
 |
 |
 |
 |
 |
Artigo
| Characterizing the mechanism of quetiapine distribution in lipid-core nanocapsules pseudo-phases using a validated LC/UV method |
|
Fernando Carreño; Karina Paese; Carolina de Miranda Silva; Sílvia S. Guterres; Teresa Dalla Costa*
Faculdade de Farmácia, Universidade Federal do Rio Grande do Sul, Avenida Ipiranga 2752, 90610-000 Porto Alegre - RS, Brasil Recebido em 12/05/2015 *e-mail: teresadc@farmacia.ufrgs.br Quetiapine is an atypical antipsychotic used to treat schizophrenia. However, despite great interest for its chronic therapeutic use, quetiapine has some important side effects such as weight gain induction. The development of a quetiapine nanocarrier can potentially target the drug into central nervous system, resulting in a reduction of systemic side effects and improved patient treatment. In the present work, a simple liquid chromatography/ultraviolet detection (LC/UV) analytical method was developed and validated for quantification of total quetiapine content in lipid core nanocapsules as well as for determination of incorporation efficiency. An algorithm proposed by Oliveira et al. (2012) was applied to characterize the distribution of quetiapine in the pseudo-phases of the nanocarrier, leading to a better understanding of the quetiapine nanoparticles produced. The analytical methodology developed was specific, linear in the range of 0.5 to 100 µg mL-1 (r2 > 0,99), and accurate and precise (R.S.D < ±5%). The absolute recovery of quetiapine from the nanoparticles was approximately 98% with an incorporation efficiency of approximately 96%. The results indicated that quetiapine was present in a type III distribution according to the algorithm, and was mainly located in the core of the nanoparticle because of its logD in the formulation pH (6.86 ± 0.4). INTRODUCTION Schizophrenia is a chronic and severe brain disorder that presets cognitive impairment in nearly all patients being very disabling.1 Quetiapine (QTP), marketed as quetiapine fumarate (Figure 1), is an atypical antipsychotic used to treat schizophrenia. It belongs to the group of the dibenzothiazepines and is structurally similar to clozapine, the prototype of the atypical antipsychotics.2
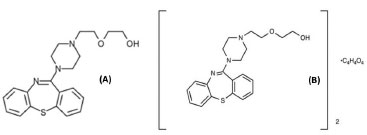 Figure 1. Molecular structure of quetiapine (A) and quetiapine fumarate (B)
QTP interacts with multiple neurotransmitter brain receptors and has a high serotonin/dopamine binding ratio, with the affinity to serotonin type 2 receptor (5-HT2) about twice as strong as the dopamine D2-receptor.3 This receptor blocking profile is in part responsible for the low incidence of extrapyramidal side-effects of quetiapine and the good response in reduction of positive (e.g: hallucinations and delusions) and negative (e.g: poverty of speech and apathy) symptoms of schizophrenia.4 However, in spite of the great therapeutic interest of this drug for chronic use, quetiapine-induced weight gain is of major clinical importance since it is associated with severe metabolic complications and increased morbidity and mortality.5 The uses of polymeric nanocapsules, submicron vesicles composed of a polymeric shell, an oil core and stabilized by surfactants, as drug carriers have been received much attention over the last years due to their biodegradability, the ability to control drug release, modulate drug's pharmacokinetics and to vetorize drugs to specific tissues in the human body reducing systemic side effects.6-8 Dimer et al.6,9 showed that administration of olanzapine loaded in a polymeric nanoparticle increased brain penetration of the drug, prolonged its therapeutic efficacy and reduced olanzapine-induced weight gain and total cholesterol in male Wistar rats comparing with those that received the free drug. In this context, the development of a polymeric systems loaded with QTP can target the drug into the central nervous system, reducing peripheral tissues distribution and consequently the occurrence of side effects potentially improving the treatment of schizhophrenic patients. Jäger et al.10 developed lipid-core nanocapsules (LNC) with a core composed by a dispersion of sorbitan monostearate (SM) and medium chain tryglicerides (MCT) (capric/caprylic acid) surrounded by poli(e-caprolactone) and polysorbate 80 (Tween-80).6,8,10 The author showed that varying the MCT and SM concentration it is possible to modulate the drug release kinetics when the drug is dispersed within the lipophilic core which is an important factor when using nanocarriers as delivery systems. Oliveira et al.7 developed an algorithm in order to determine the mechanisms of drug distribution in LNC formulation using the ultrafiltration-centrifugation technique and liquid chromatography (HPLC) analysis. The called therapeutic "nano-effect" expected from the administration of LNC formulation is dependent on the drug dispersion among the diverse pseudo-phases of the colloidal suspension (inner, interface and outer pseudo-phases of the formulation). Thus this strategy can be applied in order to select formulations intended for drug delivery systems. Due to its pharmacologic activities in the CNS3 and its physicochemical properties such as logD7.4 and ability to absorb certain wavelengths from the ultraviolet spectrum of light11 QTP is a good liphophilic drug model to be encapsulated in LNC and evaluated in terms of drug targeting to the brain. Although QTP is not official in most worldwide used pharmacopeias,12 there are publications concerning only the analysis of quetiapine fumarate in bulk and pharmaceutical dosage forms using usually liquid chromatography/mass spectrometry (LC-MS/MS methods).13,14 So it was necessary to develop and validated a simple and rapid liquid chromatography/ultraviolet detection (LC/UV) analytical method, according to the Brazilian Health Surveillance Agency (ANVISA) guidelines, for the extraction and quantification of QTP from LNC developed to determine the total drug content and encapsulation efficiency as well as to investigate QTP mechanism of distribution in the pseudo-phases of this nanocarriers.
EXPERIMENTAL Chemicals Quetiapine fumarate (purity > 99.0%) was a donation from Prati Donaduzzi Pharmaceutical Laboratory (Toledo - PR, Brazil). Acetonitrile, methanol (LC grade) and triethlamine were obtained from Tedia (Fairlfield, USA). Other chemicals used were of analytical reagent grade and purchased from commercial sources. Water was purified by a Milli-Q system (Millipore®). Quetiapine base preparation QTP base was prepared by liquid-liquid extraction (ethyl acetate/ammoniun hydroxide saturated solution) from QTP-hemifumarate. Briefly, QTP-hemifumarate (corresponding to 10 g of QTP free base) was solubilized in 100 mL of ethyl acetate aqueous solution (50% v/v). Under controlled magnetic stirring ammonium hydroxide solution (30%) was slowly added to QTP solution and the pH was set to 10. After 30 min, the solution was transferred to a separation funnel and extracted with 150 mL of distillated water. The ethyl acetate phase was concentrated under reduced pressure to remove the solvents. The prepared QTP base was a faint yellow oily material. The removal of fumaric acid was confirmed by infrared spectroscopy (data not show). Preparation of lipid-core nanocapsules QTP lipid core (QLNC - 1 mg mL-1) (n = 3 batches) and LNC blank nanocapsules were obtained by nanoprecipitation of preformed polymer.8,10,15 PCL (0.1 g), medium chain triglycerides (160 µL), sorbitan monoestearate (0.019 g) and QTP (0.01 g) were dissolved in acetone (20 mL) and ethanol (3 mL). This organic phase was injected into an aqueous phase containing polysorbate 80 (P-80) (0.078 g) in ultrapurified water (53 mL) under magnetic stirring. After 10 min, acetone was eliminated and the suspension concentrated to 10 mL under reduced pressure. Blank nanocapsules were prepared in the same manner, but the drug was not added to the organic phase. LC/UV apparatus and chromatographic conditions The method was performed on a Waters LC system equipped with a Waters® 600 pump controller, automatic injector (717 Plus, Waters®), and a Waters® 2487 dual λ absorbance detector. Analytical separation was achieved on a reversed phase C18 column (Phenomenex Luna 150 mm x 4.6 mm i.d.; particle size 5 µm) coupled to a C18 Phenomenex security guard pre-column. Chromatographic separation was accomplished by the isocratic elution of a mixture of a triethylamine solution (0.4%, v/v) adjusted to pH 3.0 ± 0.2 using phosphoric acid and acetonitrile (70:30, v/v). Before delivering the mobile phase into the system, it was degassed for 30 min by sonication and filtered through 0.45 µm filter (Sartorius, Germany) using vacuum. The flow rate was 1 mL min-1 and the injection volume was 20 µL. QTP was detected at 225 nm and the total run time was 6 min. Waters® Empower software was used for data acquisition and processing. Standard solutions, analytical curves and quality control samples QTP standard stock solution (1 mg mL-1) was prepared in volumetric flask diluting the appropriate amount of the substance in methanol and stored at -80 ± 1 ºC. QTP working solutions were prepared by subsequent dilutions of the stock solution in methanol:water (50:50, v/v) to reach the concentration range of 0.5 - 100 µg mL-1. Standard curves were generated by measuring the seven different standard concentrations (0.5; 1; 5; 7.5; 12; 16 and 20 µg mL-1) and quality control (QC) samples were prepared with low (QCL: 0.75 µg mL-1), medium (QCM: 10 µg mL-1) and high (QCH: 15 µg mL-1) concentrations. All work and standard solutions were freshly prepared every day during the ongoing analysis. Sample preparation In order to determine the total QTP concentration in QLNC formulations the nanocapsules (100 µL) were dissolved in methanol (10 mL) and placed for 20 min in an ultrasonic bath. An aliquot of 1 mL was centrifuged at 12.000 rpm for 10 min and filtering (0.45 µm) before HPLC injection. QCs were prepared using 100 µL of blank LNC (without drug) spiked with QTP solution. To obtain the encapsulation efficiency, free QTP concentration was obtained using the centrifugation-ultrafiltration technique using a Microcon® membrane device (MC Milipore 10 KDa) filled with 0.2 mL of QLNC formulation and centrifuged at 5.000 rpm for 10 min. The filtrate was collected and inject into the LC/UV system. Method validation The method was validated according to the International Conference of Harmonization guideline16 considering the following parameters: linearity, precision, repeatability, accuracy, recovery, limit of quantification and limit of detection. To assess selectivity, comparative analyses of the QLNC and blank LNC were performed in order to evaluate if any formulation components interfered with QTP quantification. Linearity determination was conducted by quantifying three different QTP standard curves (ranging from 0.5 - 20 µg mL-1) prepared daily for two consecutive days. The standard curve equation was obtained plotting QTP peak area against the respective nominal concentration. By using linear regression analysis (least square regression method) the slopes, intercepts and determination coefficients were determined. The validity of the assay was verified by means of the one-way ANOVA (α = 0.05). Intra-day precision (repeatability) was evaluated by extracting and performing six determinations of QCM (10 µg mL-1) sample (blank LNC spiked with QTP), corresponding to 100% of the test concentration, in the same day. Inter-day precision (intermediate precision) was evaluated comparing six determinations of QCM on two consecutive days. Precision was given as the relative standard deviation (R.S.D%). Accuracy of the method was determined by adding a known amount of QTP (100 µg mL-1) to the sample solution (blank LNC), resulting in six concentrations of each QC samples (0.75 µg mL-1; 10 µg mL-1and 15 µg mL-1) on two consecutive days. The assay accuracy was calculated as relative error (R.E%). Precision and accuracy values within ± 5% of the nominal concentration were considered acceptable, according to the validation guidelines used. The quantitation limit (QL) and the detection limit (DL) were estimated using the standard deviation of the response based on the calibration curve and the slope ratio multiplied by 10 and by 3, respectively. The standard solutions stability was investigated at room temperature after 6 h of preparation and after 1 week and 1 month stored at -80 ± 1 ºC. Sample stability studies were carried out in triplicate for the lower and higher QCs solutions. The samples were considered stable if the deviation from nominal concentration was within ± 5% (16,17). QTP saturation concentration in water and P-80 aqueous solution QTP saturation concentration in pure water and polysorbate 80 aqueous solution was determined by adding an excess of the drug to pure water (n = 3) or to solutions (n = 3) containing P-80 in the same concentration as in the LCN aqueous phase. The mixture was sonicated for 10 min and, after 24 hours of storage at room temperature, samples were centrifuged at 1844 x g for 10 min and the supernatant was analyzed by LC/UV. pH measurements and logD determination The pH values of the QLNC formulation were measured using a calibrated potentiometer (Digimed, Brazil) at 25 ºC. The software MarvinSketch 5.12.4 (Academic Licenses - ChemAxon,Ltda, Budapest, Hungary) was used to determine the logD values, using the experimental pH values obtained. Determination of the drug distribution into the LNC The distribution of QTP among the diverse pseudo-phases of the nanocapsules suspension was determined using an algorithm developed by Oliveira et al.7. Briefly, QLNC were diluted with ultrapure water in order to give final concentrations of 1:2, 1:10, 1:100 and 1:1000 (v/v). 200 µL of each diluted sample was transferred to centrifugal filter device (Microcon® - Millipore) and centrifuged at 1844 x g for 10 min at 25 ± 1 ºC. The filtrate was collected and 20 µL was injected into the LC/UV as describe previously. This experiment was performed in triplicate.
RESULTS AND DISCUSSION LC/UV method development Before the validation step, the chromatographic conditions were optimized for the determination of QTP in order to provide a good performance analysis within a suitable analysis time (5 min), peak parameters (symmetry, tailing), ease of preparation and low cost. The mobile phase consisting a mixture of triethylamine aqueous solution (0.4%, v/v) adjusted to pH 3.0 ± 0.2 using phosphoric acid and acetonitrile (70:30,v/v), using isocratic mode with mobile phase flow rate at 1.0 mL.min-1 was found to be satisfactory allowing a rapid determination of the drug, which is important for routine analysis. The use of a C18 column was suitable for separation of QTP and resulted in adequate peak shape and resolution. Based on QTP UV spectra (data not shown) the wavelength for UV analysis was chosen allowing the maximum absorption for the drug detection. The specificity of the analytical method is illustrated in Figure 2 by comparing the chromatograms of blank extracted LNC (Figure 2-A) and QTP extracted from QLNC formulation (10 µg mL-1) (Figure 2-B).
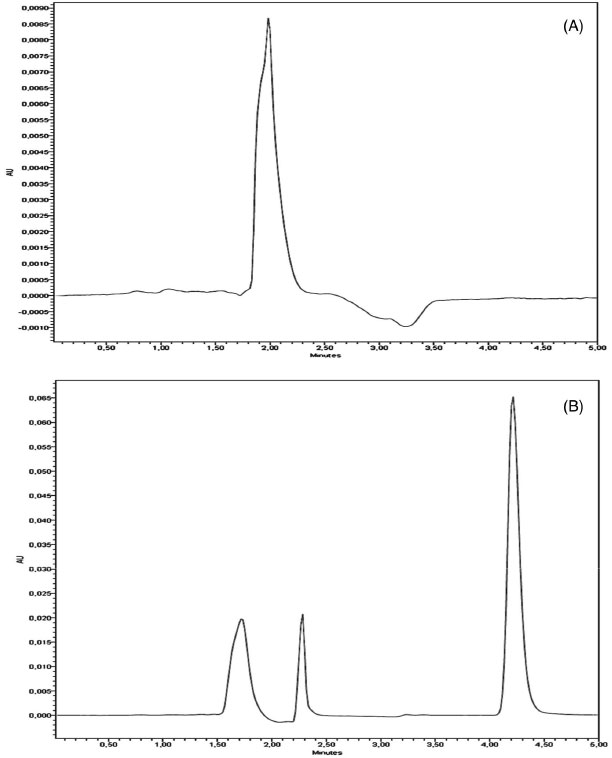 Figure 2. Representative LC chromatograms of: (A) blank extracted LNC; (B) and QTP extracted from QLNC formulation (10 µg mL-1). QTP retention time is 4.3 min
The chromatograms obtained by LC/UV for blank LNC showed a low signal response for the solvent and did not show any peak with similar retention time to QTP (4.3 min ± 5%) allowing to conclude that the developed method is selective in relation to the excipients of the final QLNC formulation. Method validation Linearity of the standard curves was checked in six different runs by the calculation of slopes, intercepts and determination coefficient of each individual curve after plotting the peak area versus nominal concentration. The method showed good linearity in the 0.5 - 20 µg mL-1 range. The representative linear equation was: y = 66519.17x - 5343.8 with a determination coefficient (r2 = 0.999) highly significant for the method (Table 1).
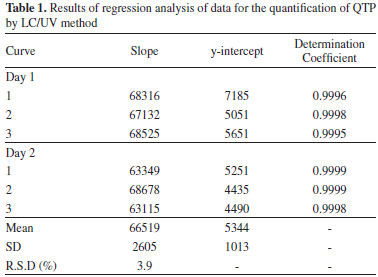
The linearity results were analyzed for statistical significance by ANOVA, using the software GraphPad Prism 5.01. A linear regression (Fcalc > Fcrit; p > 0.05) with no deviation from linearity (Fcalc < Fcrit; p < 0.05) was observed. Repeatability (intra-day) and intermediate precision (inter-day) of the method are reported in Table 2.

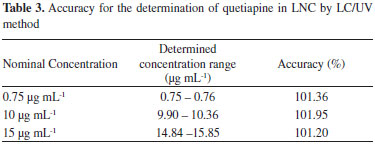
The method showed repeatability R.S.D (%) ranging between 0.53 - 1 and an intermediate precision R.S.D (%) of 1.36, lower than the officially accepted 5%, in accordance to ANVISA (16) and ICH (17) being considered precise. The accuracy of the method was determined and the mean recovery was found to be 101.5% (Table 3). According to ICH and ANVISA guidelines, for pharmaceutical formulations the recovery percent in accuracy test must be between 95% and 105%, equivalent to ± 5.0% of the relative error.
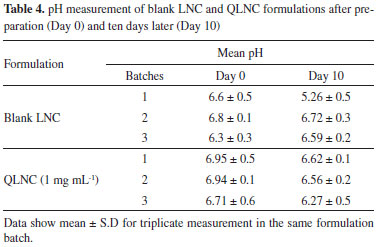
The results of the stability tests showed that QTP standard solution was stable at room temperature for 6 hours, maintaining 104.11 ± 0.19% (lower CQ) and 102.06 ± 3.80% (higher CQ) of the initial concentration and for 1 month in at the -80 ± 1 ºC freezer (lower CQ: 99.31 ± 0.19% and higher CQ: 100.20 ± 1.43%). A literature review showed that previous methods described QTP quantification using liquid chromatography in tandem with mass spectrometry, using high percentage of organic solvent or long running time (~ 9.0 min).13,14,17 The present method is simple, economical (the mobile phase is 70% water) and rapid (QTP retention time: 4.3 min) and was successfully applied for quantification of QTP associated with nanoparticles. QLNC pH measurements and logD determination In order to determine the pH of the QLNC, three different batches were prepared. The QLNC were physicochemically characterized presenting nanometric size (D4.3 = 172 ± 5 nm), negative zeta potential (-9.15 ± 0.9 mV) and low polydispersity index (PI < 0.1). Blank nanocapsules showed similar results in the physicochemical characterization (data not shown) The pH was measured in triplicate, just after the preparation of the LNC (Day 0) and ten days after (Day 10), using a calibrated pHmeter. The results are presented in Table 4.
The logarithm of the octanol-water distribution coefficient (logD) is defined as the ratio of the equilibrium concentration of a molecule (unionized) in octanol to that of the same molecule in the water phase (unionized or ionized) and is used as a tool to estimate drug's lipophilicity. According to Oliveira et al.7 the logD at the pH of the nanocapsules formulation is the best physicochemical parameter for predicting the type of drug distribution in LNC aqueous solution. Using the software MarvinSketch 5.12.4 QTP octanol-water partitioning pH-dependent logD was simulated considering the molecule ionizable groups and the different ionic forms of the drug in solution with pHs ranging between 0 and 14. Fo the simulations equation 1 was used:  The simulation plot of LogD as a function of pH is shown in Figure 3.
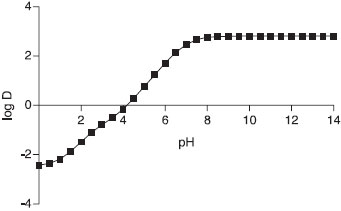 Figure 3. QTP octanol-water partitioning values (LogD) as a function of pH obtained after MarvinSketch simulation using Eq. 1
Analyzing the graph in Figure 3 it is possible to note that there is a proportional pH-dependent partitioning effect for QTP. An increasing in the pH promotes an increase in QTP unionized molecules in water increasing the drug's affinity to octanol phase. QLNC formulation pH at Day 0 was 6.86 ± 0.4, leading to a QTP logD of 2.35. QLNC drug content and incorporation efficiency The LC/UV validated method was successfully applied in order to determine the total QTP content and the drug loading into LNC following centrifugation-ultrafiltration method. The incorporation efficiency (IE%) was determined for the difference between the total QTP concentration and the concentration of free drug in the ultrafiltrate, using the following equation (Eq. 2): 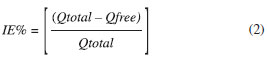 where Qtotal is the total concentration of QTP in the colloidal suspension (µg mL-1) and Qfree is the concentration of free QTP in the ultrafiltrate phase (µg mL-1). The results of incorporation efficiency are summarized in Table 5.

The method of sample extraction and analysis proved to be efficient for absolute recovery analysis, the average-absolute recovery (QTP content) was 0.976 ± 1.4 mg mL-1, which is considered very efficient. The method was also capable of determining free QTP present in the ultrafitrate samples. The encapsulation efficiency (IE%) describes the efficiency of the QTP loading process into the QLNC. After centrifugation, the ultrafiltrate could be directly analyzed by the LC/UV method developed, without any sample preparation steps. The high IE observed (95.5%) is due to QTP lipophilicity at the formulation pH which increases the dispersion of the drug in the lipid core of the nanocapsules. QTP saturation concentrations in water and polysorbate 80 aqueous solution The saturation concentration of QTP in pure water was 4.1 ± 0.6 x 10-2 mg mL-1, slightly lower than in the P-80 solution, 5.5 ± 0.3 x 10-2 mg mL-1. These results demonstrate that free drug is soluble in LNC aqueous medium of nanoparticles dispersion and that QTP concentration in the ultrafiltrate is below its saturation concentration does not allowing for the formation of insolubles crystals. Furthermore, the drug is very soluble in the oil of the nanoparticles core (> 1 mg mL-1). After LNC dilution with pure water, viewing to determine the type of drug distribution in the nanoparticles, QTP concentration in the aqueous media is even lower that the saturation concentration and no nanocrystal could be formed. Application of the algorithm to experimentally determine QTP distribution QLNC formulation We applied an algorithm proposed by Oliveira et al.7 in order to determine the distribution of quetiapine in the pseudo-phases of the nanoparticles. The algorithm is composed of six questions and the rational basis of it is the use of serial dilutions and centrifugations in order to shift the drug distribution equilibrium allowing the quantification of the drug distribution among the different pseudo-phases of the formulation. The results obtained for the dilution in QLNC are shown in Table 6.
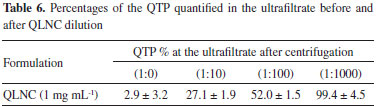
The results summarized in Table 6 were used in order to answer the questions of the algorithm. The first question is regarding the size distribution of the formulation and differentiates a suspension from a colloidal formulation (exclusively nanoscopic particles size), this distinction is important since when microscopic contamination is detected pre-formulation studies are still needed. QLNC formulation presented a volume- weighted mean diameter (D4.3) of 172 ± 5 nm and a low polydispersity index (PI < 0.1) indicating that is a real colloidal formulation. The second question "is the drug detected in the ultrafiltrate?" was answered after the centrifugation-ultrafiltration method using the formulation without any dilution. QTP was detected in the ultrafiltration which indicates that there are drug dispersed in the aqueous solution either as drug aggregates or in the colloidal phase. The small percentage of QTP detected in the ultrafiltrate without previous dilution (2.9 ± 3.2%) answers the third algorithm's question: "Is 100% of the drug detected in the ultrafiltrate?" and allows one to conclude that the drug in QLNC formulations is not only dispersed in colloidal continuous phase. The dilution of QLNC formulation promotes a shift in the drug distribution equilibrium among the colloidal pseudo-phases and was necessary in order to differentiate the drug distribution types. The fourth question of the algorithm "After diluting at 1:10 (v/v) is 100% of the drug detected in the ultrafiltrate?". The results showed that 27.1 ± 1.9% of QTP were detected in the ultrafiltrate after 1:10 (v/v) dilution which indicates that QLNC have a drug distribution in continuous phase, dispersed at the interface and dissolved in the core of the nanocapsules. Increasing the formulation's dilution (1:100 and 1: 1000 v/v) the QTP (%) in the ultrafiltrate increases and was near 100% after 1:1000 (v/v) dilution analysis. According to the algorithm proposed QTP follow a type III distribution (Figure 4) and is distributed mainly in the inner pseudo-phase of the colloidal suspension. However, there is still some drug associated to the interface of the polymeric shell as well as soluble in the outer phase of the formulation. The results obtained with QTP corroborate the existence of a strong correlation between the logD and the type of drug distribution into LNC.
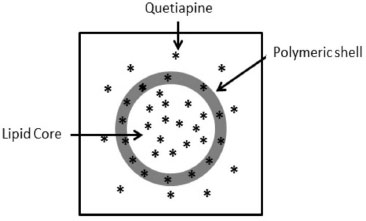 Figure 4. Schematic representation of QTP distribution in the LNC based on the algorithm described in ref. 7
CONCLUSIONS The results from our study demonstrated the development, validation and application of a new LC/UV for the quantification of QTP in lipid-core nanocapsules. The method was shown to be simple, rapid, specific, linear, accurate, sensitive and precise. The analytical procedure has a total analysis time of 5 min which allows analyzing a large number of samples in a short period of time. The low percentage of acetonitrile used reduces the cost and the damage to the environment. The method was suitable for quantifying QTP total content and incorporation efficiency in the nanocarriers, showing that QTP has a high incorporation efficiency in the LNC formulation. By using a previously described algorithm7 it was shown that QTP is mainly distributed into the core of the LNC and that this distribution is related to its logD at pH 6.8. Taking together these results indicates that QLNC can be a suitable formulation to deliver QTP to the brain assuming that the type of drug distribution influences the potential for the application of a new formulation as a drug delivery system.
ACKNOWLEDGMENTS The authors would like to thank Dr. D. Kawano (UFRGS) for the contributions during the early stages of this project and Dr. L. Brum-Junior (Prati Donaduzzi) for the donation of quetiapine fumarate. This study was financially supported by CNPq. The authors thank CNPq for the scholarships.
REFERENCES 1. Chandrappa, P.; Ther. Adv. Psychopharmacol. 2012, 2, 207. DOI: http://dx.doi.org/10.1177/2045125312451265 PMID: 23983977 2. Ogawa, N.; Kaga, M.; Endo, T.; Nagase, H.; Furuishi, T.; Yamamoto, H.; Chem. Pharm. Bull. 2013, 61, 809. DOI: http://dx.doi.org/10.1248/cpb.c13-00157 PMID: 23902863 3. Pucci, V.; Mandrioli, R.; Ferranti, A.; Furlanetto, S.; Augusta Raggi, M.; J. Pharm. Biomed. Anal. 2003, 32, 1037. DOI: http://dx.doi.org/10.1016/S0731-7085(03)00206-1 PMID: 12899991 4. Narala, A.; Veerabrahma, K.; J. Pharm. (2013), doi: 10.1155/2013/265741. DOI: http://dx.doi.org/10.1155/2013/265741 5. Rasmussen, H.; Ebdrup, B. H.; Oranje, B.; Pinborg, L. H.; Knudsen, G. M.; Glenthoj, B.; Int. J. Neuropsychopharmacol. 2014, 17, 1729. DOI: http://dx.doi.org/10.1017/S1461145714000777 PMID: 24830305 6. Dimer, F.A.; Ortiz, M.; Pase, C. S.; Roversi, K.; Friedrich, R. B.; Pohlmann, A. R.; Guterres, S. S.; J. Biomed. Nanotechnol. 2014, 10, 1137. DOI: http://dx.doi.org/10.1166/jbn.2014.1817 PMID: 24749408 7. Oliveira, C. P.; Venturini, C. G.; Donida, B.; Poletto, F. S.; Guterres, S. S.; Pohlmann, A. R.; Soft Matter 2012, 9, 1141. DOI: http://dx.doi.org/10.1039/C2SM26959G 8. Venturini, C. G.; Jäger, E.; Oliveira, C. P.; Bernardi, A.; Battastini, A. M. O.; Guterres, S. S.; Pohlmann, A. R.; Colloids Surf., A 2011, 375, 200. DOI: http://dx.doi.org/10.1016/j.colsurfa.2010.12.011 9. Dimer, F. A.; Pigatto, M. C.; Boque, C. A.; Pase, C. S.; Roversi, K.; Pohlmann, A. R.; Dalla Costa, T.; Guterres, S. S.; J. Biomed. Nanotechnol. 2015, 11, 1482. DOI: http://dx.doi.org/10.1166/jbn.2015.2082 10. Jäger, E.; Venturini, C. G.; Poletto, F. S.; Colomé, L. M.; Pohlmann J. P.; Bernardi, A.; Battastini, A. M.; Guterres, S. S.; Pohlman, A. R.; J. Biomed. Nanotechnol. 2009, 5, 130. DOI: http://dx.doi.org/10.1166/jbn.2009.1004 PMID: 20055116 11. Davis, P. C.; Wong, J.; Gefvert, O.; J. Pharm. Biomed. Anal. 1999, 20, 271. DOI: http://dx.doi.org/10.1016/S0731-7085(99)00036-9 PMID: 10704032 12. Pires-Rosa, P. C.; Rodrigues-Pires, Y. F.; Ortega, B. H.; Perazzo, F. F.; J. Appl. Pharm. Sci. 2013, 3, 6. 13. Barrett, B.; Holcapek, M.; Huclová, J.; Borek-Dohalsky, V.; Fejt, P.; Nemec, B.; J. Pharm. Biomed. Anal. 2007, 44, 498. DOI: http://dx.doi.org/10.1016/j.jpba.2007.03.034 PMID: 17499470 14. Fisher, D. S.; Handley, S. A.; Taylor, D.; Flanagan, R. J.; Biomed. Chromatogr. 2012, 26, 1125. DOI: http://dx.doi.org/10.1002/bmc.2672 PMID: 22241669 15. Fessi, H.; Puisieux, F.; Devissaguet, J. P.; Ammoury, N.; Benita, S.; Int. J. Pharm. 1989, 55, 1. DOI: http://dx.doi.org/10.1016/0378-5173(89)90270-6 16. International Conference on Harmonization (ICH); Technical Requirements for the Registration of Pharmaceutical for Human Use, Validation of Analytical Procedures: Text and Methodology Q2 (R1), 2005. 17. Pant, M.; Khatri, N. C.; Int. J. Pharm. Life Sci. 2012, 2, 95. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access