
 |
 |
 |
 |
 |
Revisão
|
|
| Sínteses totais no Brasil empregando acoplamentos cruzados mediados por paládio# Total synthesis in Brazil employing palladium cross-coupling as key step |
|
Mauricio M. Victora,b,*; Gálber S. B. da Silvaa,b
aDepartamento de Química Orgânica, Instituto de Química, Universidade Federal da Bahia, 40170-115 Salvador - BA, Brasil Recebido em 25/11/2015 *e-mail: mmvictor@ufba.br This article aims to show the selected total syntheses of organic compounds conducted by Brazilian researchers, performed in Brazil, which have employed a palladium catalyzed cross-coupling reaction as key step. We intended to thus create an account, although restricted to the use of palladium, showing the evolution and current state of art of Synthetic Organic Chemistry in Brazil, which has advanced and accept the challenge of total synthesis of increasingly complex molecules, registering growth and maturity of this area among Brazilian researchers. INTRODUÇAO O desenvolvimento da síntese orgânica deu-se rapidamente na segunda metade do século XX, devido às descobertas de muitas novas reaçoes e metodologias sintéticas, dentre as quais aquelas que possibilitavam a formaçao da ligaçao C-C e aos avanços dos reagentes organometálicos preparados a partir dos metais de transiçao.1 Os compostos organometálicos sao definidos como aqueles que contenham pelo menos uma ligaçao entre um átomo de carbono de um composto orgânico e um metal, combinando aspectos das químicas inorgânica e orgânica. Assim, compostos como Li4(CH3)4, Ti(CH3)4, Fe(CO)5, ClMgC2H5, Pb(C2H5)4 e (CH3)3Ti(C2H3O2) sao descritos como organometálicos, porque em todos eles existem ligaçoes metal carbono (M-C).2 O primeiro composto organometálico foi obtido em 1830,3 com a síntese do sal de Zeise (K[PtCl3(C2H4)].H2O), descoberto por William Christopher Zeise,4 ao fazer reagir o PtCl4 com etanol. Esse sal foi muito estudado durante a segunda metade do século XIX porque os químicos nao conseguiam explicar sua estrutura, o que só ocorreu no século XX, com o advento da difraçao de Raios-X. Outro pioneiro para o desenvolvimento da química organometálica foi o inglês E. C. Frankland (1825-1899). Foi Frankland quem criou o termo "organometálico",5 além de ter contribuído com a síntese de vários compostos desta classe como, por exemplo, o Zn(CH3)2, Zn(C2H5)2, HgI(CH3), SnI2(C2H5)2 e o B(CH3)3. Os compostos organometálicos apresentam variadas propriedades químicas e físicas, podendo ser encontrados no estado sólido, líquido ou gasoso; uns sao estáveis, outros altamente inflamáveis, sendo que alguns apresentam elevada toxidade, principalmente os voláteis.6 No geral, essas propriedades variam devido ao tipo de ligaçao estabelecida entre o carbono e os metais da cadeia, que podem ser ligaçoes covalentes ou iônicas, mono ou multinucleadas (que abrangem dois ou mais átomos). Desde os anos 60, a pesquisa exploratória em compostos organometálicos tem sido dominada por estudos dos compostos do bloco "d". Com esta evoluçao, os metais de transiçao tornaram-se um instrumento importante na síntese orgânica, devido a sua capacidade de ativar substratos orgânicos e promover várias transformaçoes químicas. Contudo, mais recentemente, foi retomado o interesse na síntese exploratória e estudos físicos dos compostos organometálicos do grupo principal, tornando possível o surgimento de novas classes de compostos e o aumento do conhecimento da variedade e do tipo de ligaçao e reaçoes dos elementos do bloco "s" e bloco 'p".2 O desenvolvimento da área dos compostos de coordenaçao ampliou o espectro das substâncias investigadas para além dos organometálicos, aqueles onde havia uma ligaçao do tipo metal carbono (M-C): desenvolveu-se a química de complexos metálicos e complexos metalorgânicos. Estes compostos sao caracterizados pela presença de um átomo central (que pode ser metal ou nao) e de ligantes que estao ao seu redor, na esfera de coordenaçao, com os quais mantém uma interaçao química, mas nao através de uma ligaçao metal carbono. Um exemplo de complexo metálico seria [PdCl4]2-, com o átomo de paládio como átomo central e os cloretos como ligantes, enquanto o composto [Pd(MeCN)2Cl2] representa um complexo metalorgânico: há ligantes com carbono, mas a coordenaçao se dá através do heteroátomo presente no ligante. O progresso no estudo dos compostos organometálicos, complexos metálicos e complexos metalorgânicos levou ao desenvolvimento e aplicaçao industrial bem sucedida de um grande número de processos catalíticos. Muitos destes processos catalíticos necessitaram do emprego de compostos de coordenaçao de metais de transiçao como catalisadores.7 As principais razoes pelas quais os metais de transiçao contribuem tao essencialmente em catálise sao: a) capacidade de ligaçao: a capacidade de formar tanto ligaçoes σ como ligaçoes π com outros grupos; b) grande variedade de ligantes: elementos de transiçao formam facilmente ligaçoes químicas com quase todos os outros elementos e com muitas moléculas orgânicas; c) efeito do ligante: um ligante pode influenciar o comportamento de um catalisador de metal de transiçao variando o ambiente estérico e/ou eletrônico no sítio ativo; d) variabilidade de estado de oxidaçao e número de coordenaçao; e) capacidade para intercambiar prontamente entre os estados de oxidaçao durante a reaçao catalítica: metais de transiçao podem ser facilmente envolvidos em processos redox. Com isso, nota-se que os compostos de coordenaçao têm contribuído bastante para mudanças de paradigmas nas reaçoes químicas, especialmente no que se refere às estruturas e às ligaçoes químicas. Isso tem ocorrido desde a síntese do primeiro organometálico por Zeise, passando pelo esclarecimento das estruturas destes compostos, até o presente, com a utilizaçao de reaçoes envolvendo metais em sistemas biológicos e nos processos catalíticos de uma forma geral.8 Dentro desta importância atual, a química dos compostos de coordenaçao centrada sobre o átomo de paládio é, sem sombra de dúvida, uma das mais robustas e com maior amplitude de utilizaçao.9 Reaçoes como os acoplamentos de Mizoroki-Heck, Sonogashira, Negishi, Stille, Suzuki-Miyaura e Hiyama estao presentes em um enorme número de sínteses totais de moléculas complexas. Além disso, o agraciamento dos professores Richard F. Heck, Ei-ichi Negishi e Akira Suzuki com o prêmio Nobel de Química em 201010 mostra a relevância e importância da química de acoplamento cruzado centrada no átomo de paládio. Assim, esta revisao tem a pretensao de mostrar a maturidade da Química Orgânica Sintética brasileira, sob o ponto de vista da preparaçao de moléculas complexas, em território nacional,11 empregando reaçoes de acoplamento cruzado mediadas por catalisadores de paládio. Algumas poucas compilaçoes têm buscado este objetivo,12 e deste modo buscamos colaborar com a divulgaçao endógena de exemplos selecionados dos importantes avanços até agora alcançados pelos pesquisadores brasileiros e seus grupos de pesquisa.
SINTESES TOTAIS NO BRASIL EMPREGANDO REAÇOES MEDIADAS POR PALADIO Reaçao de Heck A reaçao de Heck, também conhecida como reaçao Mizoroki-Heck, é uma reaçao química organometálica, em que um grupo vinila ou arila substituído reage com um alceno, na presença de uma base e um catalisador de paládio (ou catalisador à base de paládio), para formar um novo alceno substituído (Esquema 1). A reaçao foi descoberta de forma independente pelos pesquisadores T. Mizoroki e R. F. Heck.13-15 O processo descoberto mostrou que halogenetos de arila, benzila e estirila, reagiam com compostos olefínicos, a temperaturas elevadas, na presença de uma base impedida do tipo amina e uma quantidade catalítica de Pd(0) para formar grupos aril, benzil, e olefinas substituídos com estirila. Em 2010, Heck ganhou o Prêmio Nobel de Química, que compartilhou com Ei-ichi Negishi e Akira Suzuki, pela descoberta e desenvolvimento desta reaçao. A reaçao de Heck é de grande importância, pois permite fazer reaçoes de substituiçao em centros de carbono com hibridizaçao sp2. Essa reaçao também possui algumas implicaçoes como: i) deve-se evitar que os substratos utilizados na reaçao contenham átomos de hidrogênio em carbonos beta, porque seus derivados de organopaládio correspondentes tendem a sofrer uma rápida eliminaçao de β-hidreto para gerar olefinas; e ii) cloretos de arila nem sempre sao bons substratos, tipicamente porque reagem muito lentamente.
 Esquema 1. Esquema genérico das reaçoes de Heck
O ciclo catalítico para a reaçao de Heck envolve uma série de transformaçoes do catalisador de paládio (Pd0), que geralmente é preparado in situ a partir de um catalisador de paládio(II) precursor.16 O mecanismo mais aceito (Figura 1) descreve uma sequência de eventos que começam com a geraçao de Pd(0) como catalisador ativo e tem como passo determinante da velocidade a adiçao oxidativa de Pd(0) para a ligaçao C-X. Em seguida, é formado um complexo π com o alceno, seguido de uma adiçao syn. Por fim, há a rotaçao da ligaçao C-C e, no passo seguinte, ocorre a etapa de eliminaçao redutiva, regenerando o catalisador pela açao de uma base e recomeçando todo o processo.
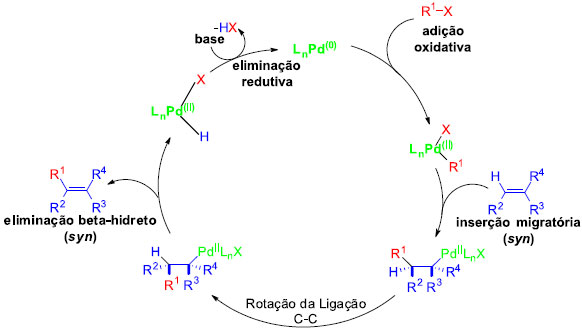 Figura 1. Ciclo catalítico geral da reaçao de Heck
Sínteses totais de produtos naturais empregando a reaçao de Heck As primeiras moléculas de origem natural que temos notícia de síntese em território brasileiro, empregando a reaçao de Heck, sao os alcaloides pirrolidínicos (-)-Codonopsina (1) e (-)-Codonopsinina (2). Essas moléculas foram isoladas das partes aéreas de Codonopsis clematidea. Estes dois alcaloides exibem atividade antibiótica e hipotensiva em estudos com animais, sem apresentar qualquer efeito sobre o sistema nervoso central.17 Correia e colaboradores, em trabalho desenvolvido no Instituto de Química da UNICAMP/SP,18 iniciaram a síntese desses alcaloides aplicando as condiçoes de arilaçao de Heck-Matsuda, empregando os sais de diazônio 3 e 4, com os enecarbamatos enantiomericamente puros 5 e 6, na presença de 2,6-di-terc-butilpiridina ou 2,6-di-terc-butil-4-metilpiridina como base. Esta condiçao levou aos desejados produtos Heck 7-9 em rendimentos razoáveis (36-85%), mas com altas régio e estereosseletividades (Esquema 2). O uso de etanol (EtOH) como solvente e a presença da base provaram ser cruciais para o sucesso da reaçao. Posteriores modificaçoes sintéticas nos compostos 7 e 9 levaram à síntese total dos alcaloides (-)-Codonopsina (1) e (-)-Codonopsinina (2).
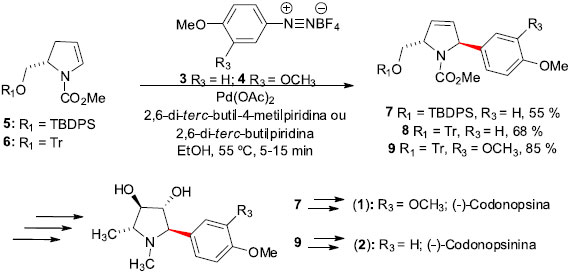 Esquema 2. Sínteses da (-)-Codonopsina (1) e (-)-Codonopsinina (2) por Correia e col.
Em 2000, Correia e colaboradores19 propuseram um aprimoramento da rota sintética para a síntese da (-)-Codonopsinina (2), com melhorias significativas sobre a arilaçao de Heck. Na tentativa de otimizar ainda mais os rendimentos do acoplamento entre o enecarbamato tritilado 6 e o sal de diazônio 3, os autores descobriram que a arilaçao de Heck pode ser realizada em rendimentos elevados e com boa estereosseletividade (90:10 anti:syn). Para tal, empregaram apenas 1% de Pd2(dba)3 e acetonitrila como solvente, à temperatura ambiente e na presença de acetato de sódio como base, levando aos produtos Heck 8a,b em rendimentos elevados (90-95%). A separaçao dos diastereoisômeros trans:cis foi realizada facilmente após a remoçao do grupo tritila (HCO2H, EtOAc, temperatura ambiente, 1 h, ou ZnBr2, CH2Cl2/MeOH), permitindo a obtençao de 10 trans (Esquema 3). Uma sequência de transformaçoes permitiu a síntese total da (-)-Codonopsinina (2) em 7 etapas e 16% de rendimento total, superior à anteriormente descrita.
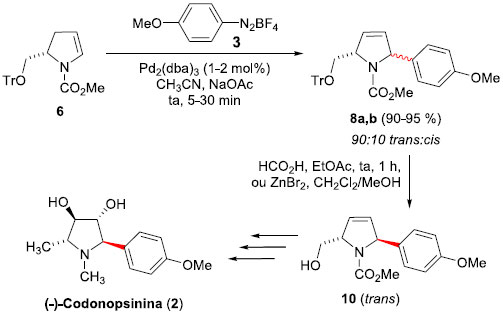 Esquema 3. Síntese da (-)-Codonopsinina (2) por Correia e col.
Aprimorando o trabalhado sobre arilaçao de Heck-Matsuda de enecarbamatos endocíclicas e pirrolinas, empregando sais de arenodiazônio como uma forma eficaz de introduçao de anéis aromáticos em uma estrutura heterocíclica, Correia e colaboradores aprofundaram sua investigaçao sobre compostos heterocíclicos bioativos naturais e nao naturais que possuem um anel aromático alfa a um nitrogênio heterocíclico em um anel de pirrolidina poli-hidroxilado.20 Deste modo, a síntese das trans- e cis-3,4-di-hidróxi-prolinas (±)-(11) e (±)-(12), respectivamente, bem como a síntese do (2R,3R,4R,5R)-2,5-di-hidróxi-metil-3,4-di-hidróxi-pirrolidina (DMDP) (+)-(13), tornaram-se um objetivo alcançável. O composto (±)-11 é um constituinte da verotoxina, isolada a partir de um fungo tóxico (Amanita virosa), popularmente conhecido como "anjo-destruidor",21 enquanto (±)-12 é encontrado como um constituinte da proteína Mefp1, produzida pelo molusco marinho Mytilus edulis.22 A síntese dos epímeros foi realizada a partir do enecarbamato de cinco membros 14, tendo como etapa chave a arilaçao de Heck-Matsuda com o sal tetra-flúor-borato de p-metóxi-benzenodiazônio (3) em acetonitrila, promovendo a formaçao da 2-arilpirrolina 15 em 83% de rendimento, sem quaisquer quantidades detectáveis do aduto Heck isomerizado (Esquema 4). A partir deste intermediário em comum, as sínteses de (±)-11 e (±)-12 foram obtidas em um total de 6 e 5 etapas em 17% e 45% de rendimento, respectivamente.
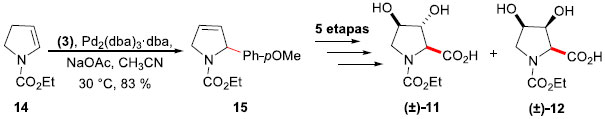 Esquema 4. Sínteses das hidróxi-prolinas (±)-11 e (±)-12 por Correia e col.
O (2R,3R,4R,5R)-2,5-di-hidróxi-metil-3,4-di-hidróxi-pirrolidina (DMDP) (+)-(13) é um potente inibidor natural de α- e β-glicosidases,23 e teve sua síntese também alcançada empregando-se a mesma estratégia descrita anteriormente. A etapa chave de arilaçao de Heck-Matsuda foi realizada entre o enecarbamato quiral 6 com o sal tetra-flúor-borato de p-metóxi-benzenodiazônio (3), fornecendo uma mistura das arilpirrolidinas 8a,b em 95% de rendimento como uma mistura diastereoisomérica trans:cis na proporçao de 87:13 (Esquema 5). Remoçao do grupo tritila permitiu a separaçao dos diastereoisômeros e a obtençao de 10 trans em 70% de rendimento. Após estas eficientes etapas de reaçao de acoplamento e purificaçao diastereoisomérica, a síntese foi alcançada com mais oito etapas, levando ao DMDP (+)-(13) em rendimento total de 12%.
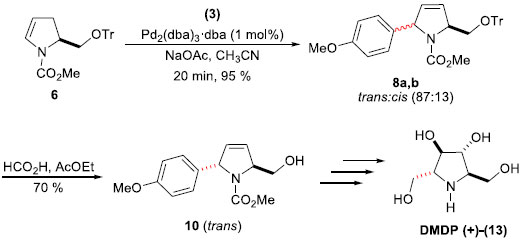 Esquema 5. Síntese do (DMDP) (+)-(13) por Correia e col.
Os alcaloides isolados de plantas da família Amaryllidaceae fazem parte de um subgrupo, onde a licorina é a substância mais conhecida. Os compostos deste subgrupo possuem propriedades biológicas significantes e apresentam como característica estrutural os núcleos pirrolo[3,2,1-de]fenantridinas ou di-hidro-pirrolo[3,2,1-de]fenantridinas.24 Por estas razoes, em 2004 o grupo de Garden e colaboradores, sediado na UFRJ/RJ, desenvolveu a síntese de uma série de alcaloides deste grupo de compostos,25 empregando reaçoes de acoplamento do tipo aril-aril catalisadas por paládio, através das condiçoes de Heck desenvolvidas por Jeffery.26 Para tal, uma série de N-benzil-isatinas 16a-g foram preparadas e submetidas à ciclizaçao na presença de 10 mol% de Pd(OAc)2 em DMF (0,1 mol L-1), contando com KOAc como base e Bu4NBr com aditivo à 100 ºC, sem preocupaçoes com meio anidro ou uso de atmosfera inerte (Esquema 6). Os produtos de acoplamento 17a,b,c foram obtidos em rendimentos na faixa de 74-94%. Posteriores manipulaçoes levaram à síntese total dos alcaloides Hippadina (18a), Pratosina (18b), Anhydrolicorin-2-ona (19a) e Oxoassoanina (19b), em um total de 7 etapas e em rendimentos médios de 12% para os dois primeiros e de 50% para os dois últimos. Ainda foram obtidos outros alcaloides da mesma família, mas que, devido às suas instabilidades, nao foram completamente isolados e caracterizados.
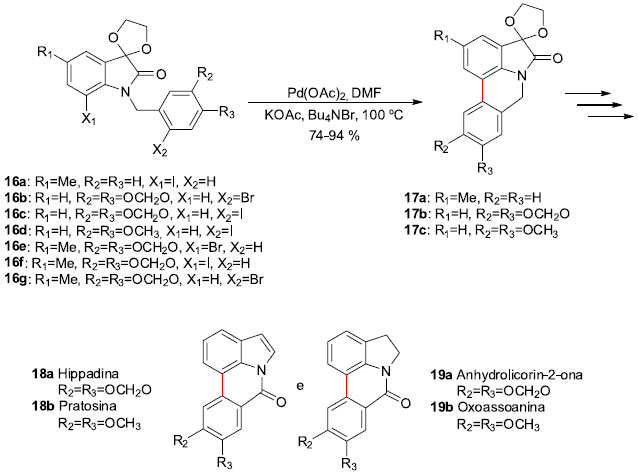 Esquema 6. Sínteses dos alcaloides Amaryllidaceae por Garden e col.
Anidridos maleicos e maleimidas ariladas estao presentes nas estruturas da Prepolicitrina A (20)27 e das Policitrinas A (21) e B (22),28 produtos naturais obtidos de ascídios marinhos do gênero Polycitor. Em 2006, Correia e colaboradores29 propuseram as sínteses totais das Prepolicitrina A (20) e da Policitrina A (21), empregando reaçoes de arilaçao de Heck-Matsuda sobre o anidrido maleico, demonstrando a versatilidade da metodologia desenvolvida no grupo de pesquisa. Contudo, posteriormente o mesmo grupo aprimorou a metodologia de preparaçao dos produtos naturais, o que permitiu sínteses mais eficientes, bem como também a síntese da Policitrina B (22), um derivado nao simétrico do anidrido maleico.30 Nestes novos eficientes protocolos, arilaçoes de Heck-Matsuda31 foram empregadas e permitiram sínteses em rendimentos mais elevados. A síntese da Prepolicitrina A (20) foi alcançada através da reaçao de arilaçao Heck-Matsuda entre o anidrido maleico (23) e o tetra-flúor-borato de 4-hidróxi-fenildiazônio (24), mediada por catalisadores do tipo complexos ácido fosfino-paládio (POPd),32 na presença de acetato de sódio em acetonitrila. A preparaçao do diaril derivado 25 em 56% de rendimento, apesar do rendimento moderado, permitiu a síntese de 20 com apenas mais uma etapa de bromaçao, empregando ácido tri-bromo-isocianúrico (TBCA) em alto rendimento. Para obter 21, a Prepolicitrina A (20) foi submetida à reaçao com tiramina, base de Hunig e fenol, sob irradiaçao com micro-ondas (90 °C, 200 W, 4 min), com rendimento superior à reaçao térmica normal. Esta rota otimizada permitiu a síntese total da Prepolicitrina A (20) em apenas 2 etapas e 50% de rendimento, e da Policitrina A (21) em 3 etapas e 37% de rendimento global, ambas a partir do anidrido maleico (Esquema 7). Os rendimentos das primeiras sínteses (2006) eram de 29% em 3 etapas e de apenas 12% em 4 etapas, respectivamente, demonstrando a evoluçao da metodologia.
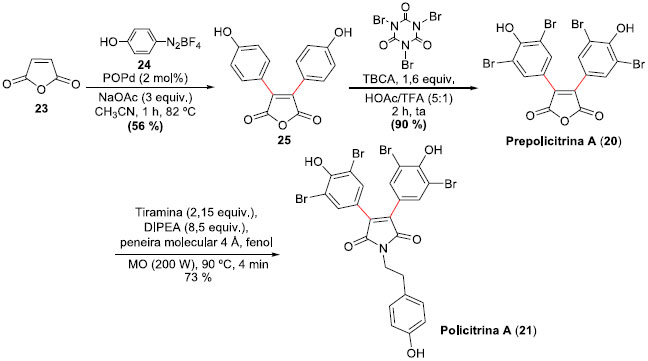 Esquema 7. Sínteses da Prepolicitrina A (20) e Policitrina A (21) por Correia e col.
Para a síntese da Policitrina B (22), uma estratégia mais arrojada foi empregada. A síntese iniciou com a reaçao do fumarato de dimetila (26) com tetra-flúor-borato de 4-metóxi-fenil-diazônio (3) através das rotas a ou b (Esquema 8). Na rota a, o acoplamento foi realizado empregando Pd(OAc)2 (10 mol%) como catalisador, em metanol e refluxo, durante 1 h. Esta condiçao forneceu a síntese estereosseletiva do aduto de Heck mono-aril maleato de dimetila (27) em um rendimento purificado de 91%. Já a rota b permitiu reduçao na quantidade do catalisador de paládio (4 mol%) e a reaçao foi feita sob irradiaçao de micro-ondas. Esta reaçao ficou completa em apenas 5 minutos, formando o aduto Heck 27 em 85% de rendimento, com uma proporçao (Z/E) de 85:15. A transformaçao de 27 ao anidrido mono-aril maleico 28 aconteceu em duas etapas e 89% de rendimento, fornecendo o novo intermediário para a segunda arilaçao. A nova reaçao de Heck-Matsuda entre 28 e tetra-flúor-borato de 4-hidróxi-benzenodiazônio (24), 2% molar de catalisador POPd, e NaOAc como a base em acetonitrila à 80 °C formou o anidrido 3,4-diaril-maleico nao simétrico 29, em um rendimento isolado de 69%. Transformaçao de 29 na Policitrina B (22) foi efetuada após mais duas etapas, o que permitiu a segunda síntese total do produto natural em 5 etapas e 47% de rendimento global.
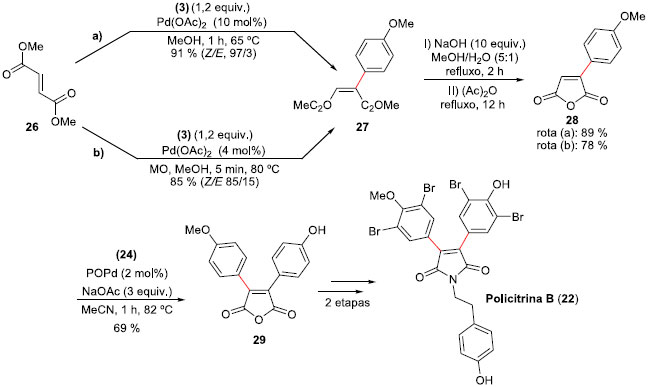 Esquema 8. Síntese da Policitrina B (22) por Correia e col.
(+)-Isoaltholactona (30) é uma furanopiranose da família das estiril-lactonas, isolada de várias espécies de árvores malasianas do gênero Goniothalamus (G. malayanus, G. Montanus e G. tapis),33 que apresenta importantes atividades biológicas, tais como antitumoral, antifúngica e antibacteriana.34 A fim de expandir as possibilidades sintéticas da estratégia de acoplamento de Heck-Matsuda, Correia e colaboradores propuseram a síntese de ent-30,35 devido ao alto custo do material de partida necessário para o produto natural. Assim, a reaçao de arilaçao foi realizada entre a di-hidrofuranona 31, obtida a partir do ácido glutâmico natural, e o tetra-flúor-borato de benzenodiazônio (32), gerando o produto de arilaçao 33. Após extensa experimentaçao, as melhores condiçoes foram aquelas em que 4 mol% de Pd2(dba)3 em acetonitrila, com NaOAc como base, forneceram 33 em 90% de rendimento como uma mistura 94:06 trans:cis (Esquema 9). A mistura pode ser separada por técnicas cromatográficas após dessililaçao, e manipulaçoes sintéticas levaram à síntese da (-)-Isoaltholactona ent-30 em 7 etapas, a partir de 31, em 25% de rendimento global.
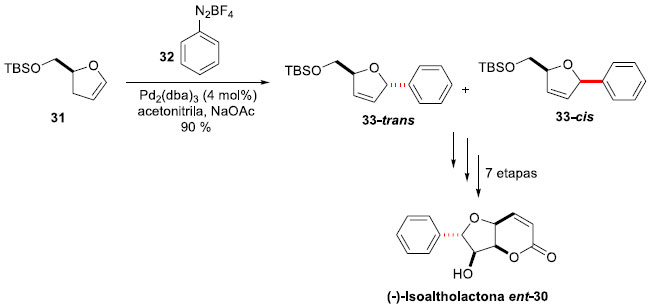 Esquema 9. Síntese da (-)-Isoaltholactona ent-30 por Correia e col.
Outro exemplo de síntese de produto natural, utilizando o acoplamento Heck, foi a síntese total racêmica diastereosseletiva da (±)-trans-Triquentrina A (34), um produto natural marinho que foi isolado a partir da esponja Trikentrion flabelliforme em 1986.36 Realizada em 2008 pelo grupo do professor Silva Jr. na USP/SP,37 o acoplamento Heck foi executado entre o bromo-indol 35 e o crotonato de etila (36), fornecendo o éster insaturado 37, com controle da configuraçao E da olefina (Esquema 10). Esta olefina foi importante para assegurar a síntese do intermediário avançado 38, o qual sofreu reaçao de contraçao de anel mediada por tálio (III), levando posteriormente à consecuçao da síntese total da (±)-trans-Triquentrina A (34) em 21 etapas e apenas 2% de rendimento global.
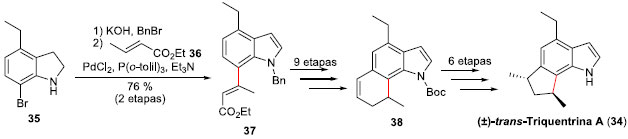 Esquema 10. Síntese da (±)-trans-Triquentrina A (34) por Silva Jr. e col.
Outro estudo desenvolvido por Correia e colaboradores está relacionado com a expansao da metodologia de acoplamento mediada por paládio, tendo como substratos ésteres alílicos empregando sais de arenodiazônio em reaçoes de arilaçao de Heck-Matsuda.38 A metodologia, publicada em 2009, é atrativa pelas condiçoes suaves empregadas e altas estéreo e regiosseletividades obtidas, e permitiu aos autores propor as sínteses totais das lactonas Iangonina (39), que apresenta atividades ansiolítica, sedativa, anti-inflamatória e analgésica,39 bem como das (±)-Metisticina (40) e (±)-di-Hidrometisticina (41), as quais sao componentes isolados de extratos de kava (Piper methysticum).40,41 Assim, a arilaçao de Heck-Matsuda com total controle em favor do isômero 43E, formado entre a lactona alílica 42 e o sal tetra-flúor-borato de 4-metóxi-benzenodiazônio (3), foi alcançada em 85% de rendimento quando realizada em PhCN sob ativaçao de micro-ondas, e sendo empregado Pd2(dba)3 como catalisador (4 mol%) (Esquema 11). A síntese do produto natural foi finalizada através de oxidaçao com DDQ em benzeno, levando à Iangonina (39) em 72% de rendimento, em 2 etapas, a partir de 42. Para a síntese das (±)-Metisticina (40) e (±)-di-Hidrometisticina (41) houve a necessidade de executar a reaçao de acoplamento empregando a mesma metodologia, mas agora entre a lactona 42 e o sal de arenodiazônio 44. O produto Heck deste acoplamento, produzido em 59% de rendimento, já é o produto natural (±)-Metisticina (40). Reaçao de hidrogenaçao regiosseletiva, efetuada em 95% de rendimento, levou à formaçao do produto natural (±)-di-Hidrometisticina (41).
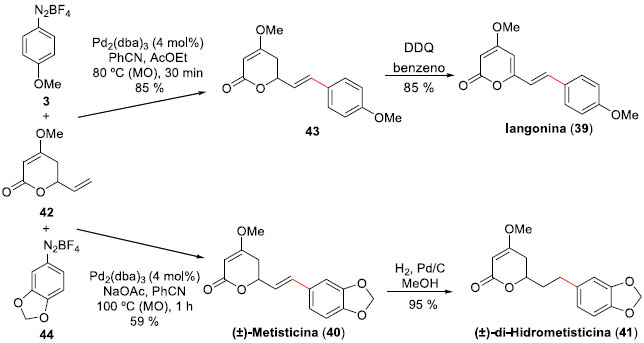 Esquema 11. Sínteses das Iangonine (39), (±)-Metisticina (40) e (±)-di-Hidrometisticina (41) por Correia e col.
Em 2012, Correia e colaboradores expandiram a metodologia de arilaçao de Heck-Matsuda e propuseram a síntese de outros compostos naturais da família das kavalactonas.42 Como alvos, além de outras lactonas isoladas de P. methysticum, em um total de 4 novos produtos naturais, os autores também prepararam três pironas bioativas isoladas de Polygala sabulosa,43,44 uma erva largamente encontrada no sul do Brasil. As kavalactonas que foram sintetizadas envolvendo uso das reaçoes de Heck-Matsuda foram a Kavalactona (45) e a 11-metóxi-Angonina (46), isoladas de P. sabulosa, além da demetóxi-Angonina (47) e da Kavalactona (48), isoladas de P. methysticum. As sínteses envolveram a reaçao de arilaçao entre a pirona 49 e os sais de arenodiazônio 4, 51, 44 e 32, respectivamente (Tabela 1). As reaçoes foram executadas em atmosfera de CO, MeCN na presença de AcONa e o catalisador empregado foi Pd(OAc)2 (5 mol%). As kavalactonas foram obtidas em rendimentos entre 25-89%. A vinil-pirona 49 foi preparada a partir do ácido desidroacético, em quatro etapas e rendimento global de 23%.
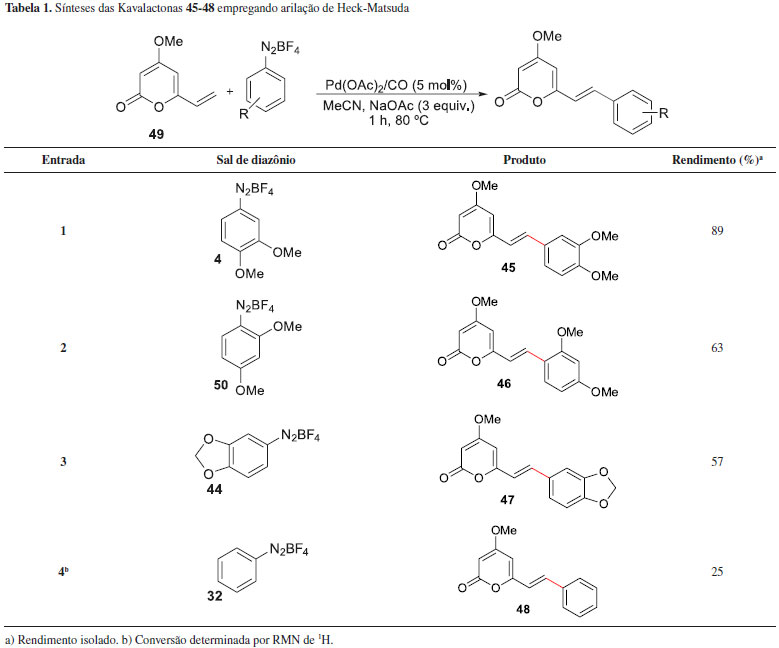
As demais kavalactonas foram preparadas a partir de reaçoes de hidrogenaçao de adutos Heck previamente preparados (Esquema 12). Assim, o aduto Heck 43E teve sua olefina exocíclica hidrogenada e gerou a Kavalactona natural (51) em 89% de rendimento. Outras kavalactonas com cadeias laterais saturadas foram preparadas a partir da hidrogenaçao catalítica das duplas exocíclicas dos adutos Heck Iangonina (39) e da Kavalactona (48), gerando as novas Kavalactonas (52) e (53) em 67% e 65%, respectivamente.
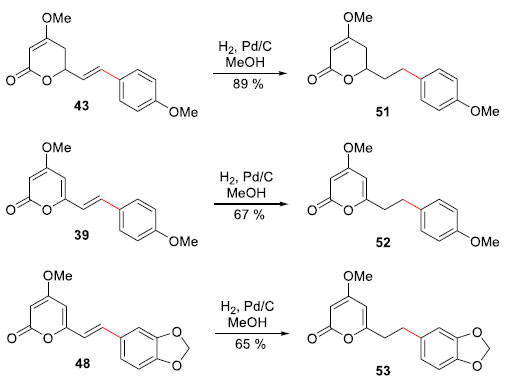 Esquema 12. Sínteses das Kavalactonas (51), (52) e (53) por Correia e col.
Marinoquinolinas sao compostos naturais de origem marinha que apresentam como característica estrutural uma unidade 3H-pirrolo[2,3-c]quinolina. Dentre estes compostos, podemos citar a Marinoquinolina A (54), um forte inibidor da acetil-colinesterase, isolada a partir da bactéria marinha Rapidithrix thailandica em 2006.45 Outras Marinoquinolinas, designadas como B (55), C (56) e E (57), foram isoladas de Ohtaekwangia kribbenses,46 e todas exibiram moderadas citotoxicidades contra linhagens de células cancerosas. Estes interessantes compostos naturais também foram alvo de investigaçao sintética pelo grupo de pesquisa do professor Correia, que desenvolveu uma síntese total concisa e divergente, apresentando a reaçao de Heck-Matsuda em água e a reaçao Pictet-Spengler como etapas-chave.47 A síntese teve início através da reaçao de arilaçao Heck-Matsuda em água pura entre o 3-pirrolina 58 e o sal orto-nitro-benzenodiazônio (59) como passo fundamental. A presença de acetonitrila como solvente levou à decomposiçao do material. O produto de acoplamento lactamol 60 precisou ser imediatamente desidratado em condiçoes ácidas, para entao fornecer o aril-enecarbamato 61 em 66% para as duas etapas (Esquema 13). Uma sequência de oxidaçao, hidrólise e reduçao permitiu a síntese do intermediário-chave pirrolil-anilina 62 em 55% de rendimento.
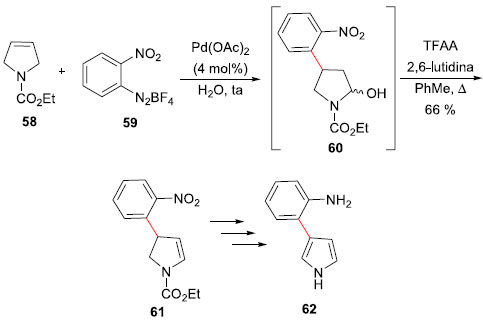 Esquema 13. Síntese do intermediário pirrolil-anilina 62 por Correia e col.
A partir da obtençao da pirrolil-anilina 62, a síntese das Marinoquinolinas pode ser obtida através da reaçao de Pictet-Spengler com os aldeídos apropriados, seguida por aromatizaçao, sendo realizadas em um único passo reacional, seguido pela aromatizaçao in situ do aduto de Pictet-Spengler (Esquema 14). Após alguma experimentaçao, o uso de TFA/CH2Cl2 em tubo selado à 60 ºC foi a melhor condiçao alcançada, ainda que os rendimentos obtidos para a síntese das Marinoquinolinas A (54), B (55), C (56) e E (57) a partir de 62 tenham sido apenas modestos.
 Esquema 14. Síntese das Marinoquinolinas A (54), B (55), C (56) e E (57) por Correia e col.
Sínteses totais de fármacos empregando a reaçao de Heck A (±)-Indatralina (Lu 19-005) (63) é um potente psicoativo terapêutico com atividade inibitória para reabsorçao de monoaminas, incluindo transportadores de dopamina e serotonina,48 e que possui efeito inibitório em consumo de cocaína por macacos.49 Para sua síntese formal, Correia e colaboradores desenvolveram um método eficaz e estereosseletivo para a preparaçao de β,β-di-aril-acrilatos em rendimentos elevados, empregando para tal uma reaçao de Heck-Matsuda entre cinamato de metila (65) com sais tetra-flúor-boratos de arenodiazônio.50 Após uma extensa investigaçao, as melhores condiçoes reacionais foram aquelas obtidas através do uso de metanol como solvente e acetato de Pd(II) como catalisador (10 mol%), sem a adiçao de base. Notou-se que as melhores diastereoseletividades foram alcançadas através da reaçao com sais de arenodiazônio contendo grupos retiradores de elétrons, demonstrando uma acentuada dependência da diastereosseletividade da reaçao de Heck-Matsuda com a natureza eletrônica do eletrófilo empregado. Desenvolvida a metodologia, a síntese formal da (±)-Indatralina (63) iniciou pela arilaçao de Heck-Matsuda entre 64 e o tetra-flúor-borato de 3,4-dicloro-benzenodiazônio (65), levando ao β,β-di-aril-acrilato 66 (Esquema 15). Uma sequência de transformaçoes (hidrogenaçao, hidrólise básica e posterior ciclizaçao em meio ácido) forneceu o conhecido precursor 67,51 já empregado para a síntese da (±)-Indatralina (63) e, portanto, representando sua síntese formal.
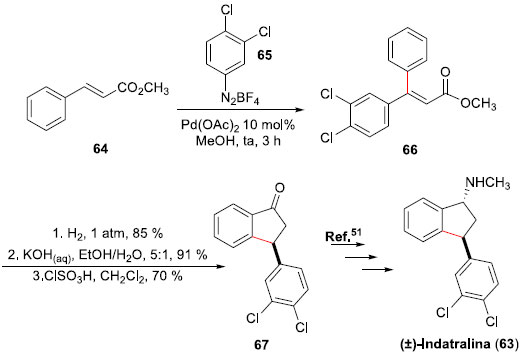 Esquema 15. Síntese formal da (±)-Indatralina (63) por Correia e col.
Empregando a mesma metodologia, Correia e colaboradores puderam obter a síntese da (±)-Sertralina (Zoloft ®) (68). O interesse da preparaçao desta substância advém do uso comercial do fármaco, empregado para tratamentos de depressao e dependência.52 O intermediário ácido di-arilado 69 foi submetido a uma homologaçao empregando o protocolo de Arndt-Eistert, fornecendo o ácido homologado 70 em 73% de rendimento para as 4 etapas. Ciclizaçao com ácido clorossulfônico permitiu a preparaçao da tetralona 71, a qual pode ser convertida à (±)-Sertralina (68) através de condiçoes descritas na literatura53 (Esquema 16).
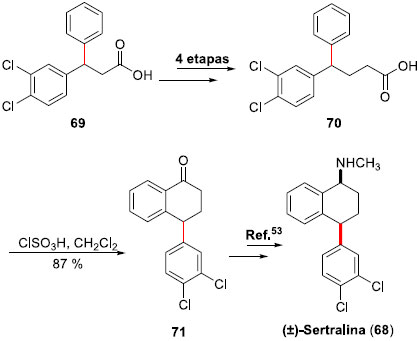 Esquema 16. Síntese formal da (±)-Sertralina (68) por Correia e col.
Outro trabalho envolvendo sais de arenodiazônios e acoplamentos Heck-Matsuda, agora com aminas alílicas,54 foi a síntese da Naftifina (72), um fármaco comercial com atividade anti-fúngica,55 e das Abamina (73) e Abamina SG (74), inibidores da biossíntese de ácido absícico,56 publicada em 2011 por Correia e colaboradores. A metodologia desenvolvida permitiu o acoplamento em condiçoes suaves, com excelentes régio e esterosseletividades. A síntese teve como etapa-chave a reaçao de acoplamento de Heck-Matsuda entre carbamato de metila da alil-amina 75 e o sal de benzenodiazônio 32, fornecendo o aduto de Heck 76 em 85% de rendimento com controle régio e estereoquímico do isômero E. Finalmente, a reduçao do grupo carbometóxi com LiAlH4, em 87% de rendimento, levou à síntese total da Naftifina (72), que foi obtida em apenas quatro etapas em um rendimento total de 68% (Esquema 17).
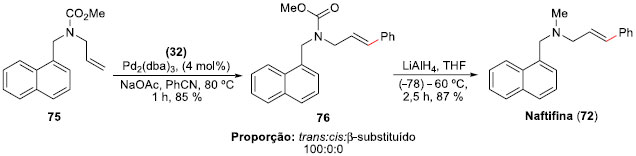 Esquema 17. Síntese total da Naftifina (72) por Correia e col.
Para a síntese das Abaminas (73) e (74), foi preparada a alil-benzamida 77, a qual foi submetida às condiçoes de arilaçao de Heck-Matsuda anteriormente otimizadas com o tetra-flúor-borato de 3,4-dimetóxi-benzenodiazônio (4). O produto desejado 78 foi obtido em um rendimento de 84%, com total controle régio e estereoquímico do isômero E (Esquema 18). A partir deste aduto de Heck, as sínteses da Abamina (73) e da Abamina SG (74) foram finalizadas em duas etapas, permitindo suas sínteses totais em cinco etapas e em 53% e 55% de rendimento global, respectivamente.
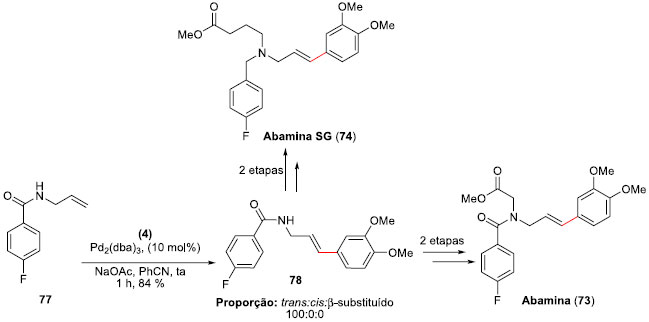 Esquema 18. Sínteses totais da Abamina (74) e da Abamina SG (75) por Correia e col.
Tendo já sido desenvolvida a arilaçao de Heck-Matsuda com derivados do tipo cinamatos, Correia e colaboradores expandiram esta metodologia para orto-hidróxi-derivados. Esta modificaçao permitiu uma eficiente estratégia de acesso a 4-aril-cumarinas,57 realizando uma concisa síntese total da (R)-Tolterodina (79), uma droga anti-muscarínica empregada em tratamento de incontinência urinária,58 em quatro etapas e em elevado rendimento global (30%) (Esquema 19). A síntese iniciou-se pela reaçao Heck-Matsuda entre o éster orto-hidróxi-cinamato (80) e o sal de arenodiazônio 81, sob condiçoes otimizadas, para fornecer a 6-metil-4-fenil-cumarina (82), em 63% de rendimento isolado após duas etapas. Instalaçao do centro estereogênico através de reduçao assimétrica, e posterior reaçao de aminaçao redutiva encerraram a preparaçao da (R)-Tolterodina (79).
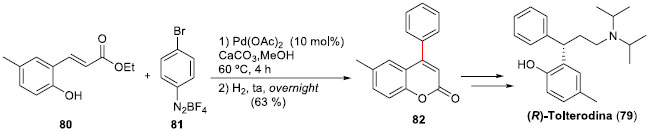 Esquema 19. Síntese total da (R)-Tolterodina (79) por Correia e col.
VPC01091 (83) é um agonista seletivo para o receptor da esfingosina-1-fosfato do subtipo 1 (S1P1),59 e pertence a uma classe de drogas importantes no tratamento de esclerose múltipla, por possuir poucos efeitos colaterais. Devido a sua complexidade, sua síntese se tornou um grande desafio, sendo necessário o desenvolvimento de uma versao diastereosseletiva da reaçao de Heck-Matsuda, o que foi alcançado pelo grupo do professor Correia em 2012.60 Carbamatos ciclopentenoicos foram arilados, fornecendo uma série de ciclopentenos em excelentes syn-diastereosseletividades (>85:15). Os autores postulam que a coordenaçao do metal na mesma face do grupo carbamato seria a origem da seletividade observada. Deste modo, a reaçao de arilaçao de Heck-Matsuda entre o carbamato cíclico 84 (obtido a partir do malonato de dimetila em 4 etapas e 61% de rendimento) e o tetra-flúor-borato de 4-octil-benzenodiazônio (85), realizada na presença de Pd2(dba)3 (4 mol%) e AcONa (3 eq.) em benzonitrila, forneceu o aduto Heck 86 em 90% de rendimento e boa seletividade syn:anti (88:12) (Esquema 20). Uma série de manipulaçoes sintéticas permitiu a transformaçao deste aduto ao VPC01091 (83) em um total de 5 etapas e 40% de rendimento total.61
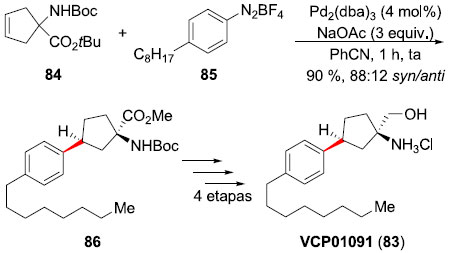 Esquema 20. Síntese total da VPC01091 (83) por Correia e col.
Reaçao de Stille A reaçao de Stille, ou o acoplamento Migita-Kosugi-Stille, é uma reaçao química de acoplamento de um composto organo-estanho com uma variedade de eletrófilos orgânicos catalisado por paládio, para formar uma nova ligaçao sigma C-C (Esquema 21).
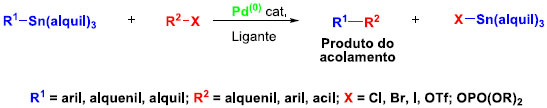 Esquema 21. Esquema genérico das reaçoes de Stille
O primeiro relato de um acoplamento de halogenetos com organo-estanho deu-se em 1976, descrito por C. Eaborn e colaboradores.62 No ano seguinte, M. Kosugi e T. Migita expandiram esse procedimento para acoplamentos entre cloretos de acila e reagentes de alquil-estanho, gerando produtos em bons rendimentos (53 a 87%).63 Já no início dos anos 80, J. K. Stille utilizou compostos de organo-estanho para a síntese de cetonas empregando condiçoes reacionais muito mais brandas do que Migita e em rendimentos significativamente superiores. O acoplamento Stille64 tornou-se uma ferramenta poderosa na síntese de novas ligaçoes C-C e uma parte desse sucesso está relacionada aos precursores do organo-estanho, pois possuem algumas características importantes: i) toleram uma ampla variedade de grupos funcionais; ii) nao sao sensíveis à umidade ou oxigênio, ao contrário de outros compostos organometálicos reativos; e iii) sao facilmente preparados, isolados e armazenados. Contudo, apresentam alguns inconvenientes, que sao sua alta toxicidade e dificuldade para remoçao de traços de subprodutos de estanho a partir da mistura reacional. O ciclo catalítico mais aceito para a reaçao de Stille envolve a adiçao oxidativa de um halogeneto a um catalisador de paládio, seguido de transmetalaçao deste com o reagente de organo-estanho. A etapa seguinte é descrita como uma eliminaçao redutiva, com a formaçao do produto de acoplamento e o catalisador de paládio sendo regenerado (Figura 2).
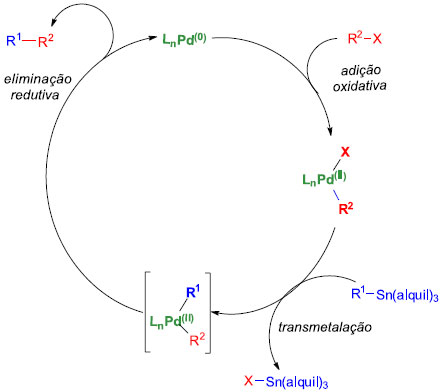 Figura 2. Ciclo catalítico geral da reaçao de Stille
A Crocacina C (87) é um, dentre quatro produtos naturais, que sao normalmente encontrados nos extratos das Chondromyces crocatus e Chondromyces pediulatus, que inibem moderadamente o crescimento de algumas bactérias Gram-positivas, bem como fungos e leveduras.65,66 Para realizar sua síntese total, no início dos anos 2000, Dias e colaboladores,67 da UNICAMP/SP, projetaram empregar o acoplamento de Stille como etapa-chave. Para a síntese dos fragmentos envolvidos na reaçao de acoplamento cruzado, a E-vinil-estanana 88 foi preparada em 3 etapas e 50% de rendimento, a partir do 2-butinoato de etila (Esquema 22). O fragmento mais complexo, o iodeto E-vinilico 89, necessitou de 14 etapas para ser sintetizado, e foi obtido em excelentes 23% de rendimento global. A reaçao de acoplamento de Stille entre a E-vinil-estanana 88 com o iodeto E-vinílico 89 foi realizada em NMP a 60 ºC, com uma quantidade catalítica de Pd2(dba)3 e na presença de AsPh3, proporcionando a (+)-Crocacina C (87) em 69% de rendimento, após purificaçao por cromatografia em coluna de sílica gel. Deste modo, a síntese do composto 88 foi alcançada em 15 etapas, em um rendimento global de 16%.
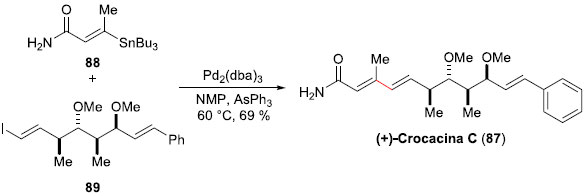 Esquema 22. Síntese total da (+)-Crocacina C (87) por Dias e col.
Para a síntese da (+)-Crocacina D (90), um membro mais complexo da família das crocacinas, Dias e colaboradores68 aproveitaram sua síntese anterior da (+)-Crocacina C (87). Como a (+)-Crocacina D (90) possui um grupo substituinte na amida presente na estrutura de 87, a síntese total se deu pela uniao entre o produto natural anteriormente preparado, através de acoplamento de Stille, agora em condiçoes otimizadas, e o fragmento iodeto Z-vinílico 91 (preparado em 10 etapas e 32% de rendimento global a partir do diol 92). A síntese assimétrica convergente de 90 se deu pelo eficiente acoplamento entre a (+)-Crocacina C (87) e o (Z)-vinil iodeto 91, mediado por cobre,69 levando à obtençao da (+)-Crocacina D (90) em 67% de rendimento (Esquema 23). A síntese total foi realizada em 16 etapas e 14% de rendimento global.
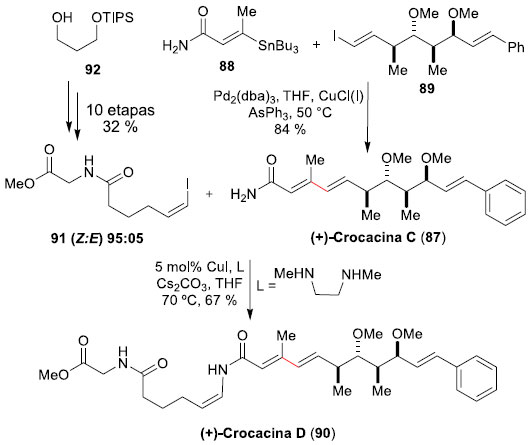 Esquema 23. Síntese total da (+)-Crocacina D (90) por Dias e col.
As (-)-Basiliskamida A (93) e (-)-Basiliskamida B (94) sao policetídeos isolados da bactéria marinha PNG-276, encontrada na costa da Papua Nova Guiné.70 Estes policetídos apresentam potente atividade antifúngica in vitro contra Cândida albicans e Aspergillus fumigatus, similar à Amfotericina B. Dias e colaboradores realizaram a síntese total dessas moléculas, empregando a reaçao de Stille como etapa-chave. A primeira destas sínteses reportada foi a da (-)-Basiliskamida B (94), em 2008, que envolveu 12 etapas produzindo o produto desejado em rendimento global de 6%.71 A etapa de acoplamento cruzado Stille deu-se entre a E-vinil-estanana 95, preparada em 2 etapas e 20% de rendimento, a partir do propiolato de etila, e o iodeto E-vinílico 96, que requereu um total de 12 etapas e foi construído em 9% de rendimento global. O acoplamento foi realizado em DMF e com uma quantidade catalítica (5 mol%) de Pd(MeCN)2Cl2, à 25 ºC, e proporcionou o aduto de Stille 97 em 69% de rendimento após purificaçao por cromatografia em coluna de sílica (Esquema 24). A conclusao da síntese foi efetuada com a remoçao do isopropilideno-cetal com AcOH 80%, fornecendo o diol 98, seguido da acilaçao seletiva do oxigênio menos impedido com cloreto de E-cinamoíla, levando à (-)-Basiliskamida B (94). Para a síntese da (-)-Basiliskamida A (93), que difere de 94 apenas pela posiçao do grupo cinamoíla, Dias e colaboradores utilizaram da maior reatividade da hidroxila em C7 para protegê-la antes da reaçao de esterificaçao.72 Assim, reaçao do diol 98 com TES-Cl permitiu a proteçao da hidroxila mais desimpedida, deixando a hidroxila em C9 livre para a posterior reaçao de esterificaçao, fornecendo o intermediário 99. Posterior remoçao do protetor de silício com HF, forneceu a (-)-Basiliskamida A (93) em 42% de rendimento e 4 etapas a partir do aduto de Stille 97.
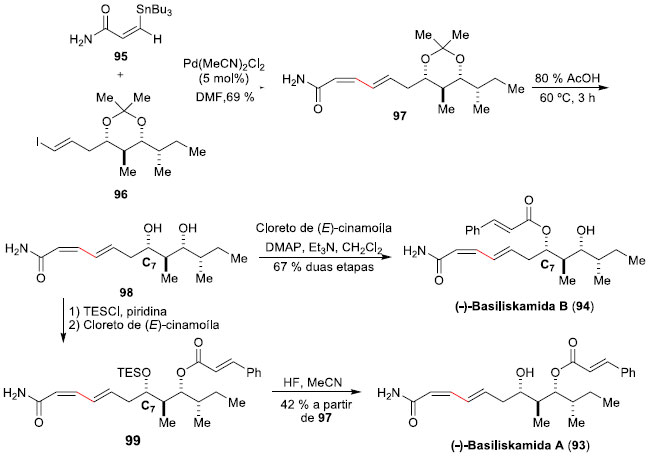 Esquema 24. Sínteses totais das (-)-Basiliskamidas B (94) e A (93) por Dias e col.
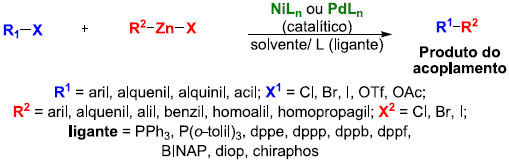
Reaçao de Negishi Em 1976, Ei-ichi Negishi e colaboradores investigaram o acoplamento cruzado envolvendo reagentes organo-alumínio, inicialmente empregando Ni e Pd como catalisadores.73 A pesquisa extensiva por Negishi mostrou que os melhores resultados sao obtidos quando os reagentes de organo-alumínio sao trocados por reagentes de organo-zinco e acoplados na presença de Ni e Pd. É uma reaçao versátil para acoplamentos cruzados de vários halogenetos de vinila (arila, alquenila e alquinila) com reagentes organo-zinco,74 que formam ligaçoes carbono-carbono (C-C) no processo. Um catalisador de paládio é geralmente mais eficiente, embora o níquel também possa ser empregado (Esquema 25).
 Esquema 25. Esquema genérico das reaçoes de Negishi
Algumas características gerais da reaçao sao as seguintes: i) tanto fosfinas de paládio como as de níquel funcionam bem como catalisadores. No entanto, os complexos de paládio tendem a fornecer rendimentos e estereosseletividade melhores, e sua tolerância com grupos funcionais é superior; ii) os catalisadores ativos de Ni(0) e Pd(0) sao relativamente instáveis, mas estes podem ser gerados in situ a partir de complexos mais estáveis de Ni(II) e Pd(II) empregando um agente redutor (por exemplo, 2 equivalentes de DIBAL-H ou n-BuLi); iii) os vários reagentes de organo-zinco podem ser preparados por reaçao direta do halogeneto orgânico com metal de zinco ou zinco metálico ativado ou por transmetalaçao do correspondente organolítio ou reagente de Grignard com um halogeneto de zinco (ZnX2); iv) outras vantagens da utilizaçao de organo-zincos incluem: elevada reatividade, elevadas régio e estereosseletividade, ampla aplicabilidade, poucas reaçoes laterais e quase nenhuma toxicidade. Uma das limitaçoes do acoplamento cruzado de Negishi é que alquil-zincos secundários e terciários podem sofrer isomerizaçao, além da reaçao com CO através de reaçoes de inserçao. O mecanismo do acoplamento é análogo aos de outros acoplamentos mediados por paládio (Figura 3), e tem como etapas distintas: i) adiçao oxidativa do haleto orgânico à espécie de Pd(0) para formar Pd(II); ii) transmetalaçao; e iii) eliminaçao redutiva para formar a ligaçao C-C e regeneraçao do catalisador de Pd(0).
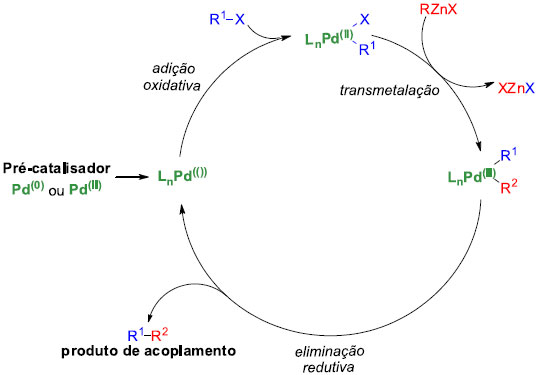 Figura 3. Ciclo catalítico geral baseado no Pd da reaçao de Stille
Um exemplo de versatilidade da reaçao de Negishi na síntese total de um produto de interesse foi descrita pelo professor Pilli e colaboradores,75 em trabalho desenvolvido na UNICAMP/SP, em seus estudos visando a síntese do (Z)-Tamoxifeno (100), um agente anti-estrogênico de uso clínico na terapia do câncer de mama.76 A síntese teve início a partir do 4-iodo-fenol (101), sendo necessárias 6 etapas (58% de rendimento global) para a síntese do intermediário dibromado 102 que seria utilizado no duplo acoplamento Negishi. A reaçao levou à síntese do desejado (Z)-Tamoxifeno (100), junto com o seu isômero (E), este último através de uma reaçao competitiva de syn-carbopaladaçao. A síntese completa envolveu 7 etapas e foi alcançada em 30% de rendimento total, com seletividade 2,3:1 dos isômeros geométricos (Z:E), respectivamente (Esquema 26).
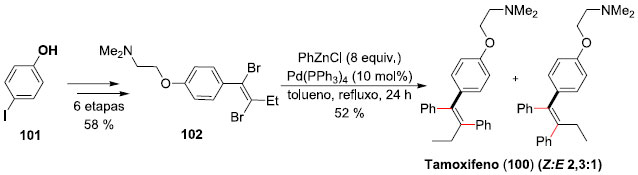 Esquema 26. Sínteses totais dos (Z) e (E)-Tamoxifenos (100) por Pilli e col.
A síntese do (Z)-Tamoxifeno (100) também foi realizada pelo grupo do prof. Monteiro e colaboradores, em estudos realizados na UFRGS/RS. Em sua estratégia sintética,77 a preparaçao do composto alvo teve início pelo trans-estilbeno (103). A olefina foi submetida a condiçoes de acoplamento de Heck, empregando um sistema catalítico composto de Pd(OAc)2 e P(o-tolil)3, com o bromobenzeno 104, fornecendo o tri-aril-alceno 105, em altos rendimento e seletividade (Esquema 27). Bromaçao de 105 levou ao bromo-tri-arilalceno, o qual foi submetido ao protocolo de acoplamento cruzado de Negishi com EtZnCl, empregando tol-BINAP como ligante do catalisador Pd(OAc)2. Este último acoplamento permitiu a síntese do (Z)-Tamoxifeno (100), junto com seu isômero (E), em 74% de rendimento e seletividade final de 65:35 (Z:E). O uso de dois acoplamentos mediados por paládio permitiu a síntese do alvo em apenas 3 etapas e 57% de rendimento global.78
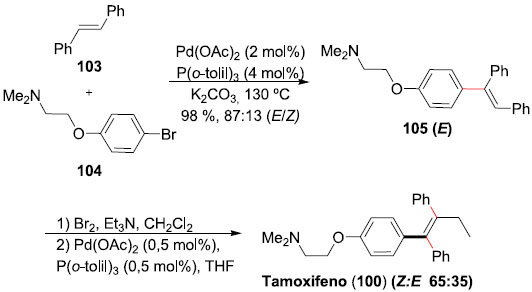 Esquema 27. Sínteses totais dos (Z) e (E)-Tamoxifenos (100) por Monteiro e col.
CONSIDERAÇOES FINAIS Os complexos de metais de transiçao têm um papel fundamental na formaçao de ligaçoes carbono-carbono em sínteses totais. Os mesmos possibilitam rápido e eficiente acesso a moléculas de alta complexidade, várias com significativas importâncias de cunho científico, econômico e/ou social. Como visto nas várias sínteses desenvolvidas por pesquisadores brasileiros, as etapas mediadas pelos catalisadores de paládio foram essenciais na produçao dos compostos em questao. Além disso, nota-se que os estudos e aplicaçao destas metodologias encontram-se bem consolidados e otimizados. Pelos vários exemplos mostrados nesta revisao, percebe-se que a comunidade sintética brasileira tem sabido aproveitar as potencialidades dos acoplamentos cruzados mediados por paládio para a síntese de estruturas complexas. Esta açao demonstra, claramente, o amadurecimento de nossos pesquisadores, que cada dia mais buscam, no arcabouço das recentes metodologias, os métodos mais eficientes de formaçao de ligaçoes carbono-carbono. Isso quando nao estao criando novas metodologias! A produçao dita "nacional" alcança os melhores periódicos, conquistou respeito além de nossas fronteiras e rivaliza, em seus feitos, com sínteses totais do mais alto nível internacional.79 Esta transformaçao, que se consolidou nos últimos 20-30 anos, nos permite projetar um futuro ainda mais brilhante para a comunidade sintética orgânica brasileira. Que os digam, por exemplo, os Brazilian Meeting on Organic Synthesis, o conhecido BMOS, um evento organizado pelos químicos orgânicos sintéticos brasileiros, e que tem alcançado o mais alto reconhecimento internacional,80,81 sendo outro excelente exemplo da capacidade desta comunidade.
AGRADECIMENTOS Os autores agradecem à FAPESB, CNPq e INCT E&A pelo suporte. G. S. B. Silva agradece à CAPES pela bolsa de doutorado. Os autores também agradecem a leitura atenta e sugestoes dos professores J. M. David e V. B. Riatto.
REFERENCIAS 1. Correia, C. R. D.; Costa, P. R. R.; Ferreira, V. F.; Quim. Nova 2002, 25, 82. 2. Crabtree, R. H.; The Organometallic Chemistry of the Transition Metals, 4ª ed., John Wiley & Sons: Hoboken, 2005. 3. Hunt, L. B.; Platinum Met. Rev. 1984, 28, 76. 4. Zeise, W. C.; Ann. Phys. (Poggendorff) 1827, 2, 632. 5. West, M. N. F.; Colenso, S. J. F.; Sketches from the Life of Edward Frankland, Spottiswoode & Company: London, 1902, p. 185. 6. Princival, J. L.; Dos Santos, A. A.; Comasseto, J. V.; J. Braz. Chem. Soc. 2010, 21, 2042. DOI: http://dx.doi.org/10.1590/S0103-50532010001100005 7. Gusevskaya, E. V.; Quim. Nova 2003, 26, 242. DOI: http://dx.doi.org/10.1590/S0100-40422003000200017 8. Masters, C.; Homogeneous Transition-metal Catalysis: a Gentle Art, Chapman and Hall: New York, 1981. 9. Batalha, P. N.; Sagrillo, F. S.; Gama, I. L.; Rev. Virtual Quim. 2014, 6, 494. 10. Brocksom, T. J.; Alves, L. C.; Wulf, G. D.; Desiderá, A. L.; Oliveira, K. T.; Química Nova na Escola 2010, 32, 233. 11. Foram consideradas apenas as sínteses totais realizadas e lideradas por pesquisadores brasileiros em laboratórios no Brasil. Há, certamente, um número bem superior de excelentes sínteses nao descritas nesta revisao, para as quais pedimos desculpas pela exclusao, mas que, devido à participaçao de grupos estrangeiros (contando com brasileiros em estágios de Pós-Doc ou como visitantes), resolvemos desconsiderar. 12. Pilli, R. A.; Comasseto, J. V.; Ferreira, J. T. B.; Organic synthesis in Brazil: an overview, EDUSP: Sao Paulo, 1994. 13. Heck, R. F.; J. Am. Chem. Soc. 1968, 90, 5518. DOI: http://dx.doi.org/10.1021/ja01022a034 14. Mirozoki, T.; Mori, K.; Ozaki, A.; Bull. Chem. Soc. Jpn. 1971, 44, 581. DOI: http://dx.doi.org/10.1246/bcsj.44.581 15. Heck, R. F.; Nolley, J. P.; J. Org. Chem. 1972, 37, 2320. DOI: http://dx.doi.org/10.1021/jo00979a024 16. Bradshaw, M.; Chem. Commun. 2011, 47, 12292. DOI: http://dx.doi.org/10.1039/c1cc14717j 17. Yoda, H.; Nakajima, T.; Takabe, K.; Tetrahedron Lett. 1996, 37, 5531. DOI: http://dx.doi.org/10.1016/0040-4039(96)01042-8 18. Oliveira, D. F.; Severino, E. A.; Correia, C. R. D.; Tetrahedron Lett. 1999, 40, 2083. DOI: http://dx.doi.org/10.1016/S0040-4039(99)00151-3 19. Severino, E. A.; Correia, C. R. D.; Org. Lett. 2000, 2, 3039. DOI: http://dx.doi.org/10.1021/ol005762d PMID: 11009340 20. Garcia, A. L. L.; Correia, C. R. D.; Tetrahedron Lett. 2003, 44, 1553. DOI: http://dx.doi.org/10.1016/S0040-4039(03)00017-0 21. Buku, A.; Faulstich, H.; Wieland, T.; Dabrowski, K.; Proc. Natl. Acad. Sci. U. S. A. 1980, 77, 2370. DOI: http://dx.doi.org/10.1073/pnas.77.5.2370 PMID: 16592813 22. Taylor, S. W.; Waite, J. H.; Ross, M. M.; Shabanowitz, J. Hunt, D. F.; J. Am. Chem. Soc. 1994, 116, 10803. DOI: http://dx.doi.org/10.1021/ja00102a063 23. Asano, N.; Nash, R. J.; Molyneux, R. J.; Fleet, G. W.; Tetrahedron: Asymmetry 2000, 11, 1645. DOI: http://dx.doi.org/10.1016/S0957-4166(00)00113-0 24. Hoshino, O. In The Amaryllidaceae alkaloids. Cordell, G. A., ed.; Academic Press: San Diego, 1998, Vol. 51, pp 323-424. 25. Torres, J. C.; Pinto, A. C.; Garden, S. J.; Tetrahedron 2004, 60, 9889. DOI: http://dx.doi.org/10.1016/j.tet.2004.08.030 26. Jeffery, T.; Tetrahedron 1996, 52, 10113. DOI: http://dx.doi.org/10.1016/0040-4020(96)00547-9 27. Rudi, A.; Evans, T.; Aknin, M.; Kashman, Y.; J. Nat. Prod. 2000, 63, 832. DOI: http://dx.doi.org/10.1021/np9905158 PMID: 10869212 28. Rudi, A.; Goldberg, I.; Stein, Z.; Frolow, F.; Benayahu, Y.; Schleyer, M.; Kashman, Y.; J. Org. Chem. 1994, 59, 999. DOI: http://dx.doi.org/10.1021/jo00084a015 29. Burtoloso, A. C. B.; Garcia, A. L. L.; Miranda, K. C.; Correia, C. R. D.; Synlett 2006, 18, 3145. DOI: http://dx.doi.org/10.1055/s-2006-951524 30. Canto, K.; da Silva Ribeiro, R.; Biajoli, A. F. P.; Correia, C. R. D.; Eur. J. Org. Chem. 2013, 8004. DOI: http://dx.doi.org/10.1002/ejoc.201301108 31. Taylor, J. G.; Moro, A. V.; Correia, C. R. D.; Eur. J. Org. Chem. 2011, 1403. DOI: http://dx.doi.org/10.1002/ejoc.201001620 32. Li, G. Y.; Angew. Chem., Int. Ed. 2001, 40, 1513. DOI: http://dx.doi.org/10.1002/1521-3773(20010417)40:8<1513::AID-ANIE1513>3.0.CO;2-C 33. Yadav, J. S.; Krishnam, R.; Purushotama, R.; Rajaiah, G.; Tetrahedron: Asymmetry 2005, 16, 3283. DOI: http://dx.doi.org/10.1016/j.tetasy.2005.08.047 34. de Fátima, A.; Modolo, L. V.; Conegero, L. S.; Pilli, R. A.; Ferreira, C. V.; Khon, L. K.; de Carvalho, J. E.; Curr. Med. Chem. 2006, 13, 3371. DOI: http://dx.doi.org/10.2174/092986706779010298 PMID: 17168711 35. Meira, P. R. R.; Moro, A. V.; Correia, C. R. D.; Synthesis 2007, 15, 2279. DOI: http://dx.doi.org/10.1055/s-2007-983781 36. Capon, R. J.; Macleod, J. K.; Scammells, P. J.; Tetrahedron 1986, 42, 6545. DOI: http://dx.doi.org/10.1016/S0040-4020(01)88117-5 37. Silva, L. F.; Craveiro, M. V.; Org. Lett. 2008, 10, 5417. DOI: http://dx.doi.org/10.1021/ol8023105 PMID: 18973334 38. Moro, A. V.; Cardoso, F. S. P.; Correia, C. R. D.; Org. Lett. 2009, 11, 3642. DOI: http://dx.doi.org/10.1021/ol901416e PMID: 19719201 39. Bilia, A. R.; Gallori, S.; Vincieri, F. F.; Life Sci. 2002, 70 , 2581. 40. Bilia, A. R.; Scalise, L.; Bergonzi, M. C.; Vincieri, F. F.; J. Chromatogr. B 2004, 812, 203. DOI: http://dx.doi.org/10.1016/S1570-0232(04)00644-0 41. Coté, C. S.; Kor, C.; Cohen, J.; Auclair, K.; Biochem. Biophys. Res. Commun. 2004, 322, 147. DOI: http://dx.doi.org/10.1016/j.bbrc.2004.07.093 PMID: 15313185 42. Soldi, C.; Moro, A. V.; Pizzolatti, M. G.; Correia, C. R. D.; Eur. J. Org. Chem. 2012, 3607. DOI: http://dx.doi.org/10.1002/ejoc.201200308 43. Pizzolatti, M. G.; Cunha, A.; Pereira, W. S.; Delle Monache, F.; Syst. Ecol. 2004, 32, 603. DOI: http://dx.doi.org/10.1016/j.bse.2003.08.011 44. Pizzolatti, M. G.; Luciano, C.; Delle Monache, F.; Phytochemistry 2000, 55, 819. DOI: http://dx.doi.org/10.1016/S0031-9422(00)00301-0 PMID: 11190403 45. Sangnoi, Y.; Sakulkeo, O.; Yuenyongsawad, S.; Kanjana-opas, A.; Ingkaninan, K.; Plubrukarn, A.; Suwanborirux, K.; Mar. Drugs 2008, 6, 578. DOI: http://dx.doi.org/10.3390/md6040578 PMID: 19172195 46. Okanya, P. W.; Mohr, K. L.; Gerth, K.; Jansen, R.; Müller, R.; J. Nat. Prod. 2011, 74, 603. DOI: http://dx.doi.org/10.1021/np100625a PMID: 21456549 47. Schwalm, C. S.; Correia, C. R. D.; Tetrahedron Lett. 2012, 53, 4836. 48. Bogeso, K. P.; Christensen, A. V.; Hyttel, J.; Liljefors, T.; J. Med. Chem. 1985, 28, 1817. DOI: http://dx.doi.org/10.1021/jm00150a012 PMID: 2999402 49. Negus, S. S.; Brandt, M. R.; Mello, N. K.; J. Pharmacol. Exp. Ther. 1999, 291, 60. PMID: 10490887 50. Pastre, J. C.; Correia, C. R. D.; Adv. Synth. Catal. 2009, 351, 1217. DOI: http://dx.doi.org/10.1002/adsc.200900032 51. Froimowitz, M.; Wu, K. M.; Moussa, A.; Haidar, R. M.; Jurayj, J.; George, C.; Gard,, ner, E. L.; J. Med. Chem. 2000, 43, 4981. DOI: http://dx.doi.org/10.1021/jm000201d PMID: 11150168 52. MacQueen, G.; Born, L.; Steiner, M.; CNS Drug Rev. 2001, 7, 1. DOI: http://dx.doi.org/10.1111/j.1527-3458.2001.tb00188.x PMID: 11420570 53. Davies, H. M. L.; Stafford, D. G.; Hansen, T.; Org. Lett. 1999, 1, 233. DOI: http://dx.doi.org/10.1021/ol9905699 PMID: 10905868 54. Prediger, P.; Barbosa, L. F.; Génisson, Y.; Correia, C. R. D.; J. Org. Chem. 2011, 76, 7737. DOI: http://dx.doi.org/10.1021/jo201105z PMID: 21877731 55. Stütz, A.; Georgepoulos, A.; Granitzer, W.; Petrany, G.; Berney, D.; J. Med. Chem. 1986, 29, 112. DOI: http://dx.doi.org/10.1021/jm00151a019 PMID: 3510297 56. Han, S. Y.; Kitahata, N.; Saito, T.; Kobayashi, M.; Shinozaki, K.; Yoshida, S.; Asami, T.; Bioorg. Med. Chem. Lett. 2004, 14, 3033. DOI: http://dx.doi.org/10.1016/j.bmcl.2004.04.035 PMID: 15149639 57. Barancelli, D. A.; Salles Jr, A. G.; Taylor, J. G.; Correia, C. R. D.; Org. Lett. 2012, 14, 6036. DOI: http://dx.doi.org/10.1021/ol302923f PMID: 23190249 58. Wefer, J.; Truss, M. C.; Jonas, U.; World J. Urol. 2001, 19, 312. DOI: http://dx.doi.org/10.1007/s003450100224 PMID: 11760779 59. Zhu, R.; Snyder, A. H.; Kharel, Y.; Schaffter, L.; Sun, Q.; Kennedy, P. C.; Lynch, K. R.; Macdonald, T. L.; J. Med. Chem. 2007, 50, 6428. DOI: http://dx.doi.org/10.1021/jm7010172 PMID: 17994678 60. Oliveira, C. C.; Santos, E. A. F.; Nunes, J. H. B.; Correia, C. R. D.; J. Org. Chem. 2012, 77, 8182. DOI: http://dx.doi.org/10.1021/jo3015209 PMID: 22924670 61. Uma síntese alternativa, envolvendo o acoplamento do carbamato cíclico 84 com o tetra-flúor-borato de 4-iodo-benzenodiazônio, foi realizada pelos autores e também levou à síntese do VPC01091 (83), após um acoplamento de Sonogashira entre o aduto formado e oct-1-ino, seguido de posteriores transformaçoes. Esta versao, apesar de ser mais flexível e de possibilitar a síntese de derivados com diferentes substituintes na posiçao 4 do grupo arila, forneceu o alvo em rendimentos inferiores e em uma sequência sintética mais longa. 62. Azarian, D.; Dua, S. S.; Eaborn, C.; Walton, D. R. M.; J. Organomet. Chem. 1976, 117, 55. DOI: http://dx.doi.org/10.1016/S0022-328X(00)91902-8 63. Kosugi, M.; Shimizu, Y.; Migita, T.; Chem. Lett. 1977, 1423. DOI: http://dx.doi.org/10.1246/cl.1977.1423 64. Milstein, D.; Stille, J. K.; J. Am. Chem. Soc. 1978, 100, 3636. DOI: http://dx.doi.org/10.1021/ja00479a077 65. Kunze, B.; Jansen, R.; Hofle, G.; Reichenbach, H.; J. Antibiot. 1994, 47, 881. DOI: http://dx.doi.org/10.7164/antibiotics.47.881 PMID: 7928674 66. Jansen, R.; Washausen, P.; Kunze, B.; Reichenbach, H.; Hofle, G.; Eur. J. Org. Chem. 1999, 1085. DOI: http://dx.doi.org/10.1021/jo981466r 67. Dias, L. C.; Oliveira, L. G.; Org. Lett. 2001, 3, 3951. DOI: http://dx.doi.org/10.1021/ol016845c PMID: 11720577 68. Dias, L. C.; Oliveira, L. G.; Vilcachagua, D.; Nigsch, F.; J. Org. Chem. 2005, 70, 2225. DOI: http://dx.doi.org/10.1021/jo047732k PMID: 15760209 69. Jiang, L.; Job, G. E.; Klapars, A.; Buchwald, S. L.; Org. Lett. 2003, 5, 3667. DOI: http://dx.doi.org/10.1021/ol035355c PMID: 14507200 70. Barsby, T.; Kelly, M. T.; Andersen, R.; J. Nat. Prod. 2002, 65, 1447. DOI: http://dx.doi.org/10.1021/np0201321 PMID: 12398541 71. Dias, L. C.; Gonçalves, C. C. S.; Adv. Synth. Catal. 2008, 350, 1017. DOI: http://dx.doi.org/10.1002/adsc.200700589 72. Dias, L. C.; Jardim, L. S. A.; Ferreira, A. A.; Soarez, H. U.; J. Braz. Chem. Soc. 2001, 12, 463. DOI: http://dx.doi.org/10.1590/S0103-50532001000400003 73. Baba, S.; Negishi, E.; J. Am. Chem. Soc. 1976, 98, 6729. DOI: http://dx.doi.org/10.1021/ja00437a067 74. Negishi, E.; King, A. O.; Okukado, N.; J. Org. Chem. 1977, 42, 1821. DOI: http://dx.doi.org/10.1021/jo00430a041 75. Pilli, R. A.; Robello, L. G.; J. Braz. Chem. Soc. 2004, 15, 938. DOI: http://dx.doi.org/10.1590/S0103-50532004000600023 76. Harper, M. J. K.; Walpole, A. L.; Nature 1966, 212, 87. DOI: http://dx.doi.org/10.1038/212087b0 PMID: 5965580 77. Monteiro, A. L.; Nunes, M. C.; Limberger, J.; Poersch, S.; Seferin, M.; Synthesis, 2009, 16, 2761. DOI: http://dx.doi.org/10.1055/s-0029-1217600 78. Os autores também realizaram a síntese do (Z)-Tamoxifeno (100) através de uma rota que envolveu bromaçao do trans-estilbeno, desidrobrominaçao, reaçao de Suzuki, bromaçao e finalmente acoplamento de Negishi. No entanto, esta rota envolveu 5 etapas e rendimento global inferior (40%). 79. Durante a submissao desta revisao, o prof. Luiz Carlos Dias publicou a síntese do (-)-Marinisporolídeo C, talvez a maior e mais complexa molécula sintetizada em território nacional, na qual foi empregada a reaçao de Stille como uma das etapas chave: Dias, L. C.; de Lucca, E. C., Jr; Org. Lett. 2015, 17, 6278. DOI: http://dx.doi.org/10.1021/acs.orglett.5b03352 PMID: 26650100 80. Comasseto, J. V.; Pilli, R. A.; Simonelli, F.; J. Braz. Chem. Soc. 2001, 12, 565. DOI: http://dx.doi.org/10.1590/S0103-50532001000500001 81. O evento Brazilian Meeting on Organic Synthesis possui uma página dedicada na internet: http://www.bmos.com.br/
#Este artigo é dedicado ao professor Ronaldo Aloise Pilli (UNICAMP) pela passagem de seu 60º aniversário. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access