
 |
 |
 |
 |
 |
Artigo
| Direct infusion esi-ms applied in the detection of byproducts due to reductive degradation of acetamiprid by zero-valent iron |
|
Jean C. CruzI; Marina F. ReisI; Marília M. SalvadorI; Marciano F. AlmeidaI; Rodinei AugustiII; Renata P. Lopes*,I
IDepartamento de Química, Universidade Federal de Viçosa, 36570-000 Viçosa - MG, Brasil Recebido em 09/02/2015 *e-mail: renata.plopes@ufv.br This study investigated the reductive degradation of acetamiprid (5 mg L-1) in aqueous medium (at pH 2.0) induced by zero-valent iron (50 mg). The process was monitored using high-performance liquid chromatography (HPLC) to determine the degradation rate as a function of reaction time, and direct infusion electrospray ionization mass spectrometry (DI-ESI-MS) to search for (and potentially characterize) any possible byproducts formed during degradation. The results obtained via HPLC showed that after 60 min, the degradation of the substrate reached nearly 100% in an acidic medium, whereas the mineralization rate (as determined by total organic carbon measurements) was as low as 3%. Data obtained by DI-ESI-MS showed that byproducts were formed mainly by insertions of hydrogen atoms into the nitrile, imine, and pyridine ring moieties, in addition to the observation of chlorine substitution by hydrogen replacement (hydrodechlorination) reactions. INTRODUCTION Pesticides have greatly contributed to the increase of yields in agriculture by controlling pests.1 However, many of these compounds are potentially toxic, hazardous and carcinogenic.2 Acetamiprid, for example, is a third-generation neonicotinoid insecticide. It can be used in a variety of crops, such as vegetables, melons, fruit trees, wheat, tobacco and cotton.3 Neonicotinoids are one of the most important commercial synthetic systemic insecticides used worldwide and are effective against several important sucking insect pests, such as aphids, whiteflies, thrips, leaf- and planthoppers.4,5 According to Goulson, these compounds were synthesized in the 1980s, and imidacloprid, the first commercially-available insecticide of this class, has been in use since the early 1990s.6 However, it is well-known that the intensive use of these insecticides displays several drawbacks, including soil and groundwater contamination, disruption of natural biological control and pollination processes, mammalian toxicity and development of pest resistance.7 Negative impacts of neonicotinoid insecticides against some predators, such as acute toxicity and physiological and behavioral trait impairments, have frequently been reported.8 The deleterious presence of neonicotinoid insecticides in aqueous systems has demanded the search for viable alternatives of remediation. Among the many suitable options, the advanced oxidative processes (AOPs) are noteworthy in spite of their high costs.9-11 Furthermore, the practical implementation of these processes on a large scale is a difficult task, mainly due to the requirement for the removal or immobilization of photocatalysts nanoparticles, the need of using ultraviolet radiation sources or of a constant adding of unstable reagents, such as ozone, peroxides, among others. Because of the drawbacks of oxidative processes, reductive processes mediated by zero-valent metals have became a promising alternative to treat contaminated water sources.12,13 Elemental iron (Fe0) and dissolved Fe2+ form a redox couple with a standard reduction potential of -0.440 V (compared to the Standard Hydrogen Electrode). Zero-valent iron (ZVI) is therefore a relatively strong reducing agent that can be used to reduce a large number of species, such as hydrogen ions, carbonates, sulfates, nitrates, oxygen and many organic compounds (mainly the electron-deficient ones that bear, for instance, halogen or nitro groups in their structures and usually react slowly with hydroxyl radicals).14 There are three basic pathways comprised in the reductive degradation promoted by ZVI in aqueous medium. The first one involves the direct transfer of electrons from the metal surface to the adsorbed organic substrates. The second path encompasses the reaction of the organic substrates with Fe2+, which is generated upon the corrosion of Fe0. The third pathway refers to the direct reaction of H2, produced from the reduction of water by Fe0, with the organic substrates. This latter process is usually catalyzed by the iron surface itself.14 Several examples have been reported regarding the reductive degradation of organic compounds by ZVI in aqueous medium. These studies include a myriad of substances, such as dyes,15-17 nitrocompounds,18-22 drugs,23-26 pesticides,27-29 among other classes. The use of ZVI is highly attractive as no extra components, such as electrical energy, light, or special chemicals, are required.30 Although the reductive degradation of organic pollutants usually results in a significant decrease in toxicity, the overall removal of dissolved organic carbon (DOC) is low. Whereas more drastic conditions are required to promote a complete mineralization, the by-products must be properly characterized due to their unknown toxicity.23 In this sense, direct infusion electrospray ionization mass spectrometry (DI-ESI-MS) has been extensively used to monitor degradation reactions9,27,31-33 and to establish routes for oxidative10,9 and reductive processes.27 ESI offers a relatively simple means to transfer non-volatile ions from the condensed to the gas phase, whereas mass spectrometry comprises a sensitive and direct detection method.34 Fast and efficient methods for the degradation of acetamiprid in aqueous medium under different conditions, such as TiO2 photococalysis,5,35 Fenton and Fenton-like oxidation3 have been reported. However, just few of these studies mention the characterization of by-products or try to propose degradation routes. Thus, this work aims to assess unprecedented information regarding the reductive degradation of acetamiprid by zero-valent iron (ZVI) in aqueous medium focusing on the monitoring and identification of by-products by employing DI-ESI-MS.
EXPERIMENTAL Chemicals and materials Acetamiprid, powdered iron, sodium acetate and glacial acetic acid were purchased from Sigma-Aldrich (Milwaukee, WI, USA) and used as received. Ortho-phenanthroline and sulfuric acid were purchased from VETEC (São Paulo, Brazil). Methanol was supplied by Merck (Darmstad, Germany). Ultrapure water, from a Millipore Milli-Q system (Milford, MA, USA), was employed to prepare the solutions for all the experiments. Samples were filtered in PTFE Millipore filters (diameter: 13 mm; pore size: 0.45 µm). Reductive degradation by Zero-Valet Iron (ZVI) All experiments were performed in a batch scale and in triplicate (the average results are reported in the subsequent sections). In a typical run, 100 mL of an aqueous solution of acetamiprid at 5 mg L-1 (or 50 mg L-1 for the DI-ESI-MS assays) had its pH adjusted to 2.0 by dropping a solution of H2SO4:water 1:1 (v/v). Powdered iron (50 mg) was added and the system kept under constant stirring at room temperature. These optimized conditions were used following the information provided in a previous study.27 To verify whether the analyte would undergo hydrolysis, control experiments were performed under the same experimental conditions but without the addition of powdered iron. Aliquots were withdrawn at assorted time intervals, filtered and kept protected from light in a refrigerator prior to analyses. Degradation rates were determined by means of the data obtained by HPLC-PDA. Finally, the process was monitored by DI-ESI-MS in order to verify the formation of by-products and their characterization. Determination of residual iron The residual iron concentration was determined in the reaction aliquot collected after 60 min reaction time. A spectrophotometric method was employed in which Fe(II) reacts with ortho-phenanthroline to form a red complex in a buffered solution (acetate buffer). Absorbance measurements were performed on a spectrophotometer (Agilent, Model: 8453) and measured at 511 nm. A calibration curve was built and the linear regression parameters calculated by the Ordinary Least Squares method. Analytical methods The analyses were carried out on a 1260 Infinity HPLC system (Agilent Technologies, Wilmington, DE, USA) using a Zorbax Eclipse Plus column C18 (4.6 × 150 mm, 5.0 µm). The following operating conditions were employed: isocratic elution of MeOH:H2O (55:45, v/v), flow rate of 0.5 mL min-1, injection volume of 20 µL, and UV-Vis detector set at 255 nm. ESI-MS analyses were conducted on an ion trap mass spectrometer (Thermo Scientific, San Jose, CA, USA; model: LCQ Fleet) operating in the positive ion mode. Aliquots were directly infused into the ESI source at a flow rate of 10 µL min-1 by a micro syringe. The ESI source conditions were as follows: heated capillary temperature 275 ºC; sheath gas (N2) flow rate 4 L min-1; spray voltage 5.0 kV; tube lens offset voltage 95 V. The m/z range employed in all experiments was 50-400. For the MS/MS events, the precursor ions were first isolated within the ion trap with an isolation width of 1 m/z unit and fragmented via collision-induced dissociation (CID) with the target gas (nitrogen). The relative collision energy was adjusted to yield product ions in assessable abundance. Total Organic Carbon (TOC) experiments were carried out on a TOC 5000A (Shimadzu, Kyoto, Japan) instrument at 680 ºC using platinum as catalyst.
RESULTS AND DISCUSSION The experiments (performed in triplicate) were designed and conducted in batches to allow for rapid measurements by HPLC-PDA. Then, the calibration curve for acetamiprid was built at the concentration range of 0.00100 to 10.0 mg L-1. The curve parameters were estimated as slope (2.70 × 107), intercept (2.97 × 107) and coefficient of determination (0.9999). The limit of detection (LOD) and quantification (LOQ) were calculated based on the standard deviation of blank samples (water samples, n = 7), i.e. LOD = 3*(blank standard deviation)/ slope and LOQ = 10*(blank standard deviation)/ slope. The LOD and LOQ were respectively 1.2 × 10-4 and 4.2 × 10-4 mg L-1. Finally, the chromatographic peak areas were integrated, the concentrations were estimated and normalized (C/C0) and finally plotted as a function of the reaction time (Figure 1).
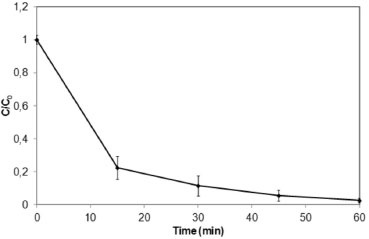 Figure 1. Normalized concentrations of acetamiprid (C/C0) monitored by HPLC-PDA as a function of reaction time. Experimental conditions: volume of acetamiprid solution = 100 mL; C0 = 5 mg L-1; pH = 2.0; 50 mg of powdered iron (n = 3)
Figure 1 reveals that 98% of the initial acetamiprid was degraded after 60 minutes, indicating that ZVI is quite effective in removing this compound under the experimental conditions tested. The result from the hydrolysis control experiment indicated that acetamiprid is stable under these conditions. These findings are in agreement with studies described in the literature that report that acetamiprid quickly hydrolyzes in basic medium but is quite stable at lower pHs (acidic conditions).3 Although approximately 100% of acetamiprid was degraded after a reaction time of 60 min (Figure 1), only 3% of the TOC removal was observed. These results clearly suggest that although the analyte is almost fully consumed, its mineralization leading to CO2, H2O, and NH3 is rather slow under these reductive conditions. Other papers have reported that reductive processes by ZVI, although very effective in causing the degradation of molecules bearing electron-deficient substituents, barely lead to a complete substrate mineralization.27 To propose a route for the degradation of acetamiprid (1) by ZVI in aqueous medium, the process was continuously analyzed by direct infusion electrospray ionization mass spectrometry in the positive ion mode, DI-ESI(+)-MS. To enable detection of a large number of by-products, the acetamiprid solution was prepared at a concentration 10-fold higher than that employed for the HPLC-PDA analysis. At this concentration level, the degradation of acetamiprid was approximately 80% after 60 min of reaction. Thus, aliquots were collected and directly infused into the ESI-MS ion source (Figure 2). The ESI(+)-MS of the initial solution of acetamiprid (1) at pH 2 (Figure 2a) shows the major presence of the ions of m/z 223/225 attributed to protonated acetamiprid, [1 + H]+. The ESI(+)-MS of the aliquot collected after 60 min of the reductive process is displayed in Figure 2b.
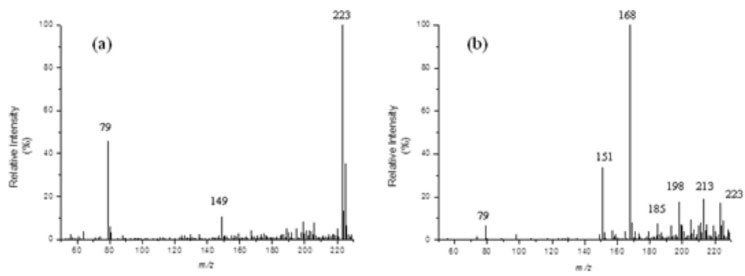 Figure 2. ESI(+)-MS of an aqueous solution (at pH 2.0) of acetamiprid (1) treated with ZVI for: (a) 0 min and (b) 60 min
The low intensity of the ion [1 + H+] in the mass spectrum displayed in Figure 2b indicates that acetamiprid (1) was almost completely consumed under these conditions. A visual inspection of this mass spectrum reveals the emergence of several ions, which probably refers to the protonated forms of by-products formed under these conditions. Hence, the presence of chlorine-containing ([2 + H]+ of m/z 213/215, [3 + H]+ of m/z 198/200 and [4 + H]+ of m/z 185/187) and chlorine-free ([5+ H]+ of m/z 168 and [6 + H]+ of m/z 151) ions can be promptly distinguished. The presence of chlorine in these ionic species is promptly verified due to the quite characteristic isotopic distribution (35Cl and 37Cl) verified on each of them. This set of data was therefore carefully evaluated and chemical structures for such by-products could thus be proposed. The chemical structures for all of them (2 to 6) and a logical route that explain their formation are proposed in Scheme 1. This possible degradation route was proposed based also on the well-known reactivity of the functional groups of acetamiprid (nitrile, imine, pyridine ring, chlorine) towards a reductive agent like metallic iron. Other positional isomers were possibly formed under these conditions but only one among all of them is displayed in Scheme 1 aiming at providing a more straightforward overview of the degradation process. Moreover, the MS/MS spectra of each precursor ion (not shown) revealed the formation of product-ions arising mainly from the release of small species, such as NH3, HCN and Cl. These MS/MS data, although not being capable to differentiate among these possible isomeric structures, were consistent with the fragmentation profiles observed for the protonated forms of each by-product, i. e. [2 + H]+, [3 + H]+, [4 + H]+, [5 + H]+ and [6 + H]+ (see discussion below).
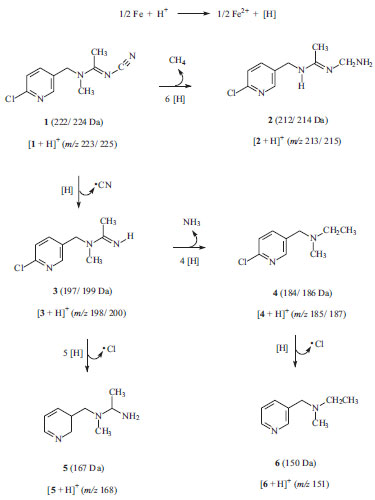 Scheme 1. Proposed route for the degradation of acetamiprid (1) promoted by zero-valent iron (ZVI) in an acidic aqueous solution (pH = 2.0)
Note that in all these pathways displayed in Scheme 1 there is the effective participation of nascent hydrogen ([H]), a reactive specie formed in situ by the reduction of H+ (acidic medium) by ZVI. Thus, a difference of 10 m/z units between the [1 + H]+ (m/z 223/225) and [2 + H]+ (m/z 213/215) ions probably indicates that 1 releases a molecule of methane (CH4) and incorporates 6 hydrogen atoms to yield by-product 2. Similarly, a Δm/z of 25 units between the [1 + H]+ (m/z 223/225) and [3 + H]+ (m/z 198/200) ions suggests that the emergence of by-product 3 takes place by the cyanide by hydrogen substitution. Analogously, the difference of 13 m/z units between the [3 + H]+ (m/z 198/200) and [4 + H]+(m/z 185/187) ions possibly indicates that by-product 3 incorporates 4 hydrogen atoms followed by the release of NH3 to yield 4. The difference of 30 m/z units between the chlorinated [4 + H]+ (m/z 185/187) and the chlorine-free [5 + H]+ (m/z 168) ions indicates that by-product 5 is probably formed by an initial Cl by H replacement (a process known as hydrodechlorination) followed by the insertion of 4 hydrogen atoms into 4. Finally, the m/z of 34 between the chlorinated [5 + H]+ (m/z 185/187) and the chlorine-free [6 + H]+ (m/z 150) ions suggests that 6 arises directly from the hydrodechlorination of 5. Although not being the main goal of the present work, the residual iron content was determined upon the application of the ZVI methodology. The residual iron concentration, which could represent an issue of concern, was determined by the spectrophotometric method, as explained in the Experimental Section. The final concentration (after 60 min. of treatment) was 13.7 ± 1.3 mg L-1, which is above the levels established by the Resolution 357 of CONAMA (National Environmental Council).36 However, it must be emphasized that Fe2+ can be readily and quantitatively removed by precipitation in basic medium.
CONCLUSIONS The efficiency of ZVI in causing the degradation of the insecticide acetamiprid in acidic aqueous solution is demonstrated herein. The results showed that the analyte is fully depleted (but not mineralized at all) after a reaction time as short as 60 min. Direct infusion ESI-MS was employed for a reliable and unprecedented characterization of the by-products formed under these conditions. The DI-ESI-MS technique showed to be very convenient in the monitoring of a degradation process because it required no sample pretreatment. Results are obtained continuously and the mass spectra roughly reflect the real reaction system composition. Other mass spectrometric techniques, particularly those coupled with chromatography, require lengthy sample pretreatment procedures, which prevents their direct use in monitoring. The data collected upon the ESI-MS monitoring was also useful to propose a detailed degradation route of acetamiprid in acidic aqueous solution induced by ZVI. It is noteworthy that hydrogen peroxide may be formed under the aerobic conditions employed herein. However, by-products arising from the reaction of acetamiprid with hydroxyl radicals (formed upon H2O2 decomposition) were not detected in the Mass Spectra of the aliquots. These results show therefore that for this substrate the reductive processes is by far favored. Then, this work presents a novel proposal to acetamiprid degradation by zero-valent iron. For the first time, the reduction of molecule with a nitrile group is monitored. Finally, this methodology can be applied in the monitoring of analogous environmentally-relevant processes.
ACKNOWLEDGMENTS The authors gratefully acknowledge Brazilian research funding agencies including FAPEMIG (Minas Gerais Research Foundation - 2014-APQ-00483-13 process), and CNPq (National Council for Scientific and Technological Development) for the financial support.
REFERENCES 1. Abhilash, P. C.; Singh, N.; J. Hazard. Mater. 2009, 165, 1. DOI: http://dx.doi.org/10.1016/j.jhazmat.2008.10.061 PMID: 19081675 2. Raut-Jadhav, S.; Saharan, V. K.; Pinjari, D.; Sonawane, S.; Saini, D.; Pandit, A.; J. Hazard. Mater. 2013, 261, 139. DOI: http://dx.doi.org/10.1016/j.jhazmat.2013.07.012 PMID: 23912079 3. Mitsika, E. E.; Christophoridis, C.; Fytianos, K.; Chemosphere. 2013, 93, 1818. DOI: http://dx.doi.org/10.1016/j.chemosphere.2013.06.033 PMID: 23871596 4. Pappas, M. L.; Migkou, F.; Broufas, G. D.; Appl. Entomol. Zool. 2013, 48, 373. DOI: http://dx.doi.org/10.1007/s13355-013-0197-z 5. Guzsvany, V. J.; Csanadi, J. J.; Lazic, S. D.; Gaal, F. F.; J. Braz. Chem. Soc. 2009, 20, 152. 6. Goulson, D.; J. Appl. Ecol. 2013, 50, 977. DOI: http://dx.doi.org/10.1111/1365-2664.12111 7. Laura Umpierrez, M.; Eugenia Lagreca, M.; Cabrera, R.; Grille, G.; Rossini, C.; Phytochem. Rev. 2013, 11, 339. DOI: http://dx.doi.org/10.1007/s11101-012-9253-5 8. He, Y.; Zhao, J.; Zheng, Y.; Desneux, N.; Wu, K.; Ecotoxicology 2012, 21, 1291. DOI: http://dx.doi.org/10.1007/s10646-012-0883-6 PMID: 22447470 9. Dalmazio, I.; Santos, L. S.; Lopes, R. P.; Eberlin, M. N.; Augusti, R.; Environ. Sci. Technol. 2005, 39, 5982. DOI: http://dx.doi.org/10.1021/es047985v PMID: 16173554 10. Cardoso da Silva, J. C.; Bispo, G. L.; Pavanelli, S. P.; Afonso, R. J. de C. F.; Augusti, R.; Rapid Commun. Mass Spectrom. 2012, 26, 1305. DOI: http://dx.doi.org/10.1002/rcm.6227 11. Dalmázio, I.; Almeida, M. O.; Augusti, R.; Alves, T. M. A.; J. Am. Soc. Mass Spectrom. 2007, 18, 679. DOI: http://dx.doi.org/10.1016/j.jasms.2006.12.001 PMID: 17234428 12. Mu, Y.; Yu, H.-Q.; Zheng, J.-C.; Zhang, S.-J.; Sheng, G.-P.; Chemosphere 2004, 54, 789. DOI: http://dx.doi.org/10.1016/j.chemosphere.2003.10.023 13. Fjordboge, A. S.; Baun, A.; Vastrup, T.; Kjeldsen, P.; Chemosphere 2013, 90, 627. DOI: http://dx.doi.org/10.1016/j.chemosphere.2012.08.058 PMID: 23021613 14. Pereira, W. S.; Freire, R. S.; Quim. Nova 2005, 28, 130. DOI: http://dx.doi.org/10.1590/S0100-40422005000100022 15. Cao, J.; Wei, L.; Huang, Q.; Wang, L.; Han, S.; Chemosphere 1999, 38, 565. DOI: http://dx.doi.org/10.1016/S0045-6535(98)00201-X PMID: 10901674 16. Fan, J.; Guo, Y.; Wang, J.; Fan, M.; J. Hazard. Mater. 2009, 166, 904. DOI: http://dx.doi.org/10.1016/j.jhazmat.2008.11.091 PMID: 19128873 17. Lin, Y.-T.; Weng, C.-H.; Chen, F.-Y.; Sep. Purif. Technol. 2008, 64, 26. DOI: http://dx.doi.org/10.1016/j.seppur.2008.08.012 18. Agrawal, A.; Tratnyek, P. G.; Environ. Sci. Technol. 1996, 30, 153. DOI: http://dx.doi.org/10.1021/es962168j 19. Keum, Y. S.; Li, Q. X.; Chemosphere 2004, 54, 255. DOI: http://dx.doi.org/10.1016/j.chemosphere.2003.08.003 PMID: 14575737 20. Mu, Y.; Yu, H. Q.; Zheng, J. C.; Zhang, S. J.; Sheng, G. P.; Chemosphere 2004, 54, 789. DOI: http://dx.doi.org/10.1016/j.chemosphere.2003.10.023 PMID: 14637335 21. Sun, X.; Wang, X.; Li, J.; Wang, L.; J. Taiwan Inst. Chem. Eng. 2014, 45, 996. DOI: http://dx.doi.org/10.1016/j.jtice.2013.09.026 22. Lai, B.; Zhang, Y.-H.; Li, R.; Zhou, Y.-X.; Wang, J.; Chem. Eng. J. 2014, 249, 143. DOI: http://dx.doi.org/10.1016/j.cej.2014.03.108 23. Bautitz, I. R.; Velosa, A. C.; Pupo Nogueira, R. F.; Chemosphere 2012, 88, 688. DOI: http://dx.doi.org/10.1016/j.chemosphere.2012.03.077 PMID: 22534198 24. Perini, J. A. S. B.; Nogueira, R. F.; Chemosphere 2014 (doi: 10.1016/j.Chemosphere.2014.07.071). DOI: http://dx.doi.org/10.1016/j.Chemosphere.2014.07.071 PMID: 25150686 25. Machado, S.; Stawinski, W.; Slonina, P.; Pinto, A. R.; Grosso, J. P.; Nouws, H. P. A.; Albergaria, J. T.; Delerue-Matos, C.; Sci. Total Environ. 2013, 461, 323. DOI: http://dx.doi.org/10.1016/j.scitotenv.2013.05.016 PMID: 23738986 26. Perini, J. A. d. L.; Silva, B. F.; Nogueira, R. F. P.; Chemosphere 2014, 117, 345. DOI: http://dx.doi.org/10.1016/j.chemosphere.2014.07.071 PMID: 25150686 27. Lopes, R. P.; de Urzedo, A. P. F. M.; Nascentes, C. C.; Augusti, R.; Rapid Commun. Mass Spectrom. 2008, 22, 3472. DOI: http://dx.doi.org/10.1002/rcm.3749 PMID: 18853392 28. Ghauch, A.; Chemosphere 2001, 43, 1109. DOI: http://dx.doi.org/10.1016/S0045-6535(00)00184-3 29. Ghauch, A.; Gallet, C.; Charef, A.; Rima, J.; Martin-Bouyer, M.; Chemosphere 2001, 42, 419. DOI: http://dx.doi.org/10.1016/S0045-6535(00)00073-4 30. Peterson, J. W.; Petrasky, L. J.; Seymour, M. D.; Burkhart, R. S.; Schuiling, A. B.; Chemosphere 2012, 87, 911. DOI: http://dx.doi.org/10.1016/j.chemosphere.2012.01.044 PMID: 22342282 31. Urzedo, A. P. F. M.; Diniz, M. E. R.; Nascentes, C. C.; Catharino, R. R.; Eberlin, M. N.; Augusti, R.; J. Mass Spectrom. 2007, 42, 1319. DOI: http://dx.doi.org/10.1002/jms.1204 PMID: 17902104 32. Berzas Nevado, J. J.; Castaneda Penalvo, G.; Rodriguez Dorado, R. M.; Rodriguez Robledo, V.; Anal. Methods 2013, 5, 3299. DOI: http://dx.doi.org/10.1039/c3ay40460a 33. Souza, A. G.; Cardeal, Z. L.; Augusti, R.; J. Environ. Sci. Health, Part B 2013, 48, 171. DOI: http://dx.doi.org/10.1080/03601234.2013.730015 34. Stewart, I. I.; Spectrochim. Acta, Part B 1999, 54, 1649. DOI: http://dx.doi.org/10.1016/S0584-8547(99)00110-X 35. Khan, A.; Haque, M. M.; Mir, N. A.; Muneer, M.; Boxall, C.; Desalination 2010, 261, 169. DOI: http://dx.doi.org/10.1016/j.desal.2010.05.001 36. Brasil. Resoluçao nº 357, de 17 de março de 2005 do CONAMA - Conselho Nacional do Meio Ambiente. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access