
 |
 |
 |
 |
 |
Artigo
|
|
| A formação de ligações de hidrogênio π∙∙∙H, F∙∙∙H e C∙∙∙H nos complexos C2H2∙∙∙(HF), C2H2∙∙∙2(HF) e C2H2∙∙∙3(HF) The formation of the π∙∙∙H, F∙∙∙H and C∙∙∙H hydrogen bonds on the C2H∙∙∙(HF), C2H2∙∙∙2(HF) and C2H2∙∙∙3(HF) complexes |
|
Boaz G. Oliveira*
Instituto de Ciências Ambientais e Desenvolvimento Sustentável, Universidade Federal da Bahia, 47801-100 Barreiras - BA, Brasil Recebido em 06/10/2015 *e-mail: boazgaldino@gmail.com In this work, a theoretical study on the basis of structural, vibrational, electronic and topological parameters of the C2H2∙∙∙(HF), C2H2∙∙∙2(HF) and C2H2∙∙∙3(HF) complexes concerning the formation of π∙∙∙H, F∙∙∙H and C∙∙∙H hydrogen bonds is presented. The main difference among these complexes is not properly the interaction strength, but the hydrogen bond type whose benchmark is ruled justly by the structure. Meanwhile, the occurrence of π∙∙∙H hydrogen bonds was unveiled in both C2H2∙∙∙(HF) dimer and C2H2∙∙∙3(HF) tetramer, although in latter, this interaction is stronger than C∙∙∙H of the C2H2∙∙∙2(HF) trimer. However, the F∙∙∙H hydrogen bonds within the subunits of hydrofluoric acid are the strongest ones, reaching a partial covalent limit, and thereby contribute decisively to the stabilization of the tetramer structure. In line with this, the largest red-shifts were observed on the hydrofluoric acid trimer of the C2H2∙∙∙3(HF) complex. INTRODUÇÃO A ligação de hidrogênio é concebida como um caso particular de interações entre dipolos, cujo portfólio apresenta a forma Y∙∙∙H em que Y representa geralmente os átomos de flúor, oxigênio, nitrogênio, enxofre ou cloro.1 Proeminentemente, se considerarmos a concepção de Pimentel e McClellan,2 Y∙∙∙H não pode ser considerada uma ligação de hidrogênio, visto que o hidrogênio interagindo com Y deva também estar ligado a outro elemento, neste caso simbolizado por X, e que, desta forma, origina o modelo Y∙∙∙H-X. Para Pauling,3 entretanto, a definição de ligação de hidrogênio recai na concepção de que o hidrogênio é atraído por estes dois outros átomos, Y e X, tornando-se um elemento mediador intermolecular sob condições de eletronegatividade propícias para ocorrência de tal interação. Mais recentemente, em 1993, Steiner e Saenger4 estabeleceram o conceito de carga pontual, em que para ocorrer à ligação de hidrogênio Y∙∙∙H-X, o hidrogênio deve estar defasado de elétrons enquanto Y acumule uma carga negativa parcial, além de X ser mais eletronicamente negativo do que o hidrogênio. Estas reflexões têm servido de base para o estudo de sistemas intra e intermoleculares formados e estabilizados por ligações de hidrogênio n∙∙∙H-C,5-9 em que n representa os pares de elétrons desemparelhados dos elementos F, O, N, S e Cl.10-13 Doravante, além da ligação de hidrogênio n∙∙∙H-C e considerando os argumentos de Steiner e Saenger,4 cargas negativas não exclusivamente provêm de átomos com pares de elétrons desemparelhados, e ligações insaturadas π e saturadas pseudo-π também acumulam concentrações de carga suficientes para interagirem com grupos do tipo H-C.14 De fato, os complexos intermoleculares mais característicos com ocorrência da ligação de hidrogênio π∙∙∙H-C são C2H2∙∙∙HCN e C2H2∙∙∙C2H2,15 nos quais há a concórdia de que sistemas oligomoleculares constituídos por mais de um doador de próton potencializam a força de interação e favorecem a estabilização em uma estrutura diferenciada. Neste discernimento, há algum tempo foi estudado o dímero C2H2∙∙∙HF juntamente com sua estrutura trimolecular, o complexo C2H2∙∙∙2(HF).16 Neste último, ao invés da ligação de hidrogênio π∙∙∙H-F, foi observado a C∙∙∙H-F, em que o complexo não apresentou forma T-shaped, mas uma forma assimétrica em que o átomo de carbono tornou-se o receptor de próton. Visto posto, este trabalho atual foi elaborado com o objetivo de investigar a formação de ligações de hidrogênio π∙∙∙H-F ou C∙∙∙H-F não simplesmente revisitando as estruturas dos complexos C2H2∙∙∙(HF) e C2H2∙∙∙2(HF), mas propondo uma nova estrutura tetramolecular com o trímero do ácido fluorídrico: C2H2∙∙∙3(HF). Conforme outros trabalhos,17-24 a investigação teórica de propriedades estruturais, eletrônicas, topológicas e vibracionais, neste caso dos complexos C2H2∙∙∙(HF), C2H2∙∙∙2(HF) e C2H2∙∙∙3(HF), esmerilha o estudo com acuracidade no torno das ligações de hidrogênio π∙∙∙H-F ou C∙∙∙H-F ou até mesmo F∙∙∙H-F no dímero 2HF e trímero 3HF. Porquanto, fazem-se necessários métodos de estrutura eletrônica que sejam capazes de capturar toda a fenomenologia destas interações. Além dos métodos ab initio serem sempre eficientes,25-27 também é bem estabelecido que a Teoria do Funcional de Densidade ou DFT (do inglês, Density Functional Theory)28-30 tem sido utilizada no estudo de propriedades de sistemas formados por ligação de hidrogênio com resultados bastante satisfatórios,31-35 e que, em alguns casos, híbridos fornecem resultados mais confiáveis do que os obtidos via métodos perturbativos. Na prática, o B3LYP é o funcional mais utilizado36-38 inegavelmente pelo histórico profícuo de resultados obtidos em diversas outras linhas de pesquisa, mas também pela aplicação em trabalhos que envolvem sistemas intermoleculares.39,40 Na zona de interstício intermolecular, a condição eletrônica que pondera a interação entre Y e H-X são os respectivos orbitais de fronteira HOMO e LUMO, cuja interação é suportada pelo fenômeno de transferência de carga.41 Como tal, esta quantificação de transferência de carga acompanhada pela determinação da carga líquida atômica e eventuais variações tornam-se parâmetros muitos importantes para o estudo das ligações de hidrogênio π∙∙∙H-F, C∙∙∙H-F e F∙∙∙H-F. Dentre os algoritmos de partição de carga mais comumente utilizados,42 as Cargas derivadas do Potencial Eletrostático baseado em uma Grade exterior a superfície de van der Waals ou ChelpG (do inglês, Charges derived from Electrostatic Potential Grid-based),43 bem como as cargas obtidas da Análise Natural de Ligação ou NBO (do inglês, Natural Bond Orbitals),44 são muito eficientes neste sentido.45,46 Pela inerência e notoriedade de suas propriedades,47 a ligação de hidrogênio pode ser considerada uma ligação com caráter covalente,14,19,48,49 e cuja caracterização teórica repousa na Teoria Quântica de Átomos em Moléculas ou QTAIM (do inglês, Quantum Theory of Atoms in Molecules),50 na partição dos termos de energia para quantificação da energia eletrônica SAPT (do inglês, Symmetry-Adapted Perturbation Theory)51 e, consequentemente, da força de interação, além da espectrosocpia de raios-X.52 Em seu formalismo mecânico-quântico, a QTAIM é munida do princípio de que os átomos são entidades de camadas abertas, e estes, por sua vez, permutam carga e momento com seus vizinhos.53 Para ligações químicas σ e π, este princípio as caracteriza como interações de camada aberta, ao passo que as ligações de hidrogênio são designadas de camadas fechadas.53 Ambos, fluxo de carga e momento, são concebidos como Caminhos de Ligação ou BP,50 nos quais são identificados Pontos Críticos de Ligação ou BCP, em que a densidade eletrônica internuclear e seu correspondente Laplaciano são determinados.53 Estes descritores QTAIM têm sido vastamente utilizados não apenas para caracterização de ligações de hidrogênio,54-56 mas também para predição de sua força de interação.14,57,58 A respeito dos complexos C2H2∙∙∙(HF), C2H2∙∙∙2(HF) e C2H2∙∙∙3(HF) objetos de estudo neste trabalho de pesquisa, pela QTAIM espera-se que as três ligações de hidrogênio π∙∙∙H-F, C∙∙∙H-F e F∙∙∙H-F sejam identificadas, e com isso possa ser possível demonstrar que os perfis destas interações são funções da estrutura intermolecular, seja dimérica, trimérica ou tetramérica, e não somente sejam oriundos dos termos eletrônicos que compõem a energia eletrônica.
PROCEDIMENTO COMPUTACIONAL As geometrias otimizadas dos complexos C2H2∙∙∙(HF), C2H2∙∙∙2(HF) e C2H2∙∙∙3(HF) foram determinadas ao nível de teoria B3LYP/6-311++G(d,p) com todos os cálculos processados pelo software GAUSSIAN 03W.59 Através deste mesmo pacote computacional foram processados os cálculos ChelpG, NBO e também do Erro de Superposição do Conjunto de Base ou BSSE (do inglês, Basis Set Superposition Error)60 de Boys e Bernardi.61 Os cálculos QTAIM foram processados pelos softwares AIM200062 e AIMAll 11.05.16.63
RESULTADOS E DISCUSSÃO Parâmetros estruturais As geometrias otimizadas dos complexos C2H2∙∙∙(HF) (I), C2H2∙∙∙2(HF) (II) e C2H2∙∙∙3(HF) (III) determinadas ao nível de teoria B3LYP/6-311++G(d,p) são apresentadas na Figura 1, enquanto na Tabela 1 são listados os valores das distâncias de ligação. Mesmo já tendo sido estudadas,16 as estruturas I e II diferenciam-se pelas ligações de hidrogênio formadas entre o acetileno e o ácido fluorídrico, seja π∙∙∙He ou Cc∙∙∙He. A primeira trata-se de uma ligação clássica T-shaped,14,64 cuja distância de 2,1921 Å maior do que 2,1050 Å aponta para uma menor força de interação na estrutura I. No complexo II, além de não usual, em que o carbono atua como receptor de próton, a ligação de hidrogênio Cc∙∙∙He é a preferencial, uma vez que Cb∙∙∙He apresenta uma distância maior de 2,1760 Å. Ao revés, o complexo III apresenta uma ligação de hidrogênio π∙∙∙He T-shaped clássica, sendo que, neste caso, sua distância de 1,9893 Å a condecora como sendo a interação mais curta e até próxima do limiar da covalência, cujo valor de referência é sempre inferior a 2,0000 Å.65 Tratam-se de ligações de hidrogênio direcionais dependentes da estrutura, mas especificamente no caso do complexo III, se tem o primeiro caso de uma ligação de hidrogênio π∙∙∙H com um perfil de força de interação superior a sistemas intermoleculares similares do tipo π.14,16,66-68 É digno de nota considerarmos a possibilidade de uma das ligações Cb∙∙∙He e Cc∙∙∙He serem formadas, embora seus respectivos valores de 2,078 e 2,050 Å sejam muito similares, mas são muito maiores do que a ligação π∙∙∙He supracitada.
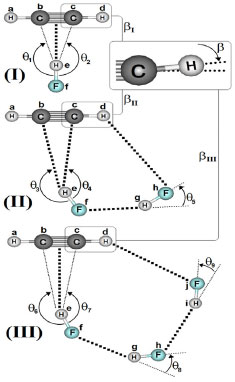 Figura 1. Geometrias otimizadas dos complexos C2H2∙∙∙(HF) (I), C2H2∙∙∙2(HF) (II) e C2H2∙∙∙3(HF) (III) determinadas ao nível de teoria B3LYP/6-311++G(d,p)

Concernente as ligações de hidrogênio F∙∙∙H nos dímero (II) e trímero (III) do ácido fluorídrico,69 a distância de 1,8313 Å em 2(HF) isolado (IV)69 é drasticamente reduzida à 1,777 Å. Com relação às duas ligações de hidrogênio Ff∙∙∙Hg e Fh∙∙∙Hi em III, na verdade estas interações praticamente se equivalem na estrutura cíclica isolada de 3(HF),69 em que cujos valores variando entre 1,7920 e 1,7990 Å estão em satisfatória concordância com o resultado de 1,7490 Å obtido por Asselin et al.,70 que utilizaram cálculos em nível coupled cluster com base de Dunning CCSD(T)-F12/AVTZ. Sabe-se que as variações nas distâncias de ligação e ângulos são reflexos da perturbação estrutural provocada pela força de interação.13,34,56,59 No acetileno, o pictograma em destaque na Figura 1 expõe o ângulo (β) formado pelo desalinhamento do átomo de hidrogênio em relação ao eixo internuclear C∞. A existência deste ângulo deve-se ao fato de que os átomos de carbono apresentam uma tendência de alteração em sua estrutura eletrônica, em particular na hibridização de sp para sp2. Conforme os valores de 0,61º para βI no complexo I, além de 1,93 (HaCbCc) e 1,46 (CbCcHd) para βII, bem como 1,75 (HaCbCc) e 0,86 (CbCcHd) para βIII, sendo estes dois últimos para os complexos II e III respectivamente, estes valores não apresentam relação direta com a distância da ligação de hidrogênio. O complexo I apresenta o mesmo valor de 164,36º para os ângulos θ1 e θ2 entre os átomos FfHeCc e FfHeCb devido a simetria T-shaped. Na estrutura II, os ângulos θ3 e θ4 se diferenciam entre os átomos FfHeCc e FfHeCb, cujos valores são 179,6 e 147,8º, respectivamente. Poder-se-ia admitir que a menor inclinação de 147,8º justificasse a formação de ligação de hidrogênio Cc∙∙∙He, mas ao analisarmos a estrutura III percebe-se que, embora a mesma tendência em relação ao ângulo θ7 e θ8 formados entre os átomos FfHeCc e FfHeCb se mantenha, cujos respectivos valores são 157 e 169º, não se pode utilizá-los para validar a existência das interações Cc∙∙∙He e Cb∙∙∙He simplesmente pelo fato da ligação de hidrogênio π∙∙∙He ser mais curta. Como vastamente estabelecido,71-73 a variação na distância de ligação no doador de próton é, juntamente com seu deslocamento vibracional, uma das principais comprovações que um sistema intermolecular é formado.8-9,12,17,41 Foram observadas variações de distância ns ligações σ (H-F e C-H) e π (C≡C), embora as mais proeminentes sejam no ácido fluorídrico. No acetileno, as variações nas distâncias de ligação estão na faixa de 0,0011 a 0,0083 Å, mas no ácido fluorídrico as variações são muito superiores e atingem valores de 0,0284 Å, neste caso, a ligação He-Ff no complexo III. Pelo fato da variação na distância de ligação do doador de próton refletir o perfil da força de interação,74 o valor de 0,0284 Å para a ligação He-Ff está condicionado à ligação de hidrogênio mais curta, a π∙∙∙H na estrutura tetramérica. Para os complexos II e I, as variações de 0,0198 e 0,009 Å correlacionam com ligações de hidrogênio com distâncias medianas e mais longas, ou seja, Cc∙∙∙He e π∙∙∙H, respectivamente. Comparando 2(HF) e 3(HF), observamos que a ligação Hg-Fh apresentou uma variação maior (0,0220 Å) no complexo III em relação a II, no qual o valor calculado foi de 0,0122 Å. Modos vibracionais Os modos vibracionais na região do espectro de infravermelho compõe uma das bases fidedignas para caracterização de um sistema intermolecular,72,74 principalmente aqueles formados por ligação de hidrogênio ou HBond (do inglês, Hydrogen Bond).75 Acerca dos novos modos vibracionais ou também reconhecidos como frequências de estiramento de ligação de hidrogênio, estas se caracterizam por valores muito baixos no espectro infravermelho acompanhado por baixas intensidades de absorção, legalmente, a depender da força de interação. Para os complexos I, II e III cujos modos vibracionais são organizados na Tabela 2, as frequências de estiramento demonstram uma total concordância com os valores de distância, conforme pode ser observado no gráfico ilustrado na Figura 2, o qual foi gerado a partir da Equação 1: 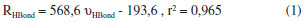

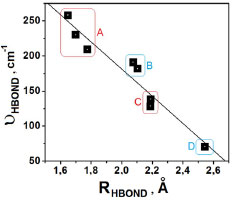 Figura 2. Relação entre os valores das frequências e distâncias das ligações de hidrogênio nos complexos C2H2∙∙∙(HF), C2H2∙∙∙2(HF) e C2H2∙∙∙3(HF)
Tem-se, novamente, que o complexo III formado por uma ligação π∙∙∙H apresenta a maior frequência de estiramento e, também, a maior intensidade de abosrção intermolecular. Há tempos que a literatura especializada dispõe de trabalhos que correlacionam a força de interação com os deslocamentos nas frequências de estiramento, especialmente dos doadores de prótons,76 e em alguns casos nos quais o carbono atua como fonte de elétrons para formar ligações de hidrogênio.77 Através dos valores listados na Tabela 3, na Figura 3 é traçado um gráfico entre os valores dos deslocamentos (Δυ) nas frequências de estiramento versus as variações nas distâncias de ligação (Δr), cuja correlação é considerada bastante satisfatória devido ao coeficiente linear r2 de 0,989: 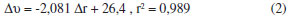

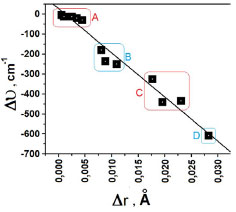 Figura 3. Relação entre os valores das variações nas frequências e distâncias de ligação nos complexos C2H2∙∙∙(HF), C2H2∙∙∙2(HF) e C2H2∙∙∙3(HF)
Como todas as distâncias de ligação sofrem aumento, sinergicamente as correspondentes frequências de estiramento são deslocadas para valores com menos energia no espectro de infravermelho, evento este denominado de batocrômico ou red-shift.78,79 Na Figura 3 são diagramatizados quatro grupos, a saber: A = efeitos red-shifts nas ligações π e C-H do acetileno; B = ligações H-F (I), He-Ff (II), Hi-Fj (III) e C-H (III); C = ligações Hg-Fh das estruturas II e III; e D que isoladamente representa o red-shift na ligação He-Ff de III. Notadamente, o maior valor red-shift de -606,5 cm-1 no complexo III manifesta-se exatamente na ligação He-Ff cuja região doadora de carga é a ligação π do acetileno, que, por sua vez, é a distância intermolecular mais curta. Uma característica também marcante dos efeitos red-shifts é a alteração na intensidade de absorção, a qual, em geral, aumenta drasticamente quando ocorre a formação do complexo de hidrogênio.19,34,41 Conforme os valores de IHF,c/IHF,m que relaciona as intensidades do complexo (IHF,c) em relação ao monômero (IHF,m), trata-se de um efeito hipercrômico em que há aumento na intensidade de absorção. Para o red-shift de -606,5 cm-1, a hipercrômia IHF,c/IHF,m fornecer um valor de 7,2 que é muito próximo de 6,8 para o oscilador HgFh cujo red-shift é -441,4 cm-1. Embora nem sempre possa ser estabelecida uma relação direta entre a magnitude dos efeitos red-shifts e a mudança hipercrômica, é por meio desta que a formação do complexo de hidrogênio torna-se melhor observável no espectro de infravermelho. Transferência de carga e energia de interação Os valores das transferências de carga computadas pelos métodos ChelpG e NBO são listados na Tabela 4.

Em I, ambos ChelpG e NBO fornecem um portfólio condizente com a teoria da ligação de hidrogênio,14,41 em que os átomos de carbono em torno da ligação π apresentam diminuição de carga ou perda eletrônica. Entretanto, a molécula de HeFf tem valores negativos de -0,068 e -0,011 u.e. para o hidrogênio e flúor, respectivamente, indicando aumento de carga a qual proveio da ligação π. Por outro lado, os valores NBO falham nesta concepção, visto que o hidrogênio apresentou balanço de carga positivo, cujo valor é 0,009 u.e. Nas estruturas subsequentes, as cargas ChelpG mostram-se muito superiores às NBO. Em II, os átomos de carbono do acetileno apresentam diminuição de carga (0,031 e 0,014 u.e.), intercedido pelos aumentos de -0,101 e -0,016 u.e. nos hidrogênios He e Hg da subparte do dímero 2(HF). Embora o valor de 0,039 u.e. aponte para uma transferência de carga do átomo de flúor Ff para a molécula HgFh, o valor de -0,027 u.e. de Fh não condiz com a expectativa de formação da ligação de hidrogênio Fh∙∙∙Hd. Com relação aos resultados NBO, estes traduzem um comportamento totalmente anômalo e contraditório, como, por exemplo, carbonos com aumento de carga (-0,024 e -0,005 u.e.), hidrogênios (0,025 e 0,016 u.e.) e flúor (-0,031 e -0,034 u.e.) com diminuição e aumento de carga, respectivamente. Com relação ao tetrâmero, a descrição do perfil de transferência de carga via algoritmo ChelpG é precisa nos átomos de hidrogênio He e Hg, cujas variações são -0,104 e -0,082 u.e., bem como nos átomos de flúor Ff e Fh, em que, nestes se contabilizou diminuição de carga devido aos resultados de 0,082 e 0,040 u.e., respectivamente. Ao restante da estrutura, entretanto, as cargas ChelpG mostram-se ineficientes, bem como a NBO. É bem estabelecido na literatura especializada que o fortalecimento das interações intermoleculares esteja sensivelmente aliada com o número e a distribuição de energia.80 Rotineiramente, a energia de interação é calculada com base na aproximação da supermolécula81 no caso do sistema I, mas para os complexos II e III, a quantificação das contribuições não cooperativas das subpartes 2(HF) e 3(HF) privam a utilização deste argumento. Diante de tal entrave, a força de interação de cada ligação de hidrogênio fica restrita em partes a energia dos orbitais NBO.82  em que -2 indica a ocupação (2 elétrons) no orbital doador, |F| é a matriz de Fock, εY(A) - εYσ*(B) corresponde a diferença de energia entre os orbitais,83 A e B representam os pares de elétrons (LP, do inglês Lone Pair) do flúor ou a ligação π do acetileno e o orbital antiligante das ligações σ (BD*(1)) F-H e C-H do doador de próton (HF ou HCCH), respectivamente. Pelo fato de independer das energias das espécies isoladas, a energia de estabilização determinada pela Equação (3) é bastante utilizada em estudos de ligações de hidrogênio intramoleculares.84,85 Para os complexos I, II e III, cujos valores são listados na Tabela 4, a energia de 55,56 kJ mol-1 da ligação de hidrogênio π∙∙∙H confirma a maior estabilização desta frente ao complexo I assim como em relação a CC∙∙∙He, a qual era até então considerada como sendo a interação preferencial no átomo de carbono ao invés da ligação π. Outrossim, comprova-se claramente que há um fortalecimento nas ligações de hidrogênio entre as unidades de ácido fluorídrico, em particular os resultados de 41,38 e 70,20 kJ mol-1 para Ff∙∙∙Hg-Fh nos complexos II e III, respectivamente. Topologia QTAIM No uso de suas atribuições delineadas na mecânica quântica,86 a QTAIM tem sido considerada uma das visões mais modernas acerca da ligação química,87 bem como na extensão a outros sistemas e questões relacionadas a estrutura eletrônica em geral.88 Para as ligações σ e π, os valores apresentados na Tabela 5 reforçam que, embora estas apresentem perfil de interação de camada aberta devido aos valores negativos do Laplaciano, as maiores variações de densidade eletrônica são observadas nas ligações H-F. Estas variações são, na verdade, reduções de concentrações de carga, aspecto este totalmente condizente com os aumentos nas distâncias de ligação e efeitos red-shifts supracitados. Contudo, a força covalente destas ligações se mantém, como pode ser constatado na relação -G/U, a qual relaciona as contribuições das energias cinéticas e potenciais de densidade eletrônica. Os valores de -G/U refletem um caráter totalmente covalente pois estão compreendidos abaixo do limite de 0,5.14,47 Conforme destacado anteriormente, estes e todos os demais parâmetros topológicos são obtidos mediante localização de BCPs internucleares, com BP de fluxo de carga e, para os complexos I, II e III, a Figura 4 fornece a visualização da topologia QTAIM com caracterização de todas as ligações químicas, inclusive as ligações de hidrogênio. Para os complexos I e III, os BPs caracterizam as ligações π∙∙∙He, ao passo que para II a ligação de hidrogênio é direcionada ao carbono como receptor de próton. Reiterando o perfil de força de interação mediante os valores apresentados na Tabela 6, mesmo que o complexo III seja formado por uma ligação de hidrogênio π∙∙∙H, a densidade eletrônica de 0,027 e.ao-3 é superior aos valores de 0,023 (II) e 0,018 e.ao-3 (I). Pelos valores positivos do Laplaciano, as ligações de hidrogênio π∙∙∙H e Cc∙∙∙He são interações de camada fechada, embora o valor de 1,066 determinado pela razão -G/U tendencia o complexo III para uma ligação de hidrogênio π∙∙∙H com força no limiar do caráter covalente. Tal condição foi atingida pelas ligações Ff∙∙∙Hg e Fh∙∙∙Hi na estrutura III, em cujos valores de -G/U são 0,975 e 0,971, respectivamente.

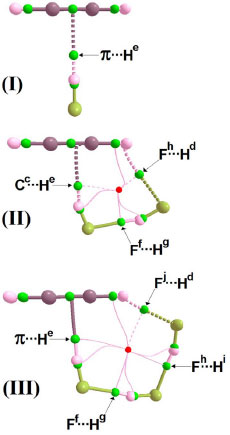 Figura 4. BCP e BP das ligações químicas e visualização das ligações de hidrogênio nos complexos C2H2∙∙∙(HF) (I), C2H2∙∙∙2(HF) (II) e C2H2∙∙∙3(HF) (III)

Através dos valores da Tabela 7, em outras ligações de hidrogênio, como por exemplo a Ff∙∙∙Hg (II), esta apresenta um valor de 1,037 para a razão -G/U, sendo, portanto, também uma tendência para a covalência. Todavia, não havendo premissa de cooperatividade pelas energias de interação determinadas pela NBO no que concerne à relação -G/U, o complexo II apresenta-se como uma estrutura intramolecular covalente, apesar do valor positivo de 0,040 e.ao-5 do Laplaciano apontar Ff∙∙∙Hg como interação de camada fechada. Igualmente, as ligações de hidrogênio Fh∙∙∙Hd e Fj∙∙∙Hd nos complexos II e III, respectivamente, são formadas com quantidades mínimas de densidade eletrônica, particularmente o valor de 0,007 e.ao-3 em II, mas que as razões -G/U de 1,200 e 1,250 testificam que tratam-se de interações de camanda fechada essencialmente não covalentes. Entrementes, a aclamação pelos parâmetros QTAIM para caracterização de ligações de hidrogênio direcionais,62,64,67 no caso dos complexos I, II e III estudados neste trabalho, observa-se uma relação discutível com as contribuições dos termos de energia87 que munem a força de interação. Recentemente, Hill e Legon89 investigaram uma série de sistemas intermoleculares formados por ligações de hidrogênio com distintos direcionamentos e inclinações em suas padronizações, e foi mostrado que as contribuicões dos termos de energia eletrônica são flexíveis mediante certa estrutura intermolecular. Em outras palavras, e enfatizando o atual trabalho, há uma dependência unívoca da estrutura para com a formação do tipo e também da força de interação intermolecular.

CONCLUSÕES Este estudo teórico das ligações de hidrogênio π∙∙∙H, C∙∙∙H e F∙∙∙H nos complexos C2H2∙∙∙(HF), C2H2∙∙∙2(HF) e C2H2∙∙∙3(HF) revelou uma dependência da estrutura intermolecular não apenas para o perfil da interação, mas também para a força de interação. Demonstrou-se uma fortificação das ligações de hidrogênio F∙∙∙H com caráter parcialmente covalente na estrutura tetramérica, na qual também se observou uma tendência da ligação de π∙∙∙H apresentar esta mesma propriedade. Nesta e no dímero, embora a nuvem eletrônica π seja a fonte eletrônica receptora de prótons, as ligações C≡C e C-H do acetileno não sofreram mudanças significativas. Ao oposto, as ligações H-F foram seriamente afetadas tanto estruturalmente como no ponto de vista vibracional, em que se teve a identificação de efeitos red-shifts superiores a -600 cm-1. Em termos de transferência de carga, o algoritmo ChelpG mostrou-se mais eficiente frente aos resultados NBO, pelo qual também foram computados as energias de interação. Consenso de que a cooperatividade nas estruturas triméricas e tetraméricas sejam imensuráveis, novamente, as ligações de hidrogênio F∙∙∙H apresentaram as maiores energias de interação NBO. A QTAIM caracterizou e diferenciou as ligações de hidrogênio nos complexos C2H2∙∙∙(HF) e C2H2∙∙∙3(HF) como sendo π∙∙∙H, enquanto para C2H2∙∙∙2(HF) observa-se a C∙∙∙H. Este panorama atesta que o tipo de ligação de hidrogênio formada não é função da força de interação, mas seguramente depende da estrutura intermolecular.
AGRADECIMENTOS CAPES, CNPq, UFBA e FAPESB.
REFERÊNCIAS 1. Desiraju, G. R.; Angew. Chem. Int. Ed. 2011, 50, 52. DOI: http://dx.doi.org/10.1002/anie.201002960 2. Pimentel, G. C.; McClellan, A. L.; The Hydrogen Bond, Freeman, W. H., San Francisco, 1960. 3. Pauling, L.; The Nature of the Chemical Bond, Cornell University Press: Ithaca, 1939. 4. Steiner, T.; Saenger, W.; J. Am. Chem. Soc. 1993, 115, 4540. DOI: http://dx.doi.org/10.1021/ja00064a016 5. Oliveira, B. G.; Lima, M. C. A.; Pitta, I. R.; Galdino, S. L.; Hernandes, M. Z.; J. Mol. Model. 2010, 16, 119. DOI: http://dx.doi.org/10.1007/s00894-009-0525-y PMID: 19517145 6. Oliveira, B. G.; Araújo, R. C. M. U.; Carvalho, A. B.; Ramos, M. N.; Quim. Nova 2007, 30, 1167. DOI: http://dx.doi.org/10.1590/S0100-40422007000500022 7. Oliveira, B. G.; Araújo, R. C. M. U.; Carvalho, A. B.; Ramos, M. N.; Hernandes, M. Z.; Cavalcante, K. R.; J. Mol. Struct. (THEOCHEM) 2007, 802, 91. DOI: http://dx.doi.org/10.1016/j.theochem.2006.09.002 8. Oliveira, B. G.; Araujo, R. C. M. U.; Carvalho, A. B.; Ramos, M. N.; Spectrochim. Acta A 2007, 68, 626. DOI: http://dx.doi.org/10.1016/j.saa.2006.12.038 9. Oliveira, B. G.; Araujo, R. C. M. U.; J. Mol. Model. 2012, 18, 2845. DOI: http://dx.doi.org/10.1007/s00894-011-1300-4 PMID: 22127607 10. Oliveira, B. G.; Araújo, R. C. M. U.; Carvalho, A. B.; Ramos, M. N.; J. Mol. Model. 2011, 17, 2847. DOI: http://dx.doi.org/10.1007/s00894-011-0969-8 PMID: 21301908 11. Oliveira, B. G.; Araújo, R. C. M. U.; Ramos, M. N.; Spectrochim. Acta A 2010, 75, 563. DOI: http://dx.doi.org/10.1016/j.saa.2009.11.017 12. Zabardasti, A.; Goudarziafshar, H.; Salehnassaj, M.; Oliveira, B. G.; J. Mol. Model. 2014, 20, 2043. 13. Oliveira, B. G.; Araújo, R. C. M. U.; Carvalho, A. B.; Ramos, M. N.; J. Mol. Model. 2009, 15, 123. DOI: http://dx.doi.org/10.1007/s00894-008-0380-2 PMID: 19037670 14. Oliveira, B.G.; Phys. Chem. Chem. Phys. 2013, 15, 37. DOI: http://dx.doi.org/10.1039/C2CP41749A PMID: 23138158 15. Oliveira, B. G.; Araújo, R. C. M. U.; Pereira, F. S.; Lima, E. F.; Silva, W. L. V.; Carvalho, A. B.; Ramos, M. N.; Quim. Nova 2008, 31, 1673. DOI: http://dx.doi.org/10.1590/S0100-40422008000100004 16. Oliveira, B. G.; Araújo, R. C. M. U.; Carvalho, A. B.; Lima, E. F.; Silva, W. L. V.; Ramos, M. N.; J. Mol. Struct. (THEOCHEM) 2006, 775, 39. DOI: http://dx.doi.org/10.1016/j.theochem.2006.06.028 17. Oliveira, B. G.; Zabardasti, A.; Goudarziafshar, H.; Salehnassaj, M.; J. Mol. Model. 2015, 21, 77. DOI: http://dx.doi.org/10.1007/s00894-015-2616-2 PMID: 25754136 18. Oliveira, B. G.; J. Theor. Comput. Chem. 2014, 13, 1450060. DOI: http://dx.doi.org/10.1142/S0219633614500606 19. Oliveira, B. G.; Struct. Chem. 2014, 25, 745. DOI: http://dx.doi.org/10.1007/s11224-013-0315-0 20. Rozenberg, M.; RSC Adv. 2014, 4, 26928. DOI: http://dx.doi.org/10.1039/c4ra03889d 21. Singh, R. N.; Kumar, A.; Tiwari, R. K.; Rawat, P.; Spectrochim. Acta A 2013, 113, 378. DOI: http://dx.doi.org/10.1016/j.saa.2013.04.121 22. Fuster, F.; Grabowski, S. J.; J. Phys. Chem. A 2011, 115, 10078. DOI: http://dx.doi.org/10.1021/jp2056859 PMID: 21777012 23. Li, L.; Wu, C.; Wang, Z.; Zhao, L.; Li, Z.; Sun, C.; Sun, T.; Spectrochim. Acta A 2015, 136, 338. DOI: http://dx.doi.org/10.1016/j.saa.2014.08.153 24. Zhu, H.; Huang, B.; Li, J.; Jiang, Z.; Wang, B.; Wang, Z.; Zhang, R. -Q.; Phys. Chem. Chem. Phys. 2015, 17, 20361. DOI: http://dx.doi.org/10.1039/C5CP02598B PMID: 26194335 25. Duvoisin Jr., S.; Lima, I. C. V.; Kuhnen, C. A.; Quim. Nova 2011, 34, 1595. DOI: http://dx.doi.org/10.1590/S0100-40422011000900020 26. Barbiellini, B.; Shukla, A.; Phys. Rev. 2002, 66, 235101. DOI: http://dx.doi.org/10.1103/PhysRevB.66.235101 27. Churakov, S. V.; Cem. Conc. Res. 2008, 38, 1359. DOI: http://dx.doi.org/10.1016/j.cemconres.2008.08.004 28. Cohen, A. J.; Mori-Sánchez, P.; Yang, W.; Chem. Rev. 2012, 112, 289. DOI: http://dx.doi.org/10.1021/cr200107z 29. van Mourik, T.; Bühl, M.; Gaigeot, M. - P.; Philos. Trans. R. Soc., A 2014, 372, 20120488. DOI: http://dx.doi.org/10.1098/rsta.2012.0488 30. Jones, R. O.; Rev. Mod. Phys. 2015, 87, 897. DOI: http://dx.doi.org/10.1103/RevModPhys.87.897 31. Srivastava, A.; Mishra, R.; Kumar, S.; Dev, K.; Tandon, P.; Maurya, R.; J. Mol. Struct. 2015, 1084, 55. DOI: http://dx.doi.org/10.1016/j.molstruc.2014.11.070 32. Nazarparvar, E.; Zahedi, M.; Klein, E.; J. Org. Chem. 2012, 77, 10093. DOI: http://dx.doi.org/10.1021/jo301612a PMID: 23078155 33. Varfolomeeva, V. V.; Terentev, A. V.; Phys. Chem. Chem. Phys. 2015, 17, 24282. DOI: http://dx.doi.org/10.1039/C5CP04295J PMID: 26327209 34. Oliveira, B. G.; Vasconcellos, M. L. A. A.; Struct. Chem. 2009, 20, 897. DOI: http://dx.doi.org/10.1007/s11224-009-9489-x 35. Oliveira, B. G.; Araújo, R. C. M. U.; Carvalho, A. B.; Ramos, M. N.; J. Mol. Model. 2009, 15, 421. DOI: http://dx.doi.org/10.1007/s00894-008-0422-9 PMID: 19083032 36. Albuquerque, A. R.; Santos, I. M. G.; Sambrano, J. R.; Quim. Nova 2014, 37, 1318. 37. Manfrini, R. M.; Teixeira, F. R.; Piló-Veloso, D.; Alcântara, A. F. C.; Nelson, D. L.; Siqueira, E. P.; Quim. Nova 2012, 35, 1294. DOI: http://dx.doi.org/10.1590/S0100-40422012000700003 38. Marana, N. L.; Sambrano, J. R.; Souza, A. R.; Quim. Nova 2010, 33, 810. DOI: http://dx.doi.org/10.1590/S0100-40422010000400009 39. Feng, G. -R.; Qi, T. -Y.; Shi, W. -J.; Guo, Y. -X.; Zhang, Y. -J.; Guo, J.; Kang, L. -X.; J. Mol. Model. 2014, 20, 2154. DOI: http://dx.doi.org/10.1007/s00894-014-2154-3 PMID: 24562861 40. DiLabio, G. A.; Johnson, E. R.; Otero-de-la-Roza, A.; Phys. Chem. Chem. Phys. 2013, 15, 12821. DOI: http://dx.doi.org/10.1039/c3cp51559a PMID: 23803877 41. Oliveira, B. G.; Araújo, R. C. M. U.; Quim. Nova 2007, 30, 1678. 42. Guadagnini, P. H.; Bruns, R. E.; Quim. Nova 1996, 19, 148. 43. Dupradeau, F. -Y.; Pigache, A.; Zaffran, T.; Savineau, C.; Lelong, R.; Grivel, N.; Lelong, D.; Rosanski, W.; Cieplak, P.; Phys. Chem. Chem. Phys. 2010, 12, 7821. DOI: http://dx.doi.org/10.1039/c0cp00111b PMID: 20574571 44. Glendening, E. D.; Landis, C. R.; Weinhold, F.; WIREs Comput. Mol. Sci. 2012, 2, 1. DOI: http://dx.doi.org/10.1002/wcms.51 45. Weinhold, F.; Klein, R. A.; Angew. Chem. Int. Ed. 2014, 53, 11214. DOI: http://dx.doi.org/10.1002/anie.201405812 46. Frenking, G.; Caramori, G. F.; Angew. Chem. Int. Ed. 2015, 54, 2596. DOI: http://dx.doi.org/10.1002/anie.201406264 47. Zhang, J.; Chen, P.; Yuan, B.; Ji, W.; Cheng, Z.; Qiu, X.; Science 2013, 342, 611. DOI: http://dx.doi.org/10.1126/science.1242603 PMID: 24072819 48. Martin, T. W.; Derewenda, Z. S.; Nat. Struct. Mol. Biol. 1999, 6, 403. DOI: http://dx.doi.org/10.1038/8195 49. Grabowski, S. J.; Chem. Rev. 2011, 111, 2597. DOI: http://dx.doi.org/10.1021/cr800346f PMID: 21322583 50. Bader, R. F. W.; Atoms in Molecules: A Quantum Theory, Oxford University Press, 1994. 51. Oliveira, B. G.; Araújo, R. C. M. U.; Quim. Nova. 2012, 35, 2002. DOI: http://dx.doi.org/10.1590/S0100-40422012001000021 52. Altaner, C. M.; Thomas, L. H.; Fernandes, A. N.; Jarvis, M. C.; Biomacromolecules 2014, 15, 791. DOI: http://dx.doi.org/10.1021/bm401616n PMID: 24568640 53. Bader, R. F. W.; Chem. Rev. 1991, 91, 893. DOI: http://dx.doi.org/10.1021/cr00005a013 54. Oliveira, B. G.; Leite, L. F. C. C.; J. Mol. Struct. (THEOCHEM) 2009, 915, 38. DOI: http://dx.doi.org/10.1016/j.molstruc.2009.05.038 55. Oliveira, B. G.; Araújo, R. C. M. U.; Monatsh. Chem. 2011, 142, 861. DOI: http://dx.doi.org/10.1007/s00706-011-0540-4 56. Oliveira, B. G.; Araújo, R. C. M. U.; Ramos, M. N.; Quim. Nova 2010, 33, 1155. DOI: http://dx.doi.org/10.1590/S0100-40422010000100023 57. Oliveira, B. G.; Vasconcellos, M. L. V. A. A.; Inorg. Chem. Commun. 2009, 12, 1142. DOI: http://dx.doi.org/10.1016/j.inoche.2009.09.010 58. Oliveira, B. G.; Araújo, R. C. M. U.; Silva, J. J.; Ramos, M. N.; Struct. Chem. 2010, 21, 221. DOI: http://dx.doi.org/10.1007/s11224-009-9567-0 59. Gaussian 03, Revision B.04, Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Montgomery Jr., J. A.; Vreven, T.; Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G. A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Gonzalez, C.; Pople, J. A.; Gaussian, Inc., Pittsburgh PA, 2003. 60. van Duijneveldt, F. B.; van Duijneveldt-van de Rijdt, J. G. C. M.; van Lenthe, J. H.; Chem. Rev. 1994, 94, 1873. DOI: http://dx.doi.org/10.1021/cr00031a007 61. Boys, S. F.; Bernardi, F.; Mol. Phys. 1970, 19, 553. DOI: http://dx.doi.org/10.1080/00268977000101561 62. AIM2000 1.0 designed by F. Biegler-König, University of Applied Sciences, Bielefeld, Germany. 63. Keith, T. A.; AIMAll Version 11.05.16, 2011. 64. Rozas, I.; Alkorta, I.; Elguero, J.; J. Phys. Chem. A 1997, 101, 9457. DOI: http://dx.doi.org/10.1021/jp971893t 65. Szatyłowicz, H.; J. Phys. Org. Chem. 2008, 21, 897. DOI: http://dx.doi.org/10.1002/poc.1394 66. Oliveira, B. G.; Araújo, R. C. M. U.; Ramos, M. N.; J. Mol. Struct. (THEOCHEM) 2009, 908, 79. DOI: http://dx.doi.org/10.1016/j.theochem.2009.05.013 67. Santos, I. T. O.; Rego, D. G.; Oliveira, B. G.; Quim. Nova 2014, 37, 624. DOI: http://dx.doi.org/10.1590/S0100-40422014000100003 68. Oliveira, B. G.; Araújo, R. C. M. U.; Ramos, M. N.; J. Mol. Struct. (THEOCHEM) 2010, 944, 168. DOI: http://dx.doi.org/10.1016/j.theochem.2009.12.032 69. Figura 1S do Material Suplementar. 70. Asselin, P.; Soulard, P.; Madebène, B.; Goubet, M.; Huet, T. R.; Georges, R.; Pirali, O.; Roy, P.; Phys. Chem. Chem. Phys. 2014, 16, 4797. DOI: http://dx.doi.org/10.1039/c3cp55047h PMID: 24469411 71. Oliveira, B. G.; Araújo, R. C. M. U.; Ramos, M. N.; J. Mol. Struct. (THEOCHEM) 2010, 944, 168. DOI: http://dx.doi.org/10.1016/j.theochem.2009.12.032 72. Oliveira, B. G.; Spectrochim. Acta A 2014, 124, 208. DOI: http://dx.doi.org/10.1016/j.saa.2013.12.029 73. Oliveira, B. G.; Chem. Phys. 2014, 443, 67. DOI: http://dx.doi.org/10.1016/j.chemphys.2014.09.001 74. Q. Pordeus, R. Q.; Rego, D. G.; Oliveira, B. G.; Spectrochim. Acta A 2015, 145, 580. DOI: http://dx.doi.org/10.1016/j.saa.2015.02.070 75. Meot-Ner (Mautner), M.; Chem. Rev. 2012, 112, PR22. 76. Hobza, P.; Havlas, Z.; Chem. Rev. 2000, 100, 4253. DOI: http://dx.doi.org/10.1021/cr9900331 PMID: 11749346 77. Compaan, K.; Vergenz, R.; Von Rague Schleyer, P.; Arreguin, I.; Int. J. Quantum Chem. 2008, 108, 2914. DOI: http://dx.doi.org/10.1002/qua.21811 78. Oliveira, B. G.; Ramos, M. N.; Int. J. Quantum Chem. 2010, 110, 307. DOI: http://dx.doi.org/10.1002/qua.21995 79. Oliveira, B. G.; Araújo, R. C. M. U.; Leite, E. S.; Ramos, M. N.; Int. J. Quantum Chem. 2011, 111, 111. 80. Sigala, P. A.; Ruben, E. A.; Liu, C. W.; Piccoli, P. M. B.; Hohenstein, E. G.; Martínez, T. J.; Schultz, A. J.; Herschlag, D.; J. Am. Chem. Soc. 2015, 137, 5730. DOI: http://dx.doi.org/10.1021/ja512980h PMID: 25871450 81. Ilnicka, A.; Sadlej, J.; Struct. Chem. 2012, 23, 1332. 82. Phipps, M. J. S.; Fox, T.; Tautermann, C. D.; Skylaris, C. K.; Chem. Soc. Rev. 2015, 44, 3177. DOI: http://dx.doi.org/10.1039/C4CS00375F PMID: 25811943 83. Oliveira, B. G.; Bueno, M. A.; Quim. Nova 2015, 38, 1. 84. Grabowski, S. J.; J. Phys. Chem. A 2011, 115, 12789. DOI: http://dx.doi.org/10.1021/jp203908n PMID: 21830803 85. Martínez-Cifuentes, M.; Weiss-López, B. E.; Santos, L. S.; Araya-Maturana, R.; Molecules 2014, 19, 9354. DOI: http://dx.doi.org/10.3390/molecules19079354 PMID: 24995921 86. Pilmé, J.; Renault, E.; Bassal, F.; Amaouch, M.; Montavon, G.; Galland, N.; J. Chem. Theory Comput. 2014, 10, 4830. DOI: http://dx.doi.org/10.1021/ct500762n PMID: 26584370 87. Hilal, R.; Aziz, S. G.; Alyoubi, A. O.; Elrobya, S.; Proc. Comput. Sci. 2015, 51, 1872. DOI: http://dx.doi.org/10.1016/j.procs.2015.05.423 88. Oliveira, B. G.; Araújo, R. C. M. U.; Quim. Nova 2012, 35, 2002. DOI: http://dx.doi.org/10.1590/S0100-40422012001000021 89. Hill, J. G.; Legon, A. C.; Phys. Chem. Chem. Phys. 2015, 17, 858. DOI: http://dx.doi.org/10.1039/C4CP03376K PMID: 25141075 |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access