York, 1988, cap. 3. 2. Ciamician, G.; Science 1912, 36, 385. DOI: http://dx.doi.org/10.1126/science.36.926.385 PMID: 17836492 3. Narayanam, J. M. R.; Stephenson, C. R. J.; Chem. Soc. Rev. 2011, 40, 102. DOI: http://dx.doi.org/10.1039/B913880N PMID: 20532341 4. Koike, T.; Akita M.; Inorg. Chem. Front. 2014, 1, 562. DOI: http://dx.doi.org/10.1039/C4QI00053F 5. Kärkäs, M. D.; Verho, O.; Johnston, E. V.; Åkermark, B.; Chem. Rev. 2014, 114, 11863; Grätzel, M.; Acc. Chem. Res. 1981, 14, 376. c) Meyer, T. J.; Acc. Chem. Res., 1989, 22, 163.. DOI: http://dx.doi.org/10.1021/cr400572f PMID: 25354019 6. Takeda, H.; Ishitani, O.; Coord. Chem. Rev. 2010, 254, 346. DOI: http://dx.doi.org/10.1016/j.ccr.2009.09.030 7. Kalyanasundaram, K.; Grätzel, M.; Coord. Chem. Rev. 1998, 77, 347. DOI: http://dx.doi.org/10.1016/S0010-8545(98)00189-1 8. Lalevée, J.; Peter, M.; Dumur, F.; Gigmes, D.; Blanchard, N.; Tehfe, M.-A.; Moerlet-Savary, F.; Fouassier, J. P.; Chem. Eur. J. 2011, 17, 15027; Fors, B. P.; Hawker, C.; Angew. Chem., Int. Ed. 2012, 51, 8850. DOI: http://dx.doi.org/10.1002/chem.201101445 9. Cano-Yelo, H.; Deronzier, A.; J. Chem. Soc. Perkin Trans. 2 1984, 6, 1093. DOI: http://dx.doi.org/10.1039/p29840001093 10. Ischay, M. A.; Anzovino, M. E.; Du, J.; Yoon, T. P.; J. Am. Chem. Soc. 2008, 130, 12886. DOI: http://dx.doi.org/10.1021/ja805387f PMID: 18767798 11. Nicewicz, D. A.; MacMillan, D. W.; Science 2008, 322, 77. DOI: http://dx.doi.org/10.1126/science.1161976 PMID: 18772399 12. Narayanam, J. M. R.; Tucker, J. W.; Stephenson, C. R. J.; J. Am. Chem. Soc. 2009, 131, 8756. DOI: http://dx.doi.org/10.1021/ja9033582 PMID: 19552447 13. Fagnoni, M.; Dondi, D.; Ravelli, D.; Albini, A.; Chem. Rev. 2007, 107, 2725. DOI: http://dx.doi.org/10.1021/cr068352x PMID: 17530909 14. Sutin, N.; Creutz, C.; J. Chem. Educ. 1983, 60, 809. DOI: http://dx.doi.org/10.1021/ed060p809 15. Prier, C. K.; Rankic, D. A.; MacMillan, D. W. C.; Chem. Rev. 2013, 113, 5322. DOI: http://dx.doi.org/10.1021/cr300503r PMID: 23509883 16. Choi, W. J.; Choi, S.; Ohkubo, K.; Fukuzumi, S.; Cho, E. J.; You, Y.; Chem. Sci. 2015, 6, 1454. DOI: http://dx.doi.org/10.1039/C4SC02537G 17. Pavlishchuk, V. V.; Addison, A. W.; Inorg. Chim. Acta 2000, 298, 97; Dixon, I. M.; Collin, J.,-P.; Sauvage, J. -P.; Flamigni, L.; Encinas, S.; Barigelletti, F.; Chem. Soc. Rev. 2000, 29, 385. DOI: http://dx.doi.org/10.1016/S0020-1693(99)00407-7 18. Tamayo, A. B.; Alleyne, B. D.; Djurovich, P. I.; Lamansky, S.; Tsyba, I., Ho, N. N.; Bau, R.; Thompson, M. E.; J. Am. Chem. Soc. 2003, 125, 7377. DOI: http://dx.doi.org/10.1021/ja034537z PMID: 12797812 19. Hass, D.; Freys, J. C.; Bernardinelli, G.; Wenger, O. S.; Eur. J. Inorg. Chem. 2009, 4850. 20. You, Y.; Nam, W.; Chem. Soc. Rev. 2012, 41, 7061. DOI: http://dx.doi.org/10.1039/c2cs35171d PMID: 22797418 21. Ischay, M. A.; Lu, Z.; Yonn, T. P.; J. Am. Chem. Soc. 2010, 132, 8572. DOI: http://dx.doi.org/10.1021/ja103934y PMID: 20527886 22. Xi, Y.; Yi, H.; Lei, A.; Org. Biomol. Chem. 2013, 11, 2387. DOI: http://dx.doi.org/10.1039/c3ob40137e PMID: 23426621 23. Rueping, M.; Zhu, S; Koenigs, R. M.; Chem. Commun. 2011, 47, 12709. DOI: http://dx.doi.org/10.1039/c1cc15643h 24. Rueping, M.; Konigs, R. M.; Poscharny, K.; Fabry, D. C.; Leonori, D.; Vila, C.; Chem. Eur. J. 2012, 18, 5170. DOI: http://dx.doi.org/10.1002/chem.201200050 25. Zeitler, K.; Angew. Chem. 2009, 121, 9969. DOI: http://dx.doi.org/10.1002/ange.200904056 26. Hari, D. P.; König, B.; Org. Lett. 2011, 13, 2682. DOI: http://dx.doi.org/10.1021/ol200779y 27. Pan, Y.; Kee, C. W.; Chen, L.; Tan, C.-H; Green Chem. 2011, 13, 2682. DOI: http://dx.doi.org/10.1039/c1gc15489c 28. Liu, Q.; Li, Y.-N; Zhang, H.,-H.; Chen, B.; Tung, C.-H., Wu, L.-Z. Chem. Eur. J. 2012, 18, 620. DOI: http://dx.doi.org/10.1002/chem.201102299 29. Tucker, J. W.; Narayanam, J. M. R.; Krabbe, S.W.; Stephenson, C. R. J.; Org. Lett. 2010, 12, 368. DOI: http://dx.doi.org/10.1021/ol902703k PMID: 20014770 30. Zhao, G.; Yang, C.; Guo, L.; Sun, H.; Chen, C.; Xia, W.; Chem. Commun. 2012, 48, 2337. DOI: http://dx.doi.org/10.1039/C1CC15443E 31. Yasu, Y.; Koike, T.; Akita, M.; Chem. Commun. 2012, 48, 5355. DOI: http://dx.doi.org/10.1039/c2cc31748f 32. Wang, Z.; Hu, M.; Huang, X.; Gong, L.; Xie, Y.; Li, J.; J. Org. Chem. 2012, 77, 8705. DOI: http://dx.doi.org/10.1021/jo301691h PMID: 22985461 33. Furst, L.; Matsuura, B. S.; Narayanam, J. M. R.; Tucker, J. W.; Stephenson, C. R. J.; Org. Lett. 2010, 12, 3104. DOI: http://dx.doi.org/10.1021/ol101146f PMID: 20518528 34. Mayty, S.; Zheng, N.; Angew. Chem. Int. Ed. 2012, 51, 1. 35. Iqbal, N.; Jung, J.; Park, S.; Cho, E. J.; Angew. Chem. 2014, 126, 549. DOI: http://dx.doi.org/10.1002/ange.201308735 36. Muller, K.; Faeh, C.; Diederich, F.; Science 2007, 317, 1881. DOI: http://dx.doi.org/10.1126/science.1131943 PMID: 17901324 37. Murphy, P. M.; Baldwin, C. S.; Buck, R. C.; J. Fluorine Chem. 2012, 138, 3. DOI: http://dx.doi.org/10.1016/j.jfluchem.2012.03.011 38. Wallentin, C. J.; Nguyen, J. D.; Finkbeiner, P.; Stephenson, C. R. J.; J. Am. Chem. Soc. 2012, 134, 8875. DOI: http://dx.doi.org/10.1021/ja300798k PMID: 22486313 39. Mizuta, S.; Verhoog, S.; Engle, K. M.; Khotavivattana, T.; O'Duill, M.; Wheelhouse K.; Rassias, G.; Medebielle, M.; Gouverneur, V.; J. Am. Chem Soc. 2013, 135, 2505. DOI: http://dx.doi.org/10.1021/ja401022x PMID: 23373772 40. Para trifluorometilação de alcinos terminais: Chu, L.; Qing, F.-L.; J. Am. Chem. Soc. 2010, 132, 7262; Luo, D.,-F.; Xu, J.; Fu, Y.; Guo, Q.,-X; Tetrahedron Lett. 2012, 53, 2769. DOI: http://dx.doi.org/10.1021/ja102175w 41. Wille, U.; Chem. Rev. 2013, 113, 813. DOI: http://dx.doi.org/10.1021/cr100359d PMID: 23121090 42. Ischay, M. A.; Lu, Z.; Yoon, T. P; Chem. Sci. 2012, 3, 2807. DOI: http://dx.doi.org/10.1039/c2sc20658g PMID: 22984640 43. Hopkinson, M. N.; Sahoo, B.; Li, J.-L.; Glorius, F.; Chem. Eur. J. 2014, 20, 3874. DOI: http://dx.doi.org/10.1002/chem.201304823 44. Osawa, M.; Nagai, H.; Akita, M.; Dalton Trans. 2007, 827. 45. Hari, D. P.; Konig, B.; Chem. Commun. 2014, 50, 6688. DOI: http://dx.doi.org/10.1039/C4CC00751D 46. Oh, S. H.; Malpani, Y. R.; Ha, N.; Jung, Y.; Han, S. B.; Org. Lett. 2014, 16, 1310. DOI: http://dx.doi.org/10.1021/ol403716t PMID: 24571334 47. Skubi, K. L.; Yoon, T. P.; Nature 2014, 515, 45. DOI: http://dx.doi.org/10.1038/515045a PMID: 25373672 48. Huo, H.; Fu, C.; Harms, K.; Meggers, E.; J. Am. Chem. Soc. 2014, 136, 2990. DOI: http://dx.doi.org/10.1021/ja4132505 PMID: 24754748 49. Huo, H.; Shen, X.; Wang, C.; Zhang, L.; Rose, P.; Chen, L.-A.; Harms, K.; Marsch,M.; Meggers, E.; Nature 2014, 515, 100. DOI: http://dx.doi.org/10.1038/nature13892 PMID: 25373679 50. Amarante, G. W.; Coelho, F.; Quim. Nova 2009, 32, 469; Ávila, E. P.; Amarante, G. W.; ChemCatChem 2012, 4, 1713; Ávila, E. P.; de Mello, A. C.; Diniz, R.; Amarante, G. W.; Eur. J. Org. Chem 2013, 10, 1881; Pereira, A. A.; De Castro, P. P.; De Melo, A. C.; Ferreira, B. R. V.; Eberlin, M. N. Amarante, G. W.; Tetrahedron 2014, 70, 3271; Ávila, E. P.; Justo, R. M. S.; Gonçalves, V. P.; Pereira, A. A.; Diniz, R.; Amarante, G. W.; J. Org. Chem. 2015, 80, 590. DOI: http://dx.doi.org/10.1590/S0100-40422009000200034 51. MacMillan, D. W. C.; Nature 2008, 455, 304. DOI: http://dx.doi.org/10.1038/nature07367 PMID: 18800128 52. Shih, H.-W.; Vander Wal, M. N.; Grange, R. L.; MacMillan, D. W. C.; J. Am. Chem. Soc. 2010, 132, 13600. DOI: http://dx.doi.org/10.1021/ja106593m PMID: 20831195 53. Cuthbertson, J. D.; MacMillan, D. W. C.; Nature 2015, 519, 74. DOI: http://dx.doi.org/10.1038/nature14255 PMID: 25739630 54. Khursan, D. A; Mikhailov; Yanborisov, V. M.; Borisov, D. I.; React. Kinet. Catal. Lett. 1997, 61, 91. DOI: http://dx.doi.org/10.1007/BF02477518 55. Qvortrup, K.; Rankic, D. A.; MacMillan, D. W. C.; J. Am. Chem. Soc. 2014, 136, 626. DOI: http://dx.doi.org/10.1021/ja411596q PMID: 24341523 56. Hanger, D.; MacMillan, D. W. C.; J. Am. Chem. Soc. 2014, 136, 16986. DOI: http://dx.doi.org/10.1021/ja5102695 57. Chan, J. M. W.; Amarante, G. W.; Toste, F. D.; Tetrahedron 2011, 67, 4306; Melhado, A. D.; Amarante, G. W.; Wang, Z. J.; Luparia, M.; Toste, F. D.; J. Am. Chem. Soc. 2011, 133, 3517. DOI: http://dx.doi.org/10.1016/j.tet.2011.04.011 PMID: 21765556 58. Basudev, S.; Hopkinson, M. N.; Glorius, F.; J. Am. Chem. Soc. 2013, 135, 5505. DOI: http://dx.doi.org/10.1021/ja400311h 59. Ye, Y.; Sanford, M. S.; J. Am. Chem. Soc. 2012, 134, 9034. DOI: http://dx.doi.org/10.1021/ja301553c PMID: 22624669 60. Zuo, Z.; Ahneman, D. T.; Chu, L.; Terrett, J. A.; Doyle, A. G.; MacMillan, D. W. C; Science 2014, 345, 6195. 61. Lang, S. B.; O'Nele, K. M.; Tunge, J. A.; J. Am. Chem. Soc. 2014, 136, 13606. DOI: http://dx.doi.org/10.1021/ja508317j PMID: 25228064 62. Thoi, V. S.; Kornienko, N.; Margarit, C. G.; Yang, P.; J. Am. Chem. Soc. 2013, 135, 14413. DOI: http://dx.doi.org/10.1021/ja4074003 PMID: 24033186 63. Ziegler, D. T.; Choi, J.; Munoz-Molina, J. M.; Bissembler, A. C.; Peters, J. C.; Fu, G. C.; J. Am. Chem. Soc. 2013, 135, 13107. DOI: http://dx.doi.org/10.1021/ja4060806 PMID: 23968565 64. Re, vol, G. McCallum, T.; Morin, M.; Gagosy, F.; Barriault, L.; Angew. Chem. 2013, 125, 13107. DOI: http://dx.doi.org/10.1002/ange.201308517 65. Do, H.-Q.; Bachman, S.; Bissember, A. C.; Peters, J. C.; Fu, G. C.; J. Am. Chem. Soc., 2014, 136, 2162. DOI: http://dx.doi.org/10.1021/ja4126609 66. Guo, W.; Lu, L.-Q.; Wang, Y.; Wang, Y.-N.; Chen, J.-R.; Xiao, W.-J.; Angew. Chem, 2014, 126, 1. DOI: http://dx.doi.org/10.1002/ange.201310509 67. Bou-Hamdan, F. R.; Seeberger, P. H.; Chem. Sci. 2012, 3, 1612. DOI: http://dx.doi.org/10.1039/c2sc01016j 68. Andrews, R. S.; Becker, J. J.; Gagne, M. R., Angew. Chem. Int. Ed. 2013, 125, 13107. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access