
 |
 |
 |
 |
 |
Nota Técnica
|
|
| Sistemas alternativos de calibração para determinação espectrofotométrica simultânea de espécies com interferência espectral Alternative calibration systems for the simultaneous spectrophotometric determination of compounds with overlapping absorption spectra |
|
Sandra StetsI; Barbara Duarte da SilvaI; Talita Maria TavaresI; Gilcélia A. CordeiroI; Noemi NagataI; Christiana Andrade PessoaII; Patricio Peralta-ZamoraI,*
IDepartamento de Química, Universidade Federal do Paraná, CP 19081, 81531-980 Curitiba - PR, Brasil Recebido em 29/04/2015 *e-mail: zamora@ufpr.br Two simple and efficient procedures have been developed for the rapid simultaneous determination of compounds with mutual spectral interference (rifampicin (RIF) and isoniazid (INH)). The first method was based on the UV-Vis spectral signal (190-600 nm) of synthetic RIF and INH aqueous solutions, whereas the second method involved the visible spectral signal registered between 350 and 800 nm after the reaction of INH with a Cu2+/neocuproine complex. Both multivariate spectrophotometric methods show excellent prevision capacity, providing results that are statistically equivalent with those provided by the standard chromatographic procedure. The methods were validated according to criteria established by ANVISA, showing precision, accuracy and robustness compatible with the requirements for new analytical methods, additionally allowing the reduction of waste generation. INTRODUÇAO Em funçao de características como elevada sensibilidade, rapidez, robustez e baixo custo, a espectroscopia eletrônica poderia fundamentar inúmeras aplicaçoes analíticas de relevância. Infelizmente, a baixa seletividade da técnica usualmente implica sérios problemas de interferência espectral, o que com frequência impede a sua aplicaçao na determinaçao de espécies de interesse em matrizes complexas e, inclusive, de misturas de substâncias em matrizes mais simples. Para contornar esses inconvenientes, sem a necessidade de técnicas de separaçao preliminar, vários sistemas de calibraçao menos convencionais têm sido propostos, dentre os quais se destacam o método de Vierordt's, fundamentado no princípio de aditividade da absorbância, métodos derivativos, geralmente associados ao ponto de anulaçao, e os sistemas de calibraçao multivariada, principalmente fundamentada em regressao por mínimos quadrados parciais (PLS). Grande parte desses métodos de calibraçao encontra importante aplicaçao no controle de qualidade de medicamentos, tal como ilustrado nos exemplos salientados a seguir. Em 1999, Dinç1 desenvolveu um método espectrofotométrico fundamentado no método de Vierordt's para a determinaçao simultânea de cafeína e paracetamol em comprimidos. Embora a interferência espectral seja severa, o método permitiu a obtençao de resultados análogos aos obtidos pelo método cromatográfico padrao. Um método similar foi proposto recentemente por Rajput et al.,2 objetivando a determinaçao simultânea de claritromicina e ranitidina em ensaios de dissoluçao a partir de cápsulas bifuncionais. Métodos fundamentados em espectrofotometria derivativa sao utilizados desde os anos 1950, tornando-se mais populares a partir da década de 1970, época em que Shibata e colaboradores3 publicaram um trabalho sobre espectrofotometria derivativa fundamentada em mediçoes em dois comprimentos de onda. A partir desta data inúmeros trabalhos reportaram a determinaçao simultânea de fármacos, destacando-se, recentemente, um estudo que objetivou a determinaçao simultânea de três antibióticos de uso oftálmico (cloranfenicol, dexametazona e nafasolina) por espectrofotometria derivativa de primeira ordem.4 Neste estudo, o método espectrofotométrico permitiu resultados muito próximos aos obtidos por um método cromatográfico de referência. Métodos quimiométricos têm sido utilizados desde a década de 1990 na área farmacêutica, principalmente para a quantificaçao de fármacos e produtos de degradaçao. Em 2014, Elkhoudary et al.5 propuseram um método espectrofotométrico multivariado para a determinaçao de quatro fármacos (metronidazol, espiramicina, diloxanida e clioquinol) por várias técnicas multivariadas, incluindo PLS. O método foi validado, mostrando sensibilidade, precisao e exatidao compatíveis com a natureza da análise, com pouca interferência por parte dos excipientes. Estudos similares têm demonstrado a excelente potencialidade dos métodos de calibraçao multivariada, nao apenas para determinaçao de misturas de fármacos,6 mas também para a determinaçao de um fármaco na presença de produtos de degradaçao.7 Em alguns casos, simples reaçoes de derivatizaçao química podem levar à formaçao de espécies que absorvem na regiao do visível, o que diminui a interferência por parte de fármacos associados ou de componentes da matriz (excipientes), que costumam absorver fortemente na regiao ultravioleta. Dentro do contexto da análise de medicamentos, as reaçoes de derivatizaçao sao relativamente comuns para facilitar a detecçao após processos de separaçao cromatográfica.8 Poucos trabalhos descrevem a derivatizaçao química como alternativa para a análise de produtos farmacêuticos por espectroscopia eletrônica. Entretanto, destacam importantes aplicaçoes que envolvem reaçoes de derivatizaçao fundamentadas em complexaçao9 e oxidaçao.10 Métodos analíticos orientados à determinaçao espectrofotométrica desta associaçao foram propostos no final da década de 1990, recorrendo-se a sistemas de calibraçao fundamentados em espectrofotometria derivativa.11 Mais recentemente têm sido propostas metodologias fundamentadas em processos de deconvoluçao e de funçoes trigonométricas de Fourier,12 assim como sistemas multivariados fundamentados em PLS.13 No trabalho de Espinosa-Mansilla et al.,14 um estudo comparativo foi realizado entre modelos elaborados por PLS-1 e uma modificaçao da análise linear híbrida (HLA/XS), objetivando a determinaçao de rifampicina, pirazinamida e isoniazida. Ambos os métodos permitiram a quantificaçao dos fármacos em medicamentos, com algumas restriçoes para isoniazida, em razao da sua baixa absortividade relativa. Neste trabalho, dois métodos PLS-2 foram avaliados em relaçao à determinaçao espectrofotométrica de rifampicina e isoniazida, sendo o primeiro desenvolvido utilizando a simples dissoluçao dos analitos em água ultra pura. Adicionalmente, foi avaliado um método colorimétrico precedido de reaçoes de derivatizaçao, mediadas por complexos formados entre íon cúprico (Cu2+) e neocuproína, objetivando-se uma maior seletividade que possibilite a aplicaçao do método na determinaçao de RIF e INH em amostras de urina.
MATERIAIS E MÉTODOS Reagentes e medicamentos Padroes de isoniazida (INH: 100,80%) e rifampicina (RIF: 99.70%) foram gentilmente fornecidos pelo Laboratório Farmacêutico Far-Manguinhos, da Fundaçao Oswaldo Cruz. Soluçoes de trabalho foram preparadas diariamente, utilizando-se água deionizada (Milli-Q® - 18,2 MΩ) e vidraria analítica previamente calibrada. Medicamentos contendo a associaçao em estudo (100 mg de INH + 150 mg de RIF e 200 mg de INH + 300 mg de RIF) foram gentilmente fornecidos pela Secretaria de Saúde do Estado do Paraná. Todos os demais reagentes utilizados foram de grau analítico. Equipamentos Espectros UV/Vis em soluçao foram adquiridos em espectrofotômetro Shimadzu (modelo 2410 PC), utilizando-se cubetas de quartzo de 1 cm de caminho ótico. Para fins comparativos, espectros foram também registrados em espectrofotômetro Hewlett Packard (modelo 8452 A). A análise por Cromatografia Líquida de Alta Eficiência foi realizada em um Cromatógrafo Varian 920-LC, equipado com coluna C18 (4,6 mm x 25 cm) e detector de arranjo de fotodiodos (238 nm). Como fase móvel foi utilizado fosfato de sódio dibásico: acetonitrila e sistema de eluiçao por gradiente, conforme método padrao de análise.15 Curvas analíticas individuais foram preparadas a partir de padroes puros, cobrindo-se a faixa de 0,08 a 0,12 mg L-1 para isoniazida (R: 0,997) e de 0,13 a 0,20 mg L-1 para rifampicina (R: 0,995). Programas computacionais Para a montagem de matrizes de dados utilizou-se o software Origin Pro 6.1® (OriginLab), enquanto que para a elaboraçao dos modelos empregou-se o programa PLS-toolbox 3.0 (Eigenvector Research, Inc.), que opera em ambiente Matlab v.6.5 (Math Work Inc.). Os gráficos de superfície de resposta foram construídos no programa Statistic 6.0 (StatSoft inc.). Desenvolvimento de modelos de calibraçao Modelos convencionais Para o desenvolvimento de modelos univariados convencionais foram elaboradas curvas analíticas individuais, no comprimento de onda de máxima absorçao apresentado por cada fármaco (INH: 262 nm, RIF: 472 nm). Cada curva analítica foi composta de 5 valores de concentraçao, cobrindo-se a faixa de 8,0 a 12,0 mg L-1 para isoniazida, e de 12,0 a 18,0 mg L-1 para rifampicina. Modelos univariados foram também elaborados no modo derivativo. Neste caso, curvas analíticas para INH foram elaboradas nos comprimentos de onda em que RIF apresenta uma derivada da absorbância igual à zero (ponto de anulaçao). O mesmo procedimento foi utilizado para a elaboraçao das curvas analíticas de rifampicina. Modelos multivariados sem reaçao colorimétrica com Cu(ll) e neocuproína Modelos multivariados de calibraçao (PLSR) foram desenvolvidos a partir de 28 misturas sintéticas, contendo 8,0 a 12,0 mg L-1 de INH e 12,0 a 18,0 mg L-1 de RIF, enquanto que outras 7 amostras foram reservadas para a fase de validaçao (Figura 1A). Nenhum tipo de planejamento de misturas foi utilizado para estabelecer o conjunto de calibraçao, sendo utilizado como único critério o teor dos fármacos nos medicamentos analisados e a variaçao máxima permitida, que costuma ser de 20%.
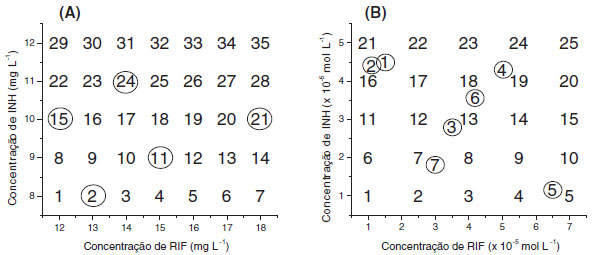 Figura 1. Misturas sintéticas utilizadas para elaboraçao dos modelos multivariados fundamentados em espectroscopia eletrônica direta (A) e com reaçao de complexaçao utilizando neocuproína (B). Amostras em destaque (circuladas) correspondem ao conjunto de validaçao externa
Os espectros foram registrados entre 190 e 600 nm, sendo processados após rotinas de centrar na média (CM), auto-escalamento (AU), alisamento pelo método Savitzky-Golay (SG) e alisamento com derivaçao (SG1ª). O número de variáveis latentes foi selecionado com base na minimizaçao do erro relativo médio na validaçao cruzada (sistema leave-one-out), enquanto que a capacidade de previsao foi avaliada com um conjunto de sete misturas sintéticas, reservadas como conjunto de validaçao externa (amostras circuladas na Figura 1A). Na análise de amostras reais (medicamentos), 20 comprimidos (ou cápsulas) foram triturados e homogeneizadas em almofariz. Massas de aproximadamente 0,06 g foram tomadas e dissolvidas em 1.000,00 mL de água destilada. Desta soluçao foram tomados volumes de 20,00 a 50,00 mL, dependendo da concentraçao dos substratos em estudo, sendo finalmente diluídos até 100,0 mL. Todos os ensaios foram realizados em triplicata. Modelos multivariados com reaçao colorimétrica com Cu(ll) e neocuproína Modelos multivariados de calibraçao (PLSR) foram desenvolvidos a partir de 25 misturas sintéticas, contendo 1,0 a 5,0 mg L-1 de INH e 1,0 a 7,0 mg L-1 de RIF, enquanto que outras 13 amostras, incluindo 3 triplicatas, foram reservadas para a fase de validaçao (Figura 1B). Em todos os ensaios foram adicionados 1000 mL das soluçoes estoque de neocuproína (1,0 x 10-2 mol L-1 em metanol 10%) e Cu(NO3)2.3H2O (1,0x10-2 mol L-1), utilizando-se tampao acetato (pH = 5,00) em todas as diluiçoes.16 Para elaboraçao dos modelos multivariados, os espectros adquiridos na regiao do visível (350 e 800 nm) foram suavizados, utilizando-se o método Savitzky-Golay. A seleçao do número de variáveis latentes foi realizada de acordo com o mesmo critério utilizado na elaboraçao dos modelos descritos no item anterior. Da mesma forma, a capacidade de previsao foi avaliada com um conjunto de sete misturas sintéticas, reservadas como conjunto de validaçao externa (amostras circuladas na Figura 1B). Na análise de amostras reais (medicamentos), 20 comprimidos (ou cápsulas) foram triturados e homogeneizadas em almofariz. Massas de aproximadamente 0,06 g foram tomadas, dissolvidas em 10,00 ml de metanol, completando-se o volume com água até 100,00 mL. A seguir, uma alíquota de 1000 µL foi transferida para balao volumétrico de 10,00 mL, adicionando-se 1000 µL de soluçao de nitrato cúprico (1,0 x 10-2 mol L-1) e 1000 µL de soluçao de neocuproina (1,0 x 10-2 mol L-1) e completando-se o volume com tampao acetato de sódio/ácido acético (pH 5,00). Validaçao de metodologias multivariadas Ambos os modelos multivariados foram validados de acordo com os critérios estabelecidos pela ANVISA,17 avaliando parâmetros de exatidao, precisao (repetibilidade e reprodutibilidade) e robustez (tempo de leitura, temperatura e pH). A precisao foi avaliada por análise em triplicata de três misturas sintéticas, contendo concentraçao baixa, média e alta. A repetibilidade foi avaliada por análises efetuadas pelo mesmo operador, no mesmo dia e utilizando-se o mesmo equipamento, enquanto que a reprodutibilidade envolveu a participaçao de um segundo analista e a utilizaçao de um equipamento diferente (espectrofotômetro HP 8452-A). A precisao foi expressa na forma de desvio padrao relativo, considerando-se aceitos valores inferiores a 5%. A exatidao foi avaliada por meio de ensaios de recuperaçao, utilizando-se as mesmas misturas sintéticas utilizadas na avaliaçao da precisao. A exatidao foi expressa na forma de erro percentual, considerando-se aceitos valores de recuperaçao entre 95 e 105%. A avaliaçao da robustez envolveu estudos orientados a verificar o efeito do tempo de leitura, da temperatura e do pH. Estes estudos foram realizados com apenas uma mistura sintética, contendo 10,00 mg L-1 de INH e 15,00 mg L-1 de RIF.
RESULTADOS E DISCUSSAO Análise univariada convencional A partir dos espectros das substâncias puras (Figura 2) é possível verificar que isoniazida apresenta apenas uma banda de absorçao, centrada em 262 nm, a qual é intensamente interferida pelo sinal da rifampicina. Para este segundo fármaco observam-se duas bandas praticamente livres de interferência, centradas em 472 e 333 nm. Em primeira análise, a quantificaçao convencional de rifampicina parece possível utilizando-se o sinal registrado em 472 nm, enquanto que para isoniazida a necessidade de métodos diferenciados é bastante evidente.
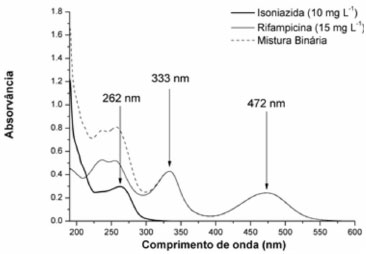 Figura 2. Espectro eletrônico de soluçoes aquosas de padroes puros de isoniazida, rifampicina e da sua mistura
Inicialmente, curvas analíticas convencionais foram elaboradas para cada fármaco, obtendo-se coeficientes de regressao (INH em 262 nm: 0,999, RIF em 472 nm: 0,998) superiores ao mínimo exigido pela ANVISA (rmin: 0,99). Na análise das 7 misturas sintéticas reservadas para validaçao o método proporcionou erros médios de previsao da ordem de 200% para INH, por conta da forte interferência espectral ocasionada pela RIF. Para rifampicina a situaçao é completamente diferente, em razao da inexistência de efeitos interferentes por parte do segundo fármaco. Nestas condiçoes, o erro médio de previsao é da ordem de 1% (Tabela 1).
Procurando aprimorar o modelo univariado, processos espectroscópicos derivativos foram avaliados. No modo derivativo, os máximos de absorçao se transformam em pontos de inflexao em que a derivada da absorbância tem valor zero, para qualquer valor de concentraçao (ponto de anulaçao). Em funçao desta característica, uma curva analítica livre de interferência pode ser elaborada para RIF e INH, em 262 nm e 295 nm, respectivamente. Embora no modo derivativo o sinal espectral diminua drasticamente, curvas analíticas de qualidade foram conseguidas, com coeficientes de correlaçao da ordem de 0,99. Finalmente, estudos de aplicabilidade do sistema fundamentado no princípio da aditividade das absorbâncias foram também desenvolvidos (método de Vierordt's), registrando-se os valores de absorbância para a mistura de substratos em 262 e 472 nm e resolvendo-se o sistema de duas equaçoes apresentado a seguir: 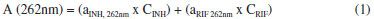 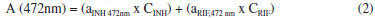 onde: A: Absorbância, a: absortividade (L mg-1 cm-1), C: concentraçao (mg L-1) O método derivativo e o método fundamentado na aditividade de absorbâncias proporcionaram excelentes resultados na previsao das misturas sintéticas (Tabela 1), com erros relativos médios inferiores a 5%. Entretanto, na análise de medicamentos contendo a associaçao em estudo, apenas o sistema fundamentado no princípio da aditividade fornece resultados coerentes, com erros relativos médios inferiores a 10%. Análise multivariada sem reaçao colorimétrica Normalmente, a primeira decisao que deve ser tomada no processo de elaboraçao de modelos multivariados está representada pela seleçao do número de variáveis latentes (VLs). Neste estudo, o critério adotado correspondeu à minimizaçao do erro de previsao no processo de validaçao cruzada (sistema leave-one-out). Para ambos os fármacos, o uso de 2 ou 3 VLs implicou significativa reduçao deste erro de previsao, ao passo que a introduçao de novas variáveis pouco contribuiu com a melhora deste parâmetro. Adicionalmente, 2 VLs conseguem representar mais do que 99% da variância da matriz de concentraçao, valendo-se de 99,97% da variância dos dados espectrais. Embora a utilizaçao de um elevado número de VLs represente riscos de superajuste, incluindo-se flutuaçoes ou ruídos instrumentais, modelos foram desenvolvidos com 2, 3, 4, 6 e 8 VLs. Adicionalmente, 4 tipos de pré-processamento de sinais foram avaliados, incluindo-se dados centrados na média, para reduzir a dimensao dos modelos, autoescalados, para evidenciar sinais de menor intensidade, suavizados pelo filtro Savitzky-Golay (janela de 5 pontos e ajuste de polinômio de primeira ordem) para a remoçao de ruídos aleatórios, e suavizados e derivados (derivada de primeira ordem) para remoçao de deslocamentos sistemáticos da linha base. Neste estudo, os menores erros de previsao foram proporcionados pelo processamento de dados centrados na média (pré-processamento 0), utilizando-se 4 a 8 VLs (Figura 3). Em funçao dos valores de erro relativo médio nao terem sido considerados estatisticamente diferentes pelo teste de significância utilizado (teste t-Student), o modelo com 4 VLs foi selecionado para estudos posteriores, modelo este que permitiu erros médios de previsao inferiores a 1% na quantificaçao de ambos fármacos nas amostras reservadas para validaçao, assim como índices de correlaçao entre valores reais e previstos superiores a 0,99.
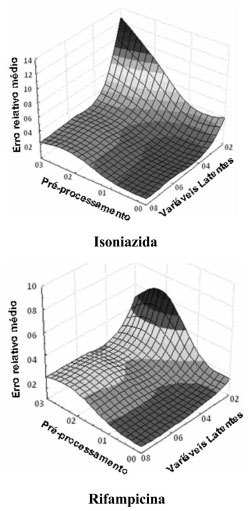 Figura 3. Variaçao do erro médio de previsao no processo de validaçao cruzada (sistema leave-one-out) em funçao do tipo de pré-processamento e do número de variáveis latentes (pré-processamento 0: dados centrados na média, pré-processamento 1: dados autoescalados, pré-processamento 2: dados suavizados, pré-processamento 3: dados suavizados e derivados (1ª ordem)
Na etapa de validaçao externa foi constatada uma excelente correlaçao entre valores reais e previstos (Figura 4), o que demonstra um bom ajuste do modelo e a ausência de erros sistemáticos.
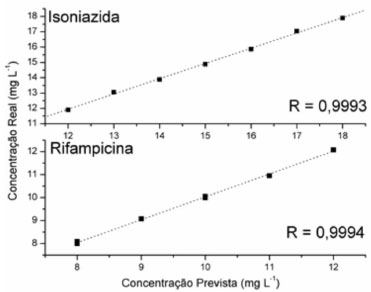 Figura 4. Gráfico de valores reais por previstos na fase de validaçao externa pelo modelo desenvolvido com espectros centrados na média e 4 VLs
Na análise de medicamentos (Tabela 2), o modelo multivariado proporcionou resultados coerentes com os resultados obtidos por aplicaçao do método cromatográfico padrao, com discrepâncias médias inferiores a 5% para ambos os fármacos.
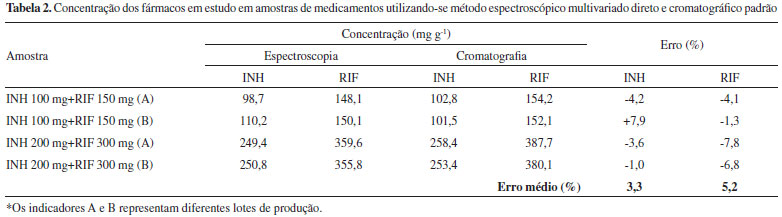
Análise multivariada após reaçao colorimétrica de RIF e INH com Cu(ll) e neocuproína A reaçao colorimétrica química da INH foi fundamentada no seu efeito redutor sobre o complexo Cu(II)-neocuproína, o que leva à formaçao do complexo Cu(I)-neocuproína que absorve intensamente em 449 nm.16 Para elaboraçao do modelo multivariado foi utilizado um conjunto de calibraçao composto por 25 misturas sintéticas e espectros na regiao do visível (350 a 800 nm), pré-processados por alisamento. O modelo de melhor desempenho foi elaborado com 3 VLs, permitindo erros médios de previsao inferiores a 2% na fase de validaçao externa, assim como ausência de erros sistemáticos. Na análise de medicamentos (Tabela 3) foi possível observar erros médios de previsao inferiores a 5%, tomando-se como referência os resultados obtidos pela técnica cromatográfica padrao. Cabe ressaltar que a utilizaçao de reaçao colorimétrica propiciou maior seletividade do método, uma vez que permite a determinaçao dos analitos na regiao visível do espectro ultravioleta, permitindo a aplicaçao com sucesso na determinaçao de RIF e INH em amostras de urina humana conforme relatado em um trabalho anteriormente publicado.16
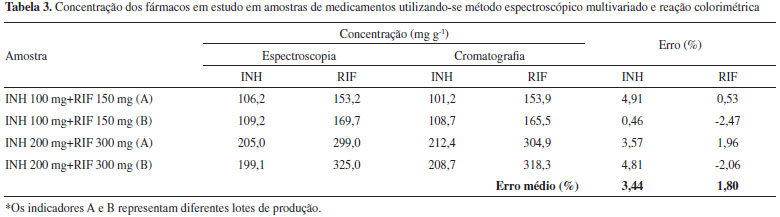
Consideraçoes sobre os modelos multivariados desenvolvidos Em primeiro lugar, é importante salientar que ambas as metodologias multivariadas foram validadas de acordo com os critérios definidos pela ANVISA, levando-se em consideraçao parâmetros de precisao, exatidao e robustez. Neste estudo, foram observados desvios padrao relativos médios inferiores a 2%, tanto nos estudos de repetibilidade como de reprodutibilidade, o que se mostra compatível com as exigências da legislaçao (máximo de 5%). A exatidao foi avaliada utilizando-se as mesmas misturas sintéticas utilizadas na avaliaçao da precisao. Os resultados indicam taxas de recuperaçao entre 99,0 e 102,7%, portanto, dentro dos limites estipulados pela legislaçao específica (5%). Adicionalmente, a exatidao foi avaliada pela análise de medicamentos, utilizando-se como referência os valores de concentraçao obtidos por aplicaçao da técnica cromatográfica padrao. Neste estudo (Tabelas 2 e 3), erros relativos médios inferiores a 5% foram observados. Nos estudos de robustez, a capacidade de previsao dos modelos multivariados foi avaliada frente a controladas variaçoes de temperatura, tempo de leitura e pH. Com relaçao aos dois primeiros fatores, é necessário antecipar que ambos os fármacos sao sensíveis à açao da luz e do calor, podendo, inclusive, decompor em meio alcalino, em funçao de reaçoes que parecem ser catalisadas por pirazinamida e etambutol, princípios comumente associados em medicamentos comercializados em outros países. Em funçao destes antecedentes, um estudo preliminar foi sistematicamente realizado, de maneira que pudessem ser definidas as condiçoes e o tempo máximo de estocagem das soluçoes utilizadas como padrao. Nesta avaliaçao preliminar se observou uma significativa decomposiçao da rifampicina nos primeiros dias de estocagem, degradaçao oxidativa que, entretanto, pode ser evitada pela adiçao de ácido ascórbico. Adicionalmente, observou-se que a açao estabilizante do ácido ascórbico pode ser substituída pela estocagem no escuro e em baixas temperaturas, condiçao que foi adotada no decorrer de todo este trabalho. Nestas condiçoes, os padroes aquosos de rifampicina se mantêm inalterados por pelo menos 7 dias, o que viabiliza a rotina de trabalho. As soluçoes de isoniazida apresentaram boa estabilidade em todas as condiçoes de estocagem ensaiadas. Mesmo assim, a sua estocagem foi feita a baixas temperaturas e na ausência de luz, respeitando-se um prazo de validade de, no máximo, uma semana. Nos estudos envolvendo a avaliaçao do efeito do tempo de leitura (0 a 6 horas) foi observada uma leve modificaçao do perfil espectroscópico da mistura de fármacos, principalmente em razao de modificaçoes associadas à rifampicina. Uma vez que referida variaçao provoca desvios relativos da ordem de 2% entre as determinaçoes realizadas no período de 6 horas, recomenda-se que as análises espectroscópicas sejam realizadas, no máximo, após 2 h do momento de preparo. Nos estudos envolvendo a avaliaçao do efeito da temperatura (5 a 70 ºC), significativas modificaçoes espectrais foram observadas somente para rifampicina em temperaturas superiores a 50 ºC. Por este motivo, recomenda-se que processos de dissoluçao sejam assistidos por ultrassom durante 30 minutos e por temperaturas nao superiores a 40 ºC. Nos estudos orientados a avaliar o efeito do pH (4,0 a 9,0) foram observadas significativas mudanças espectrais somente em pH 4,0, tanto para isoniazida (pKa 1,8; 3,5 e 9,8) como para rifampicina (pKa 1,7 e 7,9), provavelmente em razao dos respectivos equilíbrios de protonaçao/desprotonaçao. Desta forma, recomenda-se a realizaçao das análises entre pH 5 e 8, intervalo em que nao foram observadas significativas mudanças espectrais nos fármacos em estudo. Finalmente, é importante salientar que ambas as metodologias multivariadas foram comparadas entre si e em relaçao à metodologia cromatográfica padrao, utilizando-se testes para comparaçao de variâncias (teste f) e de médias (teste t). Nesta avaliaçao, diferenças estatisticamente significantes nao foram observadas, considerando-se um nível de probabilidade de 95%.
CONCLUSOES Sistemas convencionais de calibraçao fundamentados em curvas analíticas e sistemas derivativos se mostram ineficientes para a análise espectroscópica de associaçoes contendo isonizaida e rifampicina, em funçao da interferência espectral existente entre estes fármacos. Por sua vez, o sistema fundamentado no princípio de aditividade de absorbâncias permite contornar referidos problemas de interferência, permitindo resultados relativamente coerentes com as informaçoes de bula (erros médios da ordem de 9 a 10%). Os modelos multivariados desenvolvidos apresentaram uma excelente capacidade de previsao, permitindo a obtençao de resultados equivalentes aos obtidos pela técnica cromatográfica padrao. Além de atender aos requisitos da legislaçao específica, os métodos multivariados se apresentam rápidos, econômicos e de fácil implementaçao, envolvendo uma geraçao de resíduos minimizada.
REFERENCIAS 1. Dinç, E.; J. Pharm. Biomed. Anal. 1999, 21, 723. DOI: http://dx.doi.org/10.1016/S0731-7085(99)00186-7 PMID: 10701937 2. Rajput, P.; Singh, D.; Pathak, K.; Int. J. Pharm. 2014, 461, 310. DOI: http://dx.doi.org/10.1016/j.ijpharm.2013.11.053 PMID: 24309435 3. Shibata, S.; Furukawa, M.; Goto, K.; Anal. Chim. Acta 1973, 65, 49. DOI: http://dx.doi.org/10.1016/S0003-2670(01)80162-8 4. Hoang, V. D.; Hue, N. T.; Tho, N. H.; Nguyen, H. M. T.; Spectrochim. Acta, Part A 2015, 139, 20. DOI: http://dx.doi.org/10.1016/j.saa.2014.11.101 5. Elkhoudary, M. M.; Salam, R. A. A.; Hadad, G. M.; Spectrochim. Acta, Part A 2014, 130, 222. DOI: http://dx.doi.org/10.1016/j.saa.2014.04.002 6. Darwish, H. W.; Hassan, S. A.; Salem, M. Y.; El-Zeany, B. A.; Spectro chim. Acta, Part A 2013, 113, 215. DOI: http://dx.doi.org/10.1016/j.saa.2013.04.068 7. Ali, N. W.; Abbas, S. S.; Zaazaa, H. E.; Abdelrahman, M. M.; Abdelkawy, M.; J. Pharm. Anal. 2012, 2, 105. DOI: http://dx.doi.org/10.1016/j.jpha.2011.11.004 8. Dar, A. A.; Sangwan, P. L.; Khan, I.; Gupta, N.; Qaudri, A.; Tasduq, S. A.; Kitchlu, S.; Kumar, A.; Koul, S.; J. Pharm. Biomed. Anal. 2014, 100, 300. DOI: http://dx.doi.org/10.1016/j.jpba.2014.07.034 PMID: 25194343 9. Paula, C. E. R.; Almeida, V. G. K; Cassella, R. J.; J. Braz. Chem. Soc. 2010, 21, 1664. DOI: http://dx.doi.org/10.1590/S0103-50532010000900010 10. Idris, A. M.; J. Pharmacol. Toxicol. Methods 2007, 56, 330. DOI: http://dx.doi.org/10.1016/j.vascn.2007.08.005 PMID: 17897843 11. Benetton, S. A.; Kedor-Hackmann, E. R. M.; Santoro, M. I. R. M.; Borges, V. M.; Talanta 1998, 47, 639. DOI: http://dx.doi.org/10.1016/S0039-9140(98)00111-8 PMID: 18967366 12. Youssef, R. M.; Maher, H. M. Spectrochim. Acta, Part A 2008, 70, 1152. DOI: http://dx.doi.org/10.1016/j.saa.2007.10.049 13. Madan, J.; Dwivedi, A. K.; Singh, S.; Anal. Chim. Acta 2005, 538, 345. DOI: http://dx.doi.org/10.1016/j.aca.2005.02.015 14. Espinosa-Mansilla, A.; Acedo Valenzuela, M. I.; Muñoz de la Peña, A.; Salinas, F.; Cañada Cañada, F.; Anal. Chim. Acta, 2001, 427, 129. DOI: http://dx.doi.org/10.1016/S0003-2670(00)01195-8 15. http://www.anvisa.gov.br/hotsite/cd_farmacopeia/pdf/volume2.pdf, acessada em março de 2015. 16. Stets, S.; Tavares, T. M.; Peralta-Zamora, P.; Pessoa, C. A.; Nagata, N.; J. Braz. Chem. Soc. 2013, 24, 1198. 17. http://portal.anvisa.gov.br/wps/wcm/connect/4983b0004745975da005f43fbc4c6735/RE_899_2003_Determina+a+publica%C3%A7%C3%A3o+do+Guia+para+valida%C3%A7%C3%A3o+de+m%C3%A9todos+anal%C3%ADticos+e+bioanal%C3%ADticos.pdf?MOD=AJPERES, acessada em julho de 2015. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access