
 |
 |
 |
 |
 |
Artigo
|
|
| Síntese de derivados de fenantridinonas por arilação direta. Caracterização dos espectros de absorção e emissão de exemplos representativos Synthesis of phenanthridinone derivatives by direct arylation. Characterization of the absorption and emission spectra for representative examples |
|
Roberta L. da Costa; Douglas A. F. da Silva; Nanci C. de Lucas; Simon J. Garden*
Instituto de Química, Universidade Federal do Rio de Janeiro, Centro de Tecnologia, Bloco A, Cidade Universitária, 21949-900 Rio de Janeiro - RJ, Brasil Recebido em 28/09/2015 *e-mail: garden@iq.ufrj.br The phenanthridinone heterocyclic system has attracted considerable attention in recent years due to the diverse array of physical, chemical and pharmacological properties demonstrated by natural and synthetic derivatives. As a consequence there has been considerable development of synthetic methodology for the synthesis of this and related heterocyclic ring systems. The synthetic literature is discussed and is compared with a direct arylation methodology for the intramolecular cyclization of tertiary (2-iodo)benzoylamides to generate the biaryl bond of these compounds. The efficient methodology allowed the synthesis of a number of previously unknown phenanthridinone products. The photoluminescent properties of representative examples were characterized and it is proposed that the previously unknown compound 1s reveals dual fluorescence in a manner similar to the known compound 1r. INTRODUÇÃO O sistema heterocíclico das fenantridinonas é encontrado em uma grande variedade de produtos naturais e sintéticos. Esta classe de compostos tem despertado o interesse da comunidade científica devido às diversas propriedades físicas, químicas e farmacológicas. A Figura 1 apresenta alguns exemplos de produtos naturais bioativos: narciprimina,1,2 arolicoricidina,2,3 phamina,4 e subarina;5 e de estruturas sintéticas: inibidores de poli(ADP-ribose)polimerase-1 (PARP-1) para proteção contra acidente vascular cerebral (isquemia) ou para o tratamento de câncer,6 inibidores de Topoisomerase I,7 antivirais,8 anti-malarias,9 ligantes para receptores 5-HT410 e um complexo de irídio utilizado em OLEDs (organic light emitting diode, diodo orgânico emissor de luz).11
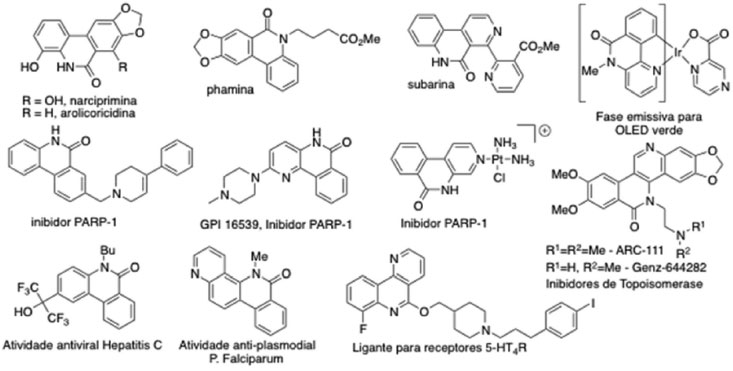 Figura 1. Exemplos de produtos naturais e sintéticos de derivados de fenantridinonas
As metodologias para a síntese de fenantridinonas (1) têm se diversificado, além dos métodos clássicos, com o desenvolvimento de métodos empregando catálise por sais de paládio e geração de radicais livres por organocatálise. A Figura 2 ilustra de forma genérica as principais abordagens para a síntese de 1.
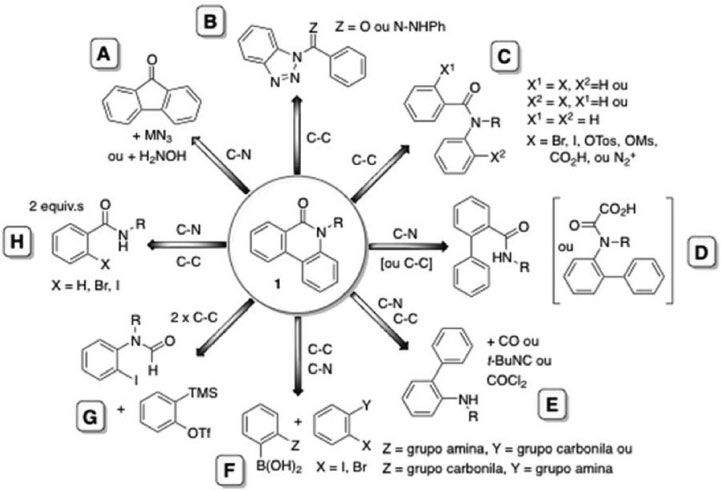 Figura 2. Retroanálise, de forma genérica, das principais abordagens para a síntese de 1
Os métodos clássicos para a preparação de 1 incluem: a reação de Schmidt12 (utilizando o ácido difênico),13 ou o rearranjo de Beckmann14 de derivados de fluorenonas (Figura 2, A) e o rearranjo de Hoffmann da monoamida de ácido difênico.15 Outros métodos empregam derivados de bifenila substituídos na posição 2, por exemplo a reação de Pictet-Hubert16 e a sua modificação de Morgan-Walls,17 aplicada à síntese de fenantridinas seguida por oxidação,18 ou a ciclização do etil carbamato de 2-aminobifenila,19 e a reação de 2-aminobifenila com fosgênio (Figura 2, E),20 ou a sequência de reações envolvendo a nitração, redução e desidratação aplicada ao ácido bifenil-2-carboxílico,21 e a oxidação de bifenil-2-carboxamidas (Figura 2, D).22 Além destes métodos, as fenantridinonas (1) podem ser obtidas pelo rearranjo térmico de 2-alcoxifenantridinas23 ou cicloadições de Diels-Alder,24 ou, fotoquimicamente, por irradiação de aril-hidrazonas de benzotriazol (Figura 2, B),25 ou por foto-desidro-halogenação de benzoilanilinas (Figura 2, C),26-28 foto-ciclo-adição de estirenos com isoquinolinonas,29 ou por pirólise de derivados de 3-arilidenoamino-1,2,3-benzotriazin-4-onas30 e de benzoilbenzotriazóis (Figura 2, B).31 Outros métodos envolvendo intermediários radicalares incluem a reação de Pschorr32 e ciclizações promovidas por reagentes como Bu3SnH,33 SmI2/HMPA34 ou TTMSS35 na presença de oxigênio (Figura 2, C). O mecanismo destas reações radicalares passa por um intermediário, um espirocicloexadieniloxindol, que pode, ou não, sofrer um rearranjo para gerar 1 dependendo das condições reacionais.36 A eliminação do uso de metais pesados e metais de transição pode ser importante dependendo da finalidade de uso dos produtos. Desta forma, a oxidação de ácidos de biaril-2-oxâmicos utilizando persulfato de sódio em DMSO (Figura 2, D)37 e a descoberta recente de que o t-BuOK na presença de diaminas pode promover a ciclização radicalar de N-2-halobenzoilanilinas38,39 ou de N-benzoil-2-haloanilinas40 (Figura 2, C) representam métodos inovadores para a síntese de 1. Por outro lado, metodologias que reduzem o grau de pre-funcionalização de substratos, como por exemplo a ciclização oxidativa de 2-fenilbenzamidas catalisada por CuI na presença de oxigênio e t-BuOK,41 são uma abordagem potencialmente mais eficiente (Figura 2, C). O uso de sais de cobre para obter derivados de 1 tem se limitado ao homoacoplamento oxidativo de reagentes de Grignard,42 a reação de Goldberg intramolecular de 2-ciano-2'-halobiarilas sob condições de hidrólise básica,43 e a reação de acoplamento cruzado, co-catalisada com Pd(0), de 2-nitrobromobenzeno com derivados de 1-bromobenzoato de metila.44 Porém, de forma mais abrangente, a investigação de métodos com catálise por sais de paládio levou ao desenvolvimento de novas metodologias e abordagens para a síntese de derivados de 1. Os pesquisadores Ames e Opalko foram os primeiros a relatar a síntese de 1 por reações de desidro-halogenação de 2-halobenzoilanilinas ou de benzoil-2-haloanilinas catalisadas por paládio (Figura 2, C).45 A abordagem utilizada é atualmente conhecida como um método de arilação direta46 (intramolecular) e é um método bastante empregado para a síntese de derivados de fenantridinonas. Os métodos de arilação direta catalisada por sais de paládio aplicados à síntese de 1 tipicamente envolvem a reação de um haloareno (bromo e iodo derivados são os mais utilizados) para formar um intermediário organopaládio via adição oxidativa de Pd(0). Este intermediário participa na funcionalização de uma ligação C-H de um segundo grupo areno para formar uma ligação biarila de forma intramolecular.47,48 Tanto derivados de N-2-halobenzoil-N-alquilanilinas quanto derivados de N-benzoil-N-alquil-2-haloanilinas podem ser empregados em reações catalisadas por diversas fontes de Pd(0) na presença ou ausência de ligantes como fosfinas.49-51 O halogênio do substrato pode ser substituído por um sal de diazônio,52 triflato,53 tosilato ou mesilato (Figura 2, C).54 Os exemplos de arilação direta envolvem a funcionalização de uma ligação C-H. Outras estratégias envolvem a funcionalização de duas ligações C-H ou uma combinação de ligações C-H e N-H. Um dos primeiros exemplos da funcionalização de duas ligações C-H aromáticas para a síntese de 1 foi a oxidação de benzanilida utilizando Pd(O2CCF3)2/Sn(OAc)2/O2 em AcOH (Figura 2, C).55 Mais recentemente, sais de Pd(II) na presença de persulfato de sódio,56 ou ácido benzóico em uma atmosfera de oxigênio,57 ou AgOAc in PivOH/AcOH foram utilizados para promover o acoplamento oxidativo para obter 1 (Figura 2, C e D).58 Exemplos da síntese de 1 por reações envolvendo a funcionalização de ligações C-H e N-H em sistemas biarilas incluem a carbonilação de biarilaminas catalisada por Pd(II),59,60 ou a carbonilação redutiva de 2-nitrobiarila catalisada por Ru3(CO)12,61 e a utilização de isocianeto de t-butila,62 ou de forma indireta via a carbonilação de derivados de 10,9-borazarofenantrenos catalizado por Pd(OAc)2 (Figura 2, E).63 Além dos métodos envolvendo reações de acoplamento intramolecular, ou que partem de sistemas biarilas funcionalizados, diversos métodos envolvendo reações de acoplamento cruzado intermoleculares tem sido descritos. A reação de Suzuki-Miyaura aplicada à síntese de derivados de 1 mostra a versatilidade desta metodologia para o acoplamento de substratos altamente funcionalizados, apesar da necessidade de utilizar dois reagentes pré-funcionalizados para a síntese da ligação biarila (Figura 2, F).64 Ácidos benzóicos podem substituir ácidos fenil borônicos em reações de acoplamento cruzado,65 e um exemplo aplicado à síntese de 1 envolveu o aquecimento do ácido N-2-bromobenzoil-N-metilantranílico em NMP na presença de Pd(OAc)2.66 Uma abordagen distinta envolvendo a ciclização de orto-halobenzamidas com arinos catalisada por paládio foi relatada por Larock.67 Subsequentemente, foi mostrado que orto-iodo-N-formilanilinas podem ser utilizados de forma análoga,68 e N-metil- ou N-metoxi- benzamidas reagem, via funcionalização de ligações C-H/N-H formando um paladaciclo, com arinos para fornecer derivados de 1 na presença de persulfato de potássio como oxidante (Figura 2, G).69 O desenvolvimento de metodologias de acoplamento intermoleculares envolvendo a funcionalização de ligações C-H/N-H tem resultado na descoberta de reações dominó, por exemplo, derivados de 2-halobenzamidas podem ser dimerizados para fornecer derivados de 1 com (ou sem dependendo no solvente) a eliminação de organoisocianeto (Figura 2, H).70-72 Derivados de N-metoxibenzoilamidas reagem com iodobenzenos em uma reação dominó catalizada por Pd(OAc)2, na presença de Ag2O em AcOH, levando à formação de derivados de 1. O processo passa por um intermediário de Pd(IV) e resulta na funcionalização de duas ligações C-H e uma ligação N-H (Figura 2, H).73 O presente trabalho desenvolveu um método de arilação direta catalisada por Pd(0), na ausência de ligantes fosfinas, para a síntese de fenantridinonas. Devido à fluorescência dos produtos, as propriedades fotofísicas de algumas das fenantridinonas foram investigadas.
RESULTADOS E DISCUSSÃO Inicialmente foram preparados derivados de N-benzilaminas secundárias através da reação de aminação redutiva. Em seguida as aminas secundárias foram transformadas em amidas terciárias por uma reação de benzoilação e finalmente as amidas terciárias foram aplicadas na reação de arilação direta para obter os derivados de fenantridinonas (Esquema 1).74
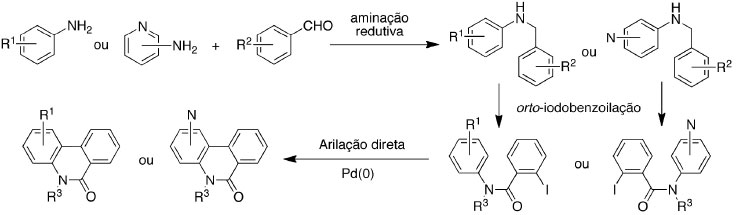 Esquema 1. Síntese das fenantridinonas a partir de anilinas e aminopiridinas
Visando investigar a versatilidade da metodologia de arilação direta para a obtenção de fenantridinonas, diversas anilinas e benzaldeídos foram empregados na reação de aminação redutiva para obter as aminas secundárias. As reações foram feitas de forma a promover a condensação da anilina (ou aminopiridina) com o derivado de benzaldeído e em seguida o redutor (NaBH4) foi acrescentado. De forma geral, obteve-se as aminas secundárias com rendimentos bons a excelentes (65 - 99%) e com pureza satisfatória para análise espectroscópica e uso na reação seguinte. Os sistemas piridínicos apresentaram uma menor reatividade frente ao aldeído. Desta forma a reação de condensação com os aldeídos foi feita em tolueno sob refluxo durante 6 h e em seguida NaBH4 foi acrescentado para promover a reação de redução. Os procedimentos são detalhados no material suplementar. A Figura 3 mostra as aminas secundárias que foram empregadas na síntese das amidas terciárias.
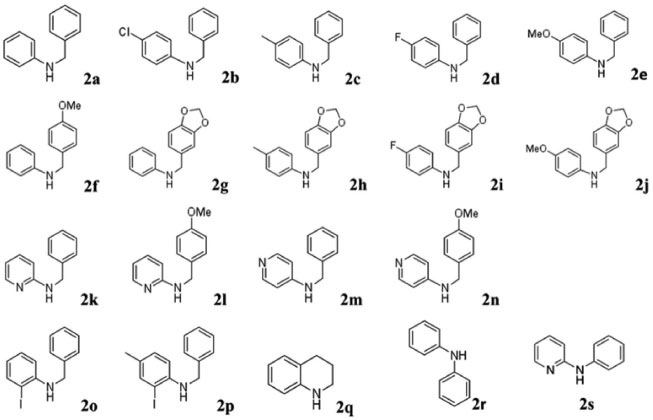 Figura 3. Estruturas das aminas secundárias utilizadas neste trabalho
Reações de benzoilação As aminas secundárias (Figura 3) foram empregadas em reações de benzoilação para obter as amidas terciárias (Esquema 1). Os substratos foram acilados utilizando cloreto de orto-iodobenzoíla (produtos 3a-n, q-s) ou cloreto de benzoíla (produtos 3o e 3p). As estruturas das amidas terciárias (3) estão ilustradas na Figura 4.
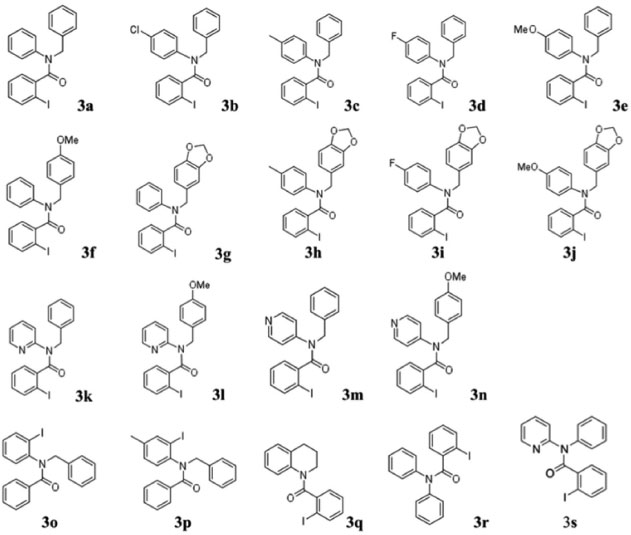 Figura 4. Estruturas das amidas terciárias que foram utilizadas como substratos na reaçao de arilaçao direta
No caso das reações de (orto-iodo)benzoilação, uma solução do cloreto de ácido em CH2Cl2 foi acrescentada lentamente a uma solução da amina secundária em solução de CH2Cl2/Et3N resfriada em banho de gelo. A reação foi relativamente rápida (~2 h). Porém, no caso dos sistemas piridínicos (2k - n), onde o nitrogênio secundário é menos básico, foi necessário utilizar uma base mais forte para promover a abstração do próton. A base utilizada foi uma mistura de Et3N e 10 mol% DMAP. A reação prosseguiu de forma mais lenta, levando aproximadamente 4 h. Os procedimentos experimentais são detalhadas no Material Suplementar. As estruturas das Figuras 3 e 4 foram caracterizadas espectroscópicamente e os dados podem ser consultados no Material Suplementar. Geralmente, os compostos N-orto-iodobenzoil-N-benzilfenilaminas apresentam sinais de RMN característicos: CH2 benzílico (~5,0 ppm); simpletos referente a presença de MeO (~3,8 ppm) ou metila (~2,0 ppm) com integração relativa equivalente a três hidrogênios ou um simpleto referente a dois hidrogênios no caso da presença de um grupo metilenodioxi (~5,8 ppm). Os metilenos do anel tetraidroquinolínico (3q) aparecem na faixa 3,0-4,5 ppm). Os derivados piridínicos (3k-m) apresentam os respectivos sinais dos hidrogênios do anel piridínico mais desblindados, tipicamente na faixa de 8,0 - 8,5 ppm, devido à maior acidez dos hidrogênios. A caracterização dos compostos por RMN de 13C mostrou, além do número de sinais esperados para cada composto, alguns sinais bastante semelhantes entre os compostos: as ligações C-I (~90ppm), C=O (~170 ppm), e o sinal CH2 benzílico (~52 ppm). Nos compostos contendo o grupo metilenodioxi, o sinal de RMN de 13C tem um deslocamento bem característico em ~100 ppm enquanto os grupos metoxi ou metila aparecem em torno de 55 e 21 ppm respectivamente. No caso das estruturas benzoiladas (3o e 3p) os sinais de 13C das ligações C-I aparecem em torno de 80 ppm. Além disso, os espectros de RMN de 13C das estruturas benzoiladas apresentam um par de sinais com intensidades bastante superiores aos outros sinais devido à presença de dois pares de carbonos equivalentes (carbonos 2 e 6, e 3 e 5) no anel aromático do grupo benzoíla. Síntese de fenantridinonas via a reação de arilação direta As reações de arilação direta tiveram como objetivo a funcionalização da ligação Csp2-H catalisada por paládio para formar a ligação biarila das fenantridinonas. A metodologia empregada48,51 para a reação de arilaçao direta consistiu no aquecimento (110 ºC sob agitação) do substrato iodado (3, 1 mmol) na presença de KOAc (5 mmol), TBAB (1 mmol), e uma quantidade catalítica de Pd(OAc)2 (10 mol%) em DMF durante 24 h. Os produtos das reações foram isolados por partição da mistura reacional entre água e um solvente orgânico (CH2Cl2). A fase orgânica foi seca com Na2SO4 anidro, a solução foi filtrada, e após a evaporação sob pressão reduzida o produto bruto foi purificado em coluna cromatográfica com sílica gel. A purificação dos produtos foi facilitada pela observação da fluorescência dos produtos, tanto no comprimento de onda longo (365 nm) quanto no comprimento de onda curto (254 nm) da lâmpada UV de revelação. A Figura 5 mostra as estruturas dos produtos das reações de arilação direta intramolecular e os respectivos rendimentos. No caso da síntese dos produtos 1a e 1c, cada produto foi obtido a partir de dois substratos diferentes mostrando uma versatilidade da metodologia: 1) utilizando os substratos orto-iodobenzoíla (3a e 3c), e 2) utilizando os substratos N-benzoil-orto-iodoanilinas (3o e 3p).
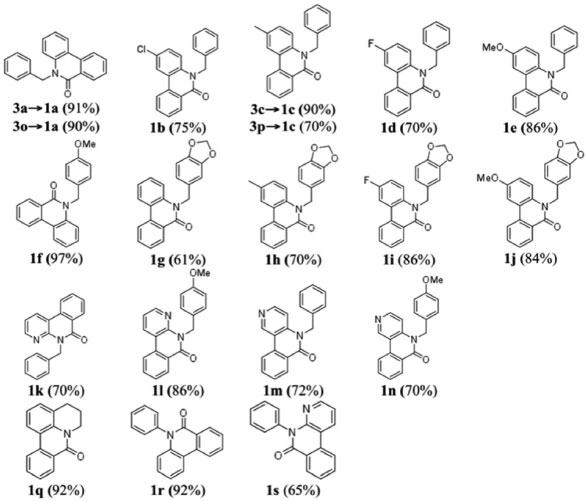 Figura 5. Resultados das reações de arilação direta intramolecular utilizando as amidas terciárias
Diversas das metodologias detalhadas na introdução foram empregadas na síntese de algumas (1a, b, f, q, e r) das fenantridinonas relatadas neste estudo. A reação de anelação de derivados de N-formil-N-benzil-2-iodoanilina com benzino catalizada por Pd(0) (Figura 2, G) resultou em 1a 93% (1b 88% e 1r 60%)68 e utilizando N-benzil-N-2-halobenzoilamidas resultou na obtenção de 1a com 82% (1f 75% a partir do bromo substrato ou em 38% a partir do iodo substrato).67 A reacão de arilacão direta (Figura 2, C) utilizando K2CO3/DPPE/Pd(OAc)2 em DMF resultou em 1a em 85% de rendimento e 1b com 76%,50 enquanto utilizando Pd(OAc)2/K2CO3/PPh3/DMAc foram obtidos 1f 93% e 1q 96%,75 e o uso de P(Tol)3/Pd(OAc)2 forneceu 1q (98%).76 Han et al. relataram a reação de arilação direta utilizando derivados do sal tetrafluoroborato de N-2-diazôniobenzoil-N-benzilanilina e obtiveram 1a (65%) e 1b (69%).52 A reação de dimerização de 2-halobenzoil-N-benzilamida por reação dominó com eliminação de um isocianeto (Figura 2, H) forneceu 1a 77% (1f 64% e 1r 23%)71 e no estudo por Donati et al. 1a (55%) e 1f (71%).70 A carbonilação de derivados de N-benzil-2-aminobifenila (Figura 2, E) forneceu 1a (65%) e 1r (52%).60 Uma reação tipo Goldberg catalisada por um sal de cobre utilizando derivados de ácido 2-clorobifenil-2'-carboxílico com anilinas forneceu 1a (72%) e 1r (96%).77 Métodos radicalares incluem: o uso de t-BuOK (Figura 2, C) fornecendo 1q (bromo substrato, 61-70%, iodo substrato, 63%),38 deidrogenação de N-fenil bifenil-2-carboxamida (Figura 2, D) utilizando persulfato de potássio para obter 1r (97%),78 ou por geração fotolítica de um íon N-acilnitrênio gerando 1r (65%),79 ou por foto-deidro-halogenação (Figura 2, C) fornecendo 1r (25%).26 Os dados de RMN das fenantridinonas conhecidas (1a,71b,68f,71q,76 e r71) são condizentes com os dados na literatutra. No caso de 1r o espectro de 13C (50 MHz) mostrou apenas 16 sinais de carbono, quando o esperado era 17 sinais de 13C, porém, quando o espectro foi obtido em um aparelho com campo magnético maior o sinal em 129,3 ppm se desdobrou, resultando em um total de 17 sinais de 13C (125 MHz). De forma geral, as respectivas áreas relativas integradas para os grupos de sinais de hidrogênio (aromático contra alifático ou benzilico) estão de acordo com os valores esperados para as respectivas fenantridinonas. Através do RMN 13C podemos confirmar as principais modificações nos grupos, como por exemplo a ausência da ligação C-I (~90 ppm), que pode indicar a ciclização ou somente a redução da ligação C-I. A carbonila dos produtos ciclizados apresenta um deslocamento químico na faixa de 161- 162 ppm, que é condizente com os dados espectroscópicos dos compostos conhecidos. No caso do produto 1s, a reação de arilação direta do substrato 3s resultou na formação de dois produtos (ciclização no anel piridínico e no anel fenila). O produto principal foi purificado da mistura, a identificação como sendo o produto majoritário foi feita por comparação com a mistura bruta por CG-MS (ambos os produtos tem o mesmo massa molecular), e a estrutura determinada por análise dos espectros de RMN. O espectro de RMN 1H mostrou um dupleto em 7,3 ppm correspondente a dois hidrogênios químicamente e magneticamente equivalentes, e um outro dupleto em 7,6 ppm, porém, sobreposto com outros sinais. O espectro de 13C mostrou 16 sinais de 13C e um par de sinais em 129,2 e 129,6 ppm com o dobro de intensidade comparados com os outros sinais das ligações C-H aromáticas. As informações são condizentes com a arilação direta do anel piridínico e a presença de um anel fenila monossubstituído. Espectroscopia de absorção e emissão das fenantridinonas A observação da fluorescência das fenantridinonas estimulou a caracterização espectroscópica de alguns exemplos destes compostos. O espectro de emissão da fenantridinona 1r é um caso interessante, uma vez que mostra um fenômeno de fluorescência dual, emissão em dois comprimentos de onda distintos.80 As bandas de emissão são atribuídas ao processo de emissão de dois estados excitado singlete diferentes, um localizado (LE) e um outro devido a transferência de carga interna (ICT). A Figura 6 mostra os espectros de absorção e a Figura 7 os espectros de emissão dos compostos 1f, 1q, 1r e 1s. Os espectros de absorção, emissão, e excitação destes compostos são apresentados no material suplementar (Figuras 164S-167S). A Tabela 1 apresenta alguns dados fotofísicos destes compostos.
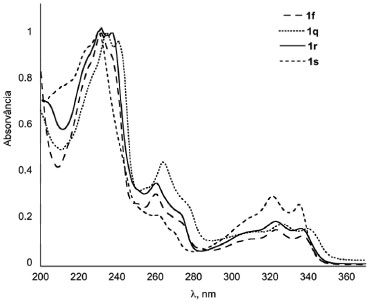 Figura 6. Espectros de absorçao normalizado de 1f, 1q, 1r e 1s em acetonitrila (A300nm = 0,1)
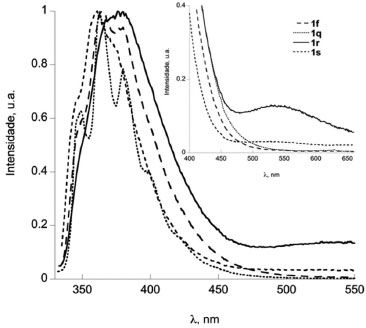 Figura 7. Espectros de fluorescência normalizado de 1f, 1q, 1r e 1s em acetonitrila (λexc= 300 nm; A300nm=0,1). Anexo - espectros em comprimento de onda mais longos
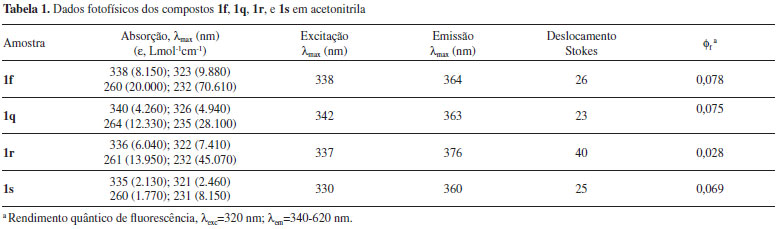
Os espectros de absorção mostraram uma banda intensa em torno de 230 nm, um ombro em torno de 260 nm e uma banda menos intensa larga em torno de 280-350 nm (Figura 6). A Tabela 1 mostra os valores exatos destas bandas com seus respectivos coeficientes de absortividade molar. Estes resultados mostraram que a azafenantridinona (1s) apresenta o espectro mais deslocado para o azul, enquanto dentre as fenantridinonas o espectro de absorção de 1q é ligeiramente deslocado para o vermelho. Os espectros de emissão mostram uma banda correspondente a emissão do estado excitado localizado (LE) com máximo em torno de 360 nm. O espectro de emissão do composto 1q é diferente dos outros espectros e mostra estrutura vibracional. Esta diferença pode ser atribuída à maior rigidez da estrutura 1q em relação as demais estruturas. Notavelmente a emissão do composto 1r mostra a presença de uma segunda banda, consistente com dados na literatura,80 em 550 nm, que é atribuída à emissão do estado de transferência de carga interna (ICT), em que o grupo N-fenila está coplanar com a fenantridinona. De forma semelhante podemos relatar a observação inédita de fluorescência dual do composto análogo 1s. Observa-se que o espectro de emissão de 1s em torno de 550 nm não volta para a linha base, ou seja, este composto também revela no seu espectro de emissão uma banda larga em comprimento de onda maior após a emissão do LE devido à emissão do estado ICT (Figura 7).
PARTE EXPERIMENTAL Os produtos foram caracterizados por espectroscopia de RMN de 1H e 13C utilizando os aparelhos Bruker DPX 200, DRX 300 e Avance 500 (IQ/UFRJ). Os espectros de RMN 1H tem seus deslocamentos químicos relatados em ppm e as multiplicidades dos sinais expressos como s (simpleto), d (dupleto), t (tripleto), dd (duplo dupleto) ou m (multipleto). Os espectros de IV foram obtidos com pastilha de KBr registrados no aparelho Nicolet 505 Magma (FTIR). Os pontos de fusão, com valores não corrigidos, foram obtidos utilizando um aparelho Mel-Temp II. Os espectros de massa de alta resolução foram obtidos utilizando um MicroTOF (IPPN/UFRJ). A técnica de ionização por eletrospray, no modo positivo, foi utilizada para analisar as amostras. A lâmpada Cole-Parmer Short/long-wave UV lamp, 4 watts, foi utilizada para revelar as placas de CCF (Merck Silica Gel 60 F254) das análises das reações. A preparação e a caracterização espectroscópica dos compostos 1 e 2 encontra-se no Material Suplementar. Os espectros de absorção foram obtidos em um espectrofotômetro Shimadzu UV-2450. Os espectros de fluorescência foram registrados em um espectrofluorímetro da Edinburgh Instruments modelo F900, com lâmpada de Xe (Xe900, 450W) e monocromador TMS300. Todos os espectros foram realizados em solução de acetonitrila a temperatura ambiente. As concentrações utilizadas foram de ~10-5 mol L-1 de modo a obter uma absorção na região de excitação (300 nm) de ~ 0,1. Para os experimentos de determinacão dos coeficientes de absortividade molar as concentrações usadas variaram de 5,0x10-6 a 3,0x10-5 mol L-1. O rendimento quântico de fluorescência foi medido pelo método do padrão relativo, em soluções saturadas com ar, usando uma solução 10-5 mol L-1 de sulfato de quinina em ácido sulfúrico 0,05 mol L-1 (φf=0,6).81 Os espectros de fluorescência do padrão e das amostras foram registrados nas mesmas condições (λexc=320 nm e λem=340-620 nm). Procedimento geral para reações de arilação direta intramolecular catalisada por paládio Em um balão de fundo redondo (10 mL) foram adicionados: substrato iodado (1,0 mmol), KOAc (0,490 g, 5 mmol), Bu4NBr (0,354 g, 1,1 mmol) e Pd(OAc)2 (22 mg, 10 mol%) em DMF (5 mL). A mistura foi aquecida a temperatura de 110 ºC durante 24 h sob agitação constante. O fim da reação foi verificado por CCF pela ausência do substrato. Foi acrescentado H2O (15 ml) e a fase orgânica extraída com CH2Cl2 (3 x 15 mL). A fase orgânica foi seca com Na2SO4 anidro, filtrada e evaporada sob pressão reduzida. O produto bruto obtido foi purificado em coluna cromatográfica utilizando sílica gel. Caracterização física, espectroscópica e espectrométrica dos produtos de arilação direta 5-Benzilfenantridin-6(5H)-ona (1a) RMN 1H (200 MHz, CDCl3): δ 5,70(s, 2H), 7,29 (m, 8H), 7,63 (t, J=8Hz, 1H), 7,80 (t, J=8Hz, 1H), 8.30 (m, 2H), 8,67 (d, J=8Hz, 1H); RMN 13C (50 MHz, CDCl3): δ 46,6; 116,2; 119,7; 121,8; 122,7; 123,4; 125,8; 126,7; 127,3; 128,2; 128,9; 129,3; 129,6; 132,8; 134,0; 136,8; 137,5; 162,7; IV (KBr, cm-1): 3067, 3029, 2968, 2930, 2872, 1640, 1607, 1584, 1493; Sólido bege; PF.: 110 ºC, Lit.:112-113 ºC.50 5-Benzil-2-clorofenantridin-6(5H)-ona (1b) RMN 1H (300 MHz, CDCl3): δ 5,61 (s, 2H), 7,28 (m, 7H), 7,63 (t, J= 9Hz,1H), 7,75 (t, J= 9Hz, 1H), 8,16 (m, 2H), 8,58 (d, J= 9Hz,1H); RMN 13C (75 MHz, CDCl3): δ 46,7; 117,5; 121,0; 121,9; 123,1; 125,7; 126,6; 127,5; 128,4; 128,8; 129,0; 129,4; 129,5; 132,7; 133,0; 135,9; 136,3; 161,0; IV (KBr, cm-1): 3079, 3030, 2959, 1643, 1606, 1579, 1497, 1173, 1109.; Sólido incolor; PF.: 165 ºC (recristalizado em isopropanol), Lit.: 167-168 ºC.50 5-Benzil-2-metilfenantridin-6(5H)-ona (1c) RMN 1H (200 MHz, CDCl3): δ 2,30 (s, 3H), 5,56 (s, 2H), 7,17 (m, 6H), 7,50 (t, J=8Hz, 1H), 7,68 (t, J=8Hz, 1H), 7,97 (s, 1H), 8,17 (d, J=8Hz, 1H), 8,50 (d, J=8Hz, 1H); RMN 13C (50 MHz, CDCl3): δ 21,0; 46,5; 116,1; 119,5; 121,8; 123,5; 125,9; 126,7; 127,2; 128,0; 128,9; 129,3; 130,7; 132,1; 132,7; 133,9; 135,3; 136,7; 162,2; IV (KBr, cm-1): 3027, 2974, 2921, 1654, 1606, 1580, 1494, 1318; ESI-MAR (m/z): calculada C21H17NO (M+H+): 322,1202; encontrada: 322,1205; Sólido incolor; PF.: 168 ºC. 5-Benzil-2-fluorofenantridin-6(5H)-ona (1d) RMN 1H (200 MHz, CDCl3): δ 5,64 (s, 2H), 7,09 (m, 1H), 7,25 (m, 6H), 7,65 (t, J=8Hz, 1H), 7,.80 (t, J=8Hz, 1H), 7,90 (t, J=8Hz, 1H), 8,15 (d, J=8Hz, 1H), 8,69 (d, J=8Hz, 1H); RMN 13C (50 MHz, CDCl3): δ 46,9; 109,2; 109,7; 116,7; 117,2; 177,6; 117,8; 122,0; 125,8; 126,7; 127,5; 128,9; 129,0; 129,5; 133,0; 136,5; 156,2; 161,7; IV (KBr, cm-1): 3072, 3063, 3040, 2963, 1636, 1591, 1499, 1428, 1309, 1282; ESI-MAR (m/z): calculada C20H14FNO (M+H+): 326,0951; encontrada: 326,0953; Sólido incolor; PF.: 160 ºC. 5-Benzil-2-metoxifenantridin-6(5H)-ona (1e) RMN 1H (200 MHz, CDCl3): δ 3,87 (s, 3H), 5,64 (s, 2H), 6,96 (d, J=8Hz, 1H), 7,25 (m, 5H), 7,62 (t, J=8Hz, 1H), 7,73 (m, 2H), 8,21 (d, J=8Hz, 1H), 8,60 (d, J=8Hz, 1H); RMN 13C (50 MHz, CDCl3): δ 46,7; 55,8; 107,4; 116,6; 117,3; 120,6; 121,9; 125,9; 126,7; 127,3; 128,3; 128,9; 129,5; 131,8; 132,7; 133,7; 136,9; 155,3; 161,6; IV(KBr, cm-1): 3043, 3003, 2937, 2911, 1633, 1580, 1493, 1428, 1300, 1174.; ESI-MAR (m/z): calculada C21H17NO2 (M+Na+): 338,1151; encontrada: 338,1151; Sólido incolor; PF.: 162 ºC. 5-(4-Metoxibenzil)fenantridin-6(5H)-ona (1f) RMN 1H (200 MHz, CDCl3): δ 3,77 (s, 3H), 5,62 (s, 2H), 6,86 (d, J=6Hz, 2H), 7,21-7,39 (m, 5H), 7,63 (t, J= 8Hz, 1H), 7,80 (t, J=8Hz, 1H), 8,32 (dd, J= 2Hz, J= 2Hz, 2H), 8,66 (d, J=6Hz, 1H); RMN 13C (50 MHz, CDCl3): δ 46,1; 55,4; 114,4; 114,8; 116,2; 119,7; 121,8; 122,6; 123,4; 125,7; 128,0; 128,1; 128,8; 129,3; 129,6; 132,8; 134,0; 137,5; 158,9; 162,0; IV (KBr, cm1): 3068, 3033, 2960, 2939, 2834, 1660, 1608, 1513, 1438, 1330, 1247, 1027; Sólido bege, PF.: 122 ºC (recristalizado em etanol), Lit.: 136-137 ºC.67 5-(Piperonil)fenantridin-6(5H)-ona (1g) RMN 1H (200 MHz, CDCl3): δ 5,61 (s, 2H), 5,92 (s, 2H), 6,81 (s, 3H), 7,40 (m, 3H), 7,65 (t, J=8Hz, 1H), 7,82 (t, J=8HZ, 1H), 8,30 (d, J=8Hz, 2H), 8,69 (d, J=8Hz, 1H); RMN 13C (50 MHz, CDCl3): δ 46,.3; 101,1; 107,4; 108,5; 116,0; 119,6; 120,0; 121,8; 122,7; 123,4; 125,5; 128,1; 129,2; 129,6; 130,6; 132,8; 133,9; 137,4; 146,8; 148,2; 161,9; IV (KBr, cm-1): 3074, 3025, 2942, 2893,1644, 1608, 1491, 1438; ESI-MAR (m/z): calculada C21H15NO3 (M+Na+): 352,0944; encontrada: 352,0939.; Sólido incolor; PF.: 160 ºC. 5-(Piperonil)-2-metilfenantridin-6(5H)-ona (1h) RMN 1H (200 MHz, CDCl3): δ 2,36 (s, 3H), 5,46 (s, 2H), 5,80 (s, 2H), 6,66 (s, 3H), 7,14 (s, 2H), 7,52 (t, J=8Hz, 1H), 7,70 (t, J=8Hz, 1H), 7,99 (s, 1H), 8,23 (d, J=8Hz, 1H), 8,55 (d, J=8Hz, 1H); RMN 13C (50 MHz, CDCl3): δ 21,1; 46,3; 101,2; 107,4; 108,6; 116,0; 119,5; 120,0; 121,8; 123,6; 125,7; 128,0; 129,3; 130,7; 130,8; 132,2; 132,7; 133,9; 135,3; 146,9; 148,3; 161,9; IV(KBr, cm-1): 3070, 3034, 2949, 2912, 2851, 1655, 1608, 1581, 1497, 1449, 1251; ESI-MAR (m/z): calculada C22H17NO3 (M+Na+): 366,1100; encontrada: 366,1095.; Sólido incolor; PF.: 137 ºC. 5-(Piperonil)-2-fluorfenantridin-6(5H)-ona (1i) RMN 1H (200 MHz, CDCl3): δ 5,54 (s, 2H), 5,89 (s, 2H), 6,73 (s, 3H), 7,11 (m , 1H), 7,29 (m, 1H), 7,64 (t, J=8Hz,1H), 7,79 (t, J=8Hz, 1H), 7,89 (d, J=8Hz, 1H), 8,15 (d, J=8Hz, 1H), 8,58 (d, J=8Hz,1H); RMN 13C (50 MHz, CDCl3): δ 46,6; 101,2; 107,4; 108,7; 109,2; 109,7; 116,7; 117,2; 117,6; 117,7; 120,0; 122,0; 125,8; 128,9; 129,5; 130,4; 133,0; 133,1; 147,0; 148,4; 161,6; IV(KBr, cm-1): 3059, 3034, 2949, 2912, 1656, 1608, 1497, 1449, 1250; ESI-Mar (m/z): calculada C21H14FNO3 (M+Na+): 370,0849; encontrada: 370,0848; Sólido incolor; PF.: 130 ºC. 5-(Piperonil)-2-metoxifenantridin-6(5H)-ona (1j) RMN 1H (300 MHz, CDCl3): δ 3,86 (s, 3H), 5,51 (s, 2H), 5,86 (s, 2H), 6,72 (s, 3H), 7,0 (d, J=6Hz, 1H), 7,24 (d, J=6Hz, 1H), 7,70 (m, 3H), 8.21 (d, J=6Hz, 1H), 8,60 (d, J=6Hz, 1H); RMN 13C (75 MHz, CDCl3): δ 46,4; 55,8; 101,2; 107,4; 108,6; 116,6; 117,2; 119,9; 120,6; 121,9; 125,8; 128,3; 129,4; 130,7; 131,6; 132,7; 133,6; 146,9; 148,3; 155,2; 161,5; IV (KBr, cm-1): 2997, 2959, 2920, 2837,1651, 1607, 1583, 1408, 1249.; ESI-MAR (m/z): calculado C21H15NO3 (M+Na+): 382,1049; encontrada: 382,1040; Sólido incolor; PF.: 135 ºC. 5-Benzilbenzo[c][1,8]naftiridin-6(5H)-ona (1k) RMN 1H (200 MHz, CDCl3): δ 5,91 (s, 2H), 7,27 (d, J=6Hz, 4H), 7,52 (d, J=6Hz, 2H), 7,64 (t, J= 8Hz, 1H), 7,79(t, J=8Hz,1 H), 8,20 (d, J=10 Hz, 1H), 8,57 (m, 3H); RMN 13C (50 MHz, CDCl3): δ 44,7; 115,1; 118,6; 121,9; 126,1; 127,2; 128,4; 128,7; 128,9; 129,3; 131,6; 132,1; 133,0; 138,1; 148.3; 148,6; 162,4; IV (KBr): 3060, 3026, 2969, 1648, 1608, 1596, 1477, 1426, 1337, 1079; ESI-MAR (m/z): calculada C19H14N2O (M+Na+): 309,0098; encontrada: 309,0096; Sólido incolor; PF.: 147 ºC (recristalizado em hexano). 5-(4-Metoxibenzil)benzo[c][1,8]naftiridin-6(5H)-ona (1l) RMN1H (200 MHz, CDCl3): δ 3,75 (s, 3H), 5,53 (s, 2H), 6,81 (d, J=8Hz, 2H), 7,22 (m, 3H), 7,66 (t, J=8Hz, 1H), 7,84 (t, J=8Hz, 1H), 8,33 (d, J=8Hz, 1H), 8,47 (d, J=8Hz, 1H), 8,57 (d, J=8Hz, 1H), 9,46 (s, 1H); RMN 13C (50 MHz, CDCl3): δ 45,8; 55,4; 110,1; 114,5; 115,4; 121,2; 125,8; 127,7; 128,1; 129,3; 129,4; 132,2; 133,5; 143,1; 145,6; 149,0; 159,2; 161,9; IV (KBr, cm-1): 3057, 2997, 2932, 2960, 2837, 1647, 1601, 1577, 1515, 1326, 1243, 1118; ESI-MAR (m/z): calculada C20H16N2O2 (M+H+): 317,1284; encontrada: 317,1287; Sólido incolor; PF.: 195 ºC (recristalizado em isopropanol). 5-Benzilbenzo[c][1,6]naftiridin-6(5H)-ona (1m) RMN 1H (200 MHz, CDCl3): δ 5,52 (s, 2H), 7,07 (d, J=6Hz, 1H), 7.18 (m, 5H), 7,60 (t, J=8Hz, 1H), 7,77 (t, J=8Hz, 1H), 8,31 (d, J=8Hz, 1H), 8,42 (d, J=4Hz,1H), 8,54 (d, J=8Hz,1H), 9,4 (s,1H); RMN 13C (50 MHz, CDCl3): δ 46,3; 110,7; 115,4; 121,3; 125,7; 126,7; 127,8; 129,1; 129,2; 129,.5; 132,.1; 133,6; 135,6; 143,2; 145,4; 148,9; 161,9; IV(KBr, cm-1): 3066, 2962, 1654, 1600, 1577, 1495, 1414, 1313; ESI-MAR (m/z): calculada C19H14N2O (M+H+): 287,1178; encontrada: 287,1174; Sólido incolor; PF.: 158 ºC (recristalizado em isopropanol). 5-(4-Metoxibenzil)benzo[c][1,6]naftiridin-6(5H)-ona (1n) RMN 1H (200 MHz, CDCl3): δ 3,75 (s, 3H), 5,53 (s, 2H), 6,81 (d, J=8Hz, 2H), 7,22 (m, 3H), 7,66 (t, J=8Hz, 1H), 7,84 (t, J=8Hz, 1H), 8,33 (d, J=8Hz, 1H), 8,47 (d, J=8Hz, 1H), 8,57 (d, J=8Hz, 1H), 9,46 (s, 1H); RMN 13C (50 MHz, CDCl3): δ 45,8; 55,4; 110,1; 114,5; 115,4; 121,2; 125,8; 127,7; 128,1; 129,3; 129,4; 132,2; 133,5; 143,1; 145,6; 149,0; 159,2; 161,9; IV (KBr, cm-1): 3057, 2997, 2932, 2960, 2837, 1647, 1601, 1577, 1515, 1326, 1243, 1118; ESI-MAR (m/z): calculada C20H16N2O2 (M+H): 317,1284; encontrada: 317,1287; Sólido incolor; PF.: 195 ºC (recristalizado em isopropanol). 5,6-Diidropirido[3,2,1-de]fenantridin-8(4H)-ona (1q) RMN 1H (200 MHz, CDCl3): δ 2,02 (m, 2H), 2,90 (t, J=6Hz, 2H), 4,21 (t, J=6Hz, 2H), 7,16 (m, 2H), 7,46 (t, J=8Hz, 1H), 7,63 (t, J=10Hz, 1H), 7,98 (d, J=8Hz, 1H), 8,12 (d, J=8Hz, 1H), 8,42 (d, J=6Hz, 1H).; RMN 13C (50 MHz, CDCl3): δ 20,9; 28,4; 42,9; 119,3; 121,4; 121,9; 122,1; 125,5; 125,7; 127,9; 128,7; 129,6; 132,4; 133,8; 134,7; 161,3; IV (KBr, cm1): 3071, 2950, 2898, 1645, 1587, 1500, 1483, 1417, 1357, 1286 ; Sólido bege; PF.:183 ºC, Lit.: 180-182 ºC.27 5-Fenilfenantridin-6(5H)-ona (1r) RMN 1H (200 MHz, CDCl3): δ 6,62 (m, 1H), 7,24 (m, 4H), 7,53 (m , 4H), 7,73 (t, J=8Hz, 1H), 8,24 (m, 2H), 8,47 (d, J=8Hz, 1H); RMN 13C (50 MHz, CDCl3): δ 117,2; 119,2; 121,9; 122,8; 123,2; 126,1; 128,3; 128,9; 129,2; 129,3; 130,3; 133,0; 134,2; 138,5; 139,4; 161,9; RMN 13C (125 MHz, CDCl3): δ 117,24; 119,24; 122,01; 122,88; 123,21; 126,06; 128,36; 129,01; 129,24; 129,30; 129,32; 130,43; 133,06; 134,22; 138,49; 139,37; 161,94; O espectro de RMN de 13C em campo maior permitiu a resolução de dois sinais sobrepostos em 129,3ppm; IV (KBr, cm1): 3055, 1655, 1603, 1582, 1494, 1454, 1320; Sólido incolor, PF.: 136 ºC (recristalizado em isopropanol), Lit.: 116-117 ºC.82 5-Fenilbenzo[c][1,8]naftiridin-6(5H)-ona (1s) RMN 1H (200 MHz, CDCl3): δ 7,28 (m, 1H), 7,37 (d, 2H), 7,59 (d, J=8Hz, 2H), 7,87 (t, J=8Hz, 1H), 8,29 (d, J=8Hz, 1H), 8,43 (d, J=6Hz, 1H), 8,57 (d, J=10 Hz, 2H); RMN 13C (50 MHz, CDCl3): δ 114,7; 118,6; 121,9; 126,2; 128,4; 129,0; 129,1; 129,4; 129,5; 131,3; 132,4; 133,2; 137,8; 148,9; 149,8; 162,9; IV (KBr, cm-1): 3056, 2921, 2854, 1658,1583, 1492, 1415, 1324, 1284; ESI-MAR (m/z): calculado C18H12N2O (M+Na): 295,0841; encontrado: 295,0844: Sólido amarelo claro; PF.: 205 ºC.
CONCLUSÃO Neste trabalho utilizamos uma metodologia simples e eficiente para a funcionalização de uma ligação Csp2-H aromática via a reação de arilação direta intramolecular catalisada por paládio(0). Diversos derivados de fenantridinonas foram obtidos com rendimentos de bons a excelentes (61 - 97%). Dos produtos obtidos, doze são inéditos na literatura (1c-e, g-n, s). Todas as fenantridinonas deste estudo mostraram emissão de fluorescência. Desta forma, exemplos representativos das fenantridinonas foram caracterizados por espectroscopia de absorção, de excitação e de emissão. A emissão dual do composto 1r foi caracterizada e foi proposto, para o composto inédito 1s, um efeito equivalente para explicar a emissão estendida de baixa intensidade em comprimento de onda maior em relação a banda de emissão principal.
MATERIAL SUPLEMENTAR O material suplementar encontra-se disponível em http://quimicanova.sbq.org.br, na forma de arquivo PDF, com acesso livre, e apresenta os espectros de RMN de 1H e 13C e infravermelho, espectrômetros de massas de alta resolução dos compostos sintetizados e a caracterização dos compostos 1f, 1q, 1r, e 1s por espectroscopia de absorção, excitação e emissão.
AGRADECIMENTOS O SJG agradece ao CNPq e à FAPERJ pela assistência financeira e ao laboratório Labmass (IPPN-UFRJ) pelas análises de massa de alta resolução. RLC agradece à CAPES pela bolsa de estudos e DAFS agradece ao CNPq pela bolsa de IC.
REFERÊNCIAS 1. Nair, J. J.; Rarova, L.; Strnad, M.; Bastida, J.; van Staden, J.; Bioorg. Med. Chem. Lett. 2012, 22, 6195; Nair, J. J.; Aremu, A. O.; van Staden, J.; J. Ethnopharmacol. 2011, 137, 1102. DOI: http://dx.doi.org/10.1016/j.bmcl.2012.08.005 PMID: 22921081 2. Bozkurt Sarikaya, B.; Zencir, S.; Unver Somer, N.; Kaya, G. I.; Onur, M. A.; Bastida, J.; Berenyi, A.; Zupko, I.; Topcu, Z.; Rec. Nat. Prod. 2012, 6, 381. 3. Kaya, G. I.; Sarikaya, B.; Onur, M. A.; Somer, N. U.; Viladomat, F.; Codina, C.; Bastida, J.; Lauinger, I. L.; Kaiser, M.; Tasdemir, D.; Phytochem. Lett. 2011, 4, 301; Ohta, S.; Kimoto, S.; Chem. Pharm. Bull. 1976, 24, 2977. DOI: http://dx.doi.org/10.1016/j.phytol.2011.05.008 4. Doepke, W.; Lam, H. P.; Gruendemann, E.; Bartoszek, M.; Flatau, S.; Pharmazie 1995, 50, 511; Doepke, W.; Pham, L. H.; Gruendemann, E.; Bartoszek, M.; Flatau, S.; Planta Med. 1995, 61, 564. 5. Lotter, M.; Bracher, F.; Sci. Pharm. 2009, 77, 1; Bijeire, L.; Legentil, L.; Bastide, J.; Darro, F.; Rochart, C.; Delfourne, E.; Eur. J. Org. Chem. 2004, 1891; Nilar; Sidebottom, P. J.; Carte, B. K.; Butler, M. S.; J. Nat. Prod. 2002, 65, 1198. DOI: http://dx.doi.org/10.3797/scipharm.0901-14 6. Wang, B. L.; Qian, H.; Yiu, S. M.; Sun, J. W.; Zhu, G. Y.; Eur. J. Med. Chem. 2014, 71, 366; Zhang, S.; Liao, L. Y.; Zhang, F.; Duan, X. F.; J. Org. Chem. 2013, 78, 2720; Ferraris, D. V.; J. Med. Chem. 2010, 53, 4561. DOI: http://dx.doi.org/10.1016/j.ejmech.2013.10.062 PMID: 24361480 7. Zhou, W.; Dai, Z.; Chen, Y.; Yuan, Z.; Med. Chem. Res. 2013, 22, 278; Thai, K.-M.; Bui, Q.-H.; Tran, T.-D.; Huynh, T.-N.-P.; Molecules 2012, 17, 5690; Kurtzberg, L. S.; Roth, S.; Krumbholz, R.; Crawford, J.; Bormann, C.; Dunham, S.; Yao, M.; Rouleau, C.; Bagley, R. G.; Yu, X.-J.; Wang, F.; Schmid, S. M.; LaVoie, E. J.; Teicher, B. A.; Clin. Cancer Res. 2011, 17, 2777; Sooryakumar, D.; Dexheimer, T. S.; Teicher, B. A.; Pommier, Y.; Mol. Cancer Ther. 2011, 10, 1490; Pommier, Y.; Leo, E.; Zhang, H.-L.; Marchand, C.; Chem. Biol. 2010, 17, 421; Teicher, B. A.; Biochem. Pharmacol. 2008, 75, 1262; Ruchelman, A. L.; Houghton, P. J.; Zhou, N.; Liu, A.; Liu, L. F.; LaVoie, E. J.; J. Med. Chem. 2005, 48, 792. DOI: http://dx.doi.org/10.1007/s00044-012-0034-x 8. Aoyama, H.; Sugita, K.; Nakamura, M.; Aoyama, A.; Salim, M. T. A.; Okamoto, M.; Baba, M.; Hashimoto, Y.; Bioorg. Med. Chem. 2011, 19, 2675; Nakamura, M.; Aoyama, A.; Salim, M. T. A.; Okamoto, M.; Baba, M.; Miyachi, H.; Hashimoto, Y.; Aoyama, H.; Bioorg. Med. Chem. 2010, 18, 2402. DOI: http://dx.doi.org/10.1016/j.bmc.2011.03.002 PMID: 21458278 9. Matsumoto, K.; Choshi, T.; Hourai, M.; Zamami, Y.; Sasaki, K.; Abe, T.; Ishikura, M.; Hatae, N.; Iwamura, T.; Tohyama, S.; Nobuhiro, J.; Hibino, S.; Bioorg. Med. Chem. Lett. 2012, 22, 4762; Yapi, A.-D.; Desbois, N.; Chezal, J.-M.; Chavignon, O.; Teulade, J.-C.; Valentin, A.; Blache, Y. Eur. J. Med. Chem. 2010, 45, 2854; Cedron, J. C.; Gutierrez, D.; Flores, N.; Ravelo, A. G.; Estevez-Braun, A.; Bioorg. Med. Chem. 2010, 18, 4694; Nyangulu, J. M.; Hargreaves, S. L.; Sharples, S. L.; Mackay, S. P.; Waigh, R. D.; Duval, O.; Mberu, E. K.; Watkins, W. M.; Bioorg. Med. Chem. Lett. 2005, 15, 2007. DOI: http://dx.doi.org/10.1016/j.bmcl.2012.05.064 PMID: 22727670 10. Dubost, E.; Dumas, N.; Fossey, C.; Magnelli, R.; Butt-Gueulle, S.; Ballandonne, C.; Caignard, D. H.; Dulin, F.; Santos, J. S. D.; Millet, P.; Charnay, Y.; Rault, S.; Cailly, T.; Fabis, F.; J. Med. Chem. 2012, 55, 9693. DOI: http://dx.doi.org/10.1021/jm300943r PMID: 23102207 11. Jou, J. H.; Li, C. J.; Shen, S. M.; Peng, S. H.; Chen, Y. L.; Jou, Y. C.; Hong, J. H.; Chin, C. L.; Shyue, J. J.; Chen, S. P.; Li, J. Y.; Wang, P. H.; Chen, C. C.; J. Mater. Chem. C 2013, 1, 4201. DOI: http://dx.doi.org/10.1039/c3tc30606b 12. Chen, Y.; Liu, B.; Liu, X.; Yang, Y.; Ling, Y.; Jia, Y.; Org. Process Res. Dev. 2014, 18, 1589; Smith, P. A. S.; J. Am. Chem. Soc. 1948, 70, 320. DOI: http://dx.doi.org/10.1021/op500269j 13. Ruediger, E. H.; Gandhi, S. S.; Gibson, M. S.; Farcasiu, D.; Uncuta, C.; Can. J. Chem. 1986, 64, 577. DOI: http://dx.doi.org/10.1139/v86-093 14. Naidu, K. M.; Nagesh, H. N.; Singh, M.; Sriram, D.; Yogeeswari, P.; Gowri Chandra Sekhar, K. V.; Eur. J. Med. Chem. 2015, 92, 415; Nagesh, H. N.; Naidu, K. M.; Rao, D. H.; Sridevi, J. P.; Sriram, D.; Yogeeswari, P.; Chandra Sekhar, K. V. G.; Bioorg. Med. Chem. Lett. 2013, 23, 6805; Meseroll, L. M. N.; McKee, J. R.; Zanger, M.; Synth. Commun. 2011, 41, 2557; Horning, E. C.; Stromberg, V. I.; Lloyd, H. A.; J. Am. Chem. Soc. 1952, 74, 5153. DOI: http://dx.doi.org/10.1016/j.ejmech.2014.12.037 PMID: 25590862 15. Oyster, L.; Adkins, H.; J. Am. Chem. Soc. 1921, 43, 208. DOI: http://dx.doi.org/10.1021/ja01434a030 16. Pictet, A.; Hubert, A.; Chem. Ber. 1896, 29, 1182. DOI: http://dx.doi.org/10.1002/cber.18960290206 17. Morgan, G. T.; Walls, L. P.; J. Chem. Soc. 1931, 2447. DOI: http://dx.doi.org/10.1039/JR9310002447 18. Lion, C.; Boukou-Poba, J. P.; Charvy, C.; Bull. Soc. Chim. Belg. 1991, 100, 169; DeRuiter, J.; Swearingen, B. E.; Wandrekar, V.; Mayfield, C. A.; J. Med. Chem. 1989, 32, 1033. DOI: http://dx.doi.org/10.1002/bscb.19911000210 19. Hollingsworth, B. L.; Petrow, U.; J. Chem. Soc. 1961, 3771. DOI: http://dx.doi.org/10.1039/jr9610003771 20. Mosby, W. L. J. Am. Chem. Soc. 1954, 76, 936; Taylor, E. C., Jr.; Kalenda, N. W.; J. Am. Chem. Soc. 1954, 76, 1699. DOI: http://dx.doi.org/10.1021/ja01632a103 21. Migachev, G. I.; Zh. Org. Khim. 1979, 15, 567; Muth, C. W.; Elkins, J. R.; DeMatte, M. L.; Chiang, S. T.; J. Org. Chem. 1967, 32, 1106. 22. Hey, D. H.; Jones, G. H.; Perkins, M. J.; J. Chem. Soc., Perkin Trans. 1 1972, 118; Perkins, M. J.; Hey, D. H.; Jones, G. H.; J. Chem. Soc. D. 1971, 998; Brown, P. M.; Dewar, P. S.; Forrester, A. R.; Ingram, A. S.; Thomson, R. H.; J. Chem. Soc. D 1970, 849. DOI: http://dx.doi.org/10.1039/p19720000118 23. Lister, T.; Prager, R. H.; Tsaconas, M.; Wilkinson, K. L.; Aust. J. Chem. 2003, 56, 913. DOI: http://dx.doi.org/10.1071/CH03044 24. Read, M. L.; Gundersen, L.-L.; J. Org. Chem. 2013, 78, 1311; Fujita, R.; Yoshisuji, T.; Wakayanagi, S.; Wakamatsu, H.; Matsuzaki, H.; Chem. Pharm. Bull. 2006, 54, 204; Fujita, R.; Wakayanagi, S.; Wakamatsu, H.; Matsuzaki, H.; Chem. Pharm. Bull. 2006, 54, 209; Padwa, A.; Brodney, M. A.; Liu, B.; Satake, K.; Wu, T.; J. Org. Chem. 1999, 64, 3595. DOI: http://dx.doi.org/10.1021/jo3027033 PMID: 23305184 25. Al-Jalal, N. A.; Ibrahim, M. R.; Al-Awadi, N. A.; Elnagdi, M. H.; Molecules 2011, 16, 10256. DOI: http://dx.doi.org/10.3390/molecules161210256 PMID: 22158592 26. Datta, I.; Das, T. K.; Ghosh, S.; Tetrahedron 1990, 46, 6821. DOI: http://dx.doi.org/10.1016/S0040-4020(01)87869-8 27. Hoshino, O.; Ogasawara, H.; Hirokawa, A.; Umezawa, B.; Chem. Lett. 1988, 1767. DOI: http://dx.doi.org/10.1246/cl.1988.1767 28. Grimshaw, J.; De Silva, A. P.; J. Chem. Soc., Chem. Commun. 1980, 302; Thyagarajan, B. S.; Kharasch, N.; Lewis, H. B.; Wolf, W.; Chem. Commun. 1967, 614. DOI: http://dx.doi.org/10.1039/c39800000302 29. Li, B.; Han, B.; Shi, Z.-j.; Ren, Y.-w.; Lu, S.-c.; Zhang, W.; Tetrahedron Lett. 2010, 51, 3748. DOI: http://dx.doi.org/10.1016/j.tetlet.2010.05.037 30. Paterson, T. M.; Smalley, R. K.; Suschitzky, H.; Barker, A. J.; J. Chem. Soc., Perkin Trans. 1 1980, 633. DOI: http://dx.doi.org/10.1039/p19800000633 31. Al-Awadi, N. A.; George, B. J.; Dib, H. H.; Ibrahim, M. R.; Ibrahim, Y. A.; El-Dusouqui, O. M. E.; Tetrahedron 2005, 61, 8257. DOI: http://dx.doi.org/10.1016/j.tet.2004.10.058 32. Hey, D. H.; Perkins, M. J.; Jones, G. H.; J. Chem. Soc., Perkin Trans. 1 1972, 113; Nagarajan, K.; Pillai, P. M.; Bhute, R. S. Indian J. Chem. 1969, 7, 848; Heacock, R. A.; Hey, D. H.; J. Chem. Soc. 1952, 1508. DOI: http://dx.doi.org/10.1039/p19720000113 33. Hoshino, Y.; Hoshino, O.; Heterocycles 2005, 66, 659; Crich, D.; Hao, X.; Lucas, M. A.; Org. Lett. 1999, 1, 269; Crich, D.; Hwang, J.-T.; J. Org. Chem. 1998, 63, 2765; Bowman, W. R.; Heaney, H.; Jordan, B. M.; Tetrahedron 1991, 47, 10119; Motherwell, W. B.; Pennell, A. M. K.; J. Chem. Soc., Chem. Commun. 1991, 877. DOI: http://dx.doi.org/10.3987/COM-05-S(K)64 34. Ohno, H.; Iwasaki, H.; Eguchi, T.; Tanaka, T.; Chem. Commun. 2004, 2228. DOI: http://dx.doi.org/10.1039/b410457a 35. Curran, D. P.; Keller, A. I.; J. Am. Chem. Soc. 2006, 128, 13706. DOI: http://dx.doi.org/10.1021/ja066077q PMID: 17044696 36. Rousseau, G.; Robert, F.; Landais, Y.; Chem. - Eur. J. 2009, 15, 11160; Iwasaki, H.; Eguchi, T.; Tsutsui, N.; Ohno, H.; Tanaka, T.; J. Org. Chem. 2008, 73, 7145. DOI: http://dx.doi.org/10.1002/chem.200901434 37. Yuan, M.; Chen, L.; Wang, J.; Chen, S.; Wang, K.; Xue, Y.; Yao, G.; Luo, Z.; Zhang, Y.; Org. Lett. 2015, 17, 346. DOI: http://dx.doi.org/10.1021/ol503459s PMID: 25569786 38. De, S.; Mishra, S.; Kakde, B. N.; Dey, D.; Bisai, A.; J. Org. Chem. 2013, 78, 7823; De, S.; Ghosh, S.; Bhunia, S.; Sheikh, J. A.; Bisai, A.; Org. Lett. 2012, 14, 4466. DOI: http://dx.doi.org/10.1021/jo400890k PMID: 23924301 39. Bhakuni, B. S.; Shrimali, K.; Kumar, A.; Kumar, S.; Org. Synth. 2013, 90, 164; Bhakuni, B. S.; Kumar, A.; Balkrishna, S. J.; Sheikh, J. A.; Konar, S.; Kumar, S.; Org. Lett. 2012, 14, 2838. DOI: http://dx.doi.org/10.15227/orgsyn.090.0164 40. Roman, D. S.; Takahashi, Y.; Charette, A. B.; Org. Lett. 2011, 13, 3242. DOI: http://dx.doi.org/10.1021/ol201160s PMID: 21568277 41. Gui, Q.; Yang, Z.; Chen, X.; Liu, J.; Tan, Z.; Guo, R.; Yu, W.; Synlett 2013, 24, 1016. DOI: http://dx.doi.org/10.1055/s-0032-1316898 42. Hua, S.-K.; Hu, Q.-P.; Ren, J.; Zeng, B.-B.; Synthesis 2013, 45, 518. DOI: http://dx.doi.org/10.1055/s-0032-1316841 43. Chen, Y.-F.; Wu, Y.-S.; Jhan, Y.-H.; Hsieh, J.-C.; Org. Chem. Front. 2014, 1, 253. DOI: http://dx.doi.org/10.1039/c3qo00082f 44. Banwell, M. G.; Lupton, D. W.; Ma, X.; Renner, J.; Sydnes, M. O.; Org. Lett. 2004, 6, 2741. DOI: http://dx.doi.org/10.1021/ol0490375 PMID: 15281758 45. Ames, D. E.; Opalko, A.; Tetrahedron 1984, 40, 1919. DOI: http://dx.doi.org/10.1016/S0040-4020(01)91149-4 46. Alguns exemplos de revisões da literatura de relevância que contemplam aspectos da reação de arilação direta: Hussain, I.; Singh, T.; Adv. Synth. Catal. 2014, 356, 1661; Yamaguchi, J.; Yamaguchi, A. D.; Itami, K.; Angew. Chem. Int. Ed. 2012, 51, 8960; de Azambuja, F.; Correia, C. R. D.; Quim. Nova 2011, 34, 1779; Lyons, T. W.; Sanford, M. S.; Chem. Rev. 2010, 110, 1147; Chiusoli, G. P.; Catellani, M.; Costa, M.; Motti, E.; Della Ca, N.; Maestri, G.; Coord. Chem. Rev. 2010, 254, 456; Chen, X.; Engle, K. M.; Wang, D. H.; Yu, J. Q.; Angew. Chem., Int. Ed. 2009, 48, 5094; Alberico, D.; Scott, M. E.; Lautens, M.; Chem. Rev. 2007, , 107, 174. DOI: http://dx.doi.org/10.1002/adsc.201400178 47. Lapointe, D.; Fagnou, K.; Chem. Lett. 2010, 39, 1119; Livendahl, M.; Echavarren, A. M.; Isr. J. Chem. 2010, 50, 630. 48. Torres, J. C.; Pinto, A. C.; Garden, S. J.; Tetrahedron 2004, 60, 9889. DOI: http://dx.doi.org/10.1016/j.tet.2004.08.030 49. Zhang, G.; Zhao, X.; Yan, Y.; Ding, C.; Eur. J. Org. Chem. 2012, 2012, 669; Zhang, L.; Geng, M.; Teng, P.; Zhao, D.; Lu, X.; Li, J.-X.; Ultrason. Sonochem. 2012, 19, 250; Majumdar, K. C.; De, N.; Chakravorty, S.; Synth. Commun. 2011, 41, 121; Harayama, T.; Nagura, C.; Miyagoe, T.; Kawata, Y.; Abe, H.; Takeuchi, Y.; Heterocycles 2010, 81, 2609; Harayama, T.; Kawata, Y.; Nagura, C.; Sato, T.; Miyagoe, T.; Abe, H.; Takeuchi, Y.; Tetrahedron Lett. 2005, 46, 6091; Parisien, M.; Valette, D.; Fagnou, K.; J. Org. Chem. 2005, 70, 7578; Campeau, L.-C.; Thansandote, P.; Fagnou, K.; Org. Lett. 2005, 7, 1857; Harayama, T.; Akiyama, T.; Akamatsu, H.; Kawano, K.; Abe, H.; Takeuchi, Y.; Synthesis 2001, 444; Harayama, T.; Akiyama, T.; Nakano, Y.; Chem. Pharm. Bull. 1997, 45, 1723; Harayama, T.; Akiyama, T.; Kawano, K.; Chem. Pharm. Bull. 1996, 44, 1634. DOI: http://dx.doi.org/10.1021/jo201413b 50. Bernini, R.; Cacchi, S.; Fabrizi, G.; Sferrazza, A.; Synthesis 2008, 729. DOI: http://dx.doi.org/10.1055/s-2008-1032169 51. Garden, S. J.; Torres, J. C.; Pinto, A. C.; J. Braz. Chem. Soc. 2000, 11, 441. DOI: http://dx.doi.org/10.1590/S0103-50532000000500002 52. Han, P.; Zhou, J.; Zhang, C.-C.; Chen, K.; Du, Z.-T.; Heterocycles 2014, 89, 2151. DOI: http://dx.doi.org/10.3987/COM-14-13051 53. Nishioka, H.; Shoujiguchi, Y.; Abe, H.; Takeuchi, Y.; Harayama, T.; Heterocycles 2004, 64, 463; Harayama, T.; Toko, H.; Kubota, K.; Nishioka, H.; Abe, H.; Takeuchi, Y.; Heterocycles 2002, 58, 175; Harayama, T.; Hori, A.; Nakano, Y.; Akiyama, T.; Abe, H.; Takeuchi, Y.; Heterocycles 2002, 58, 159; Harayama, T.; Akiyama, T.; Nakano, Y.; Nishioka, H.; Abe, H.; Takeuchi, Y.; Chem. Pharm. Bull. 2002, 50, 519. DOI: http://dx.doi.org/10.3987/COM-04-S(P)11 54. Nervig, C. S.; Waller, P. J.; Kalyani, D.; Org. Lett. 2012, 14, 4838. DOI: http://dx.doi.org/10.1021/ol302166n PMID: 22974229 55. Hagelin, H.; Oslob, J. D.; Akermark, B.; Chem. - Eur. J. 1999, 5, 2413. DOI: http://dx.doi.org/10.1002/(SICI)1521-3765(19990802)5:8<2413::AID-CHEM2413>3.0.CO;2-3 56. Yeung, C. S.; Zhao, X.; Borduas, N.; Dong, V. M.; Chem. Sci. 2010, 1, 331; Borduas, N.; Lough, A. J.; Dong, V. M.; Inorg. Chim. Acta 2011, 369, 247. DOI: http://dx.doi.org/10.1039/c0sc00231c 57. Ishida, N.; Nakanishi, Y.; Moriya, T.; Murakami, M.; Chem. Lett. 2011, 40, 1047. DOI: http://dx.doi.org/10.1246/cl.2011.1047 58. Laha, J. K.; Jethava, K. P.; Dayal, N.; J. Org. Chem. 2014, 79, 8010. DOI: http://dx.doi.org/10.1021/jo5011334 PMID: 25121579 59. Liang, Z.; Zhang, J.; Liu, Z.; Wang, K.; Zhang, Y.; Tetrahedron 2013, 69, 6519; Lu, S.-M.; Alper, H.; J. Am. Chem. Soc. 2005, 127, 14776. DOI: http://dx.doi.org/10.1016/j.tet.2013.05.025 60. Liang, D.; Hu, Z.; Peng, J.; Huang, J.; Zhu, Q.; Chem. Commun. 2013, 49, 173. DOI: http://dx.doi.org/10.1039/C2CC36817J 61. Pizzotti, M.; Cenini, S.; Quici, S.; Tollari, S.; J. Chem. Soc., Perkin Trans. 2 1994, 913. DOI: http://dx.doi.org/10.1039/p29940000913 62. Albert, J.; D'Andrea, L.; Granell, J.; Zafrilla, J.; Font-Bardia, M.; Solans, X.; J. Organomet. Chem. 2007, 692, 4895. DOI: http://dx.doi.org/10.1016/j.jorganchem.2007.07.002 63. Zhou, Q. J.; Worm, K.; Dolle, R. E.; J. Org. Chem. 2004, 69, 5147. DOI: http://dx.doi.org/10.1021/jo049343w PMID: 15255755 64. Tanimoto, K.; Nakagawa, N.; Takeda, K.; Kirihata, M.; Tanimori, S.; Tetrahedron Lett. 2013, 54, 3712; Dubost, E.; Magnelli, R.; Cailly, T.; Legay, R.; Fabis, F.; Rault, S.; Tetrahedron 2010, 66, 5008; Cailly, T.; Fabis, F.; Rault, S.; Tetrahedron 2006, 62, 5862; Siddiqui, M. A.; Snieckus, V. Tetrahedron Lett. 1988, 29, 5463. DOI: http://dx.doi.org/10.1016/j.tetlet.2013.05.022 65. Rodriguez, N.; Goossen, L. J.; Chem. Soc. Rev. 2011, 40, 5030; Goossen, L. J.; Rodriguez, N.; Melzer, B.; Linder, C.; Deng, G.; Levy, L. M.; J. Am. Chem. Soc. 2007, 129, 4824; Goossen, L. J.; Deng, G.; Levy, L. M.; Science 2006, 313, 662. DOI: http://dx.doi.org/10.1039/c1cs15093f PMID: 21792454 66. Shen, Z.; Ni, Z.; Mo, S.; Wang, J.; Zhu, Y.; Chem. - Eur. J. 2012, 18, 4859. DOI: http://dx.doi.org/10.1002/chem.201103438 67. Lu, C.; Dubrovskiy, A. V.; Larock, R. C.; J. Org. Chem. 2012, 77, 8648. DOI: http://dx.doi.org/10.1021/jo3016192 PMID: 23013049 68. Yang, Y.; Huang, H.; Wu, L.; Yun, L.; Org. Biomol. Chem. 2014, 12, 5351. DOI: http://dx.doi.org/10.1039/c4ob00997e PMID: 24935256 69. Pimparkar, S.; Jeganmohan, M.; Chem. Commun. 2014, 50, 12116. DOI: http://dx.doi.org/10.1039/C4CC05252H 70. Donati, L.; Leproux, P.; Prost, E.; Michel, S.; Tillequin, F.; Gandon, V.; Poree, F.-H.; Chem. - Eur. J. 2011, 17, 12809. DOI: http://dx.doi.org/10.1002/chem.201101354 71. Furuta, T.; Kitamura, Y.; Hashimoto, A.; Fujii, S.; Tanaka, K.; Kan, T.; Org. Lett. 2007, 9, 183. DOI: http://dx.doi.org/10.1021/ol062599z PMID: 17217260 72. Ferraccioli, R.; Carenzi, D.; Motti, E.; Catellani, M.; J. Am. Chem. Soc. 2006, 128, 722. DOI: http://dx.doi.org/10.1021/ja0566127 PMID: 16417353 73. Wang, G.-W.; Yuan, T.-T.; Li, D.-D.; Angew. Chem. Int. Ed. 2011, 50, 1380. DOI: http://dx.doi.org/10.1002/anie.201005911 74. Baxter, E. W.; Reitz, A. B.; Reductive aminations of carbonyl compounds with borohydride and borane reducing agents, John Wiley & Sons, Inc.: New York, 2002; Vol. 59. 75. Mishra, S.; De, S.; Nakde, B. N.; Dey, D.; Bisai, A.; Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem. 2013, 52A, 1093. 76. Harayama, T.; Sato, T.; Hori, A.; Abe, H.; Takeuchi, Y.; Heterocycles 2005, 66, 527. DOI: http://dx.doi.org/10.3987/COM-05-S(K)2 77. Boonya-udtayan, S.; Yotapan, N.; Woo, C.; Bruns, C. J.; Ruchirawat, S.; Thasana, N.; Chem. - Asian J. 2010, 5, 2113. DOI: http://dx.doi.org/10.1002/asia.201000237 PMID: 20677319 78. Giese, B.; Kopping, B.; Gobel, T.; Dickhaut, J.; Thoma, G.; Kulicke, K. J.; Trach, F.; Radical cyclization reactions, John Wiley & Sons, Inc.: New Jersey, 1996; Vol. 48. 79. Abramovitch, R. A.; Shi, Q.; Heterocycles 1994, 37, 1463. DOI: http://dx.doi.org/10.3987/COM-93-S155 80. Duan, X. H.; Pei, C. H.; Ma, Y. J.; Sci. China, Ser. B: Chem. 2008, 51, 632; Demeter, A.; Berces, T.; Hinderberger, J.; Timari, G.; Photochem. Photobiol. Sci. 2003, 2, 273; Demeter, A.; Berces, T.; Zachariasse, K. A.; J. Phys. Chem. A 2001, 105, 4611. DOI: http://dx.doi.org/10.1007/s11425-008-0063-6 81. Brouwer, A. M.; Pure Appl. Chem. 2011, 83, 2213. 82. Eisch, J. J.; Kovacs, C. A.; Chobe, P.; J. Org. Chem. 1989, 54, 1275. DOI: http://dx.doi.org/10.1021/jo00267a011 |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access