
 |
 |
 |
 |
 |
Artigo
| Fluorous supported synthesis of pyrazolone derivatives from allylic alcohols using a palladium-catalyzed strategy |
|
Nicolas GouaultI,*; Alexandre CariouI; Jean-François CupifI; Patrícia de Aguiar AmaralII; Michèle DavidI
IEquipe PNSCM, UMR 6226, ISCR, Université de Rennes1, 35000 Rennes, France Recebido em 23/12/2015 *e-mail: nicolas.gouault@univ-rennes1.fr Ten substituted pyrazolone derivatives were easily synthesized from perfluorinated allylic alcohols, via a straightforward two steps reaction involving a Heck-coupling/isomerization reaction and subsequent condensation of hydrazines. This fluorous supported methodology, that combines a cyclo-release approach and F-SPE purification allows for the obtaining of the final products with a good purity. INTRODUCTION Pyrazolones represent an important class of molecules that have been well-studied because of their relevant biological activities.1-4 They were first extensively developed for their anti-inflammatory,5 analgesic6 and antipyretic7 properties. More recently, great attention has been paid to their versatile role as antimicrobial,8 antiparasitic,9 antiviral10 and antineoplastic11 agents. For instance, edaravone (1) is a potent drug used in the treatment of brain ischemia12 and was shown to alleviate Alzheimer's disease-type pathologies,13 metamidole (2) is known to have antipyretic and analgesic activities,7 the pyrazole (3) is known to possess HIV-1 integrase inhibitory properties14 and (4) has idiopathic pulmonary fibrosis activity15 (Figure 1).
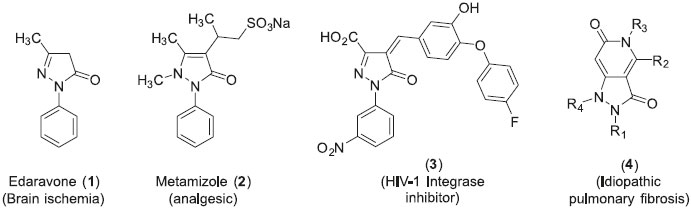 Figure 1. Representative examples of biologically active pyrazolone derivatives
The most common strategy (scheme 1) to obtain pyrazolones is the Knorr condensation of substituted hydrazines with β-ketoesters,16 the latter generally being obtained by Claisen condensation17 or from Meldrum's acid.18,19 A more recent approach to such β-ketoesters intermediates is the conversion of allylic alcohols to ketone in a Heck coupling process.20,21 It is obvious that described methods produce unwanted by-products (competition between 2 β-hydride eliminations in this process). In this respect, the development of an approach that allows an easy elimination of these would be of significance.
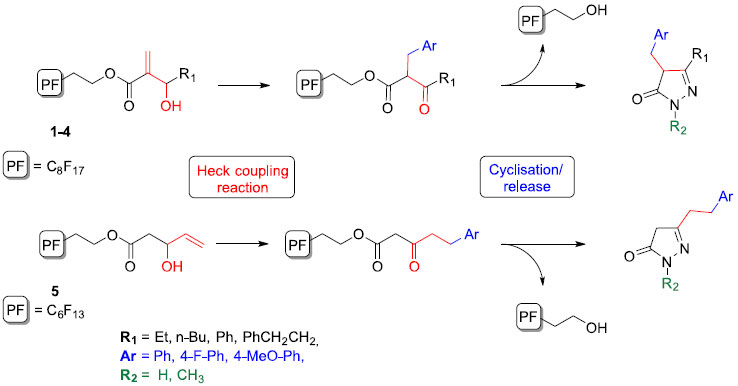 Scheme 1. Synthetic approach to pyrazolone derivatives from fluorous-supported allylic alcohols
Nowadays, in the field of combinatorial chemistry, one most attractive and versatile tool for the solid-phase parallel synthesis of pharmacologically active heterocyclic compounds is the cyclo-elimination strategy.22 Such an approach involves the simultaneous cyclization and release of the heterocyclic compound from the solid support resulting in the obtaining of the final product with the adequate purity for a biological evaluation, intermediates or by-products remaining on the support. We have previously reported the synthesis of diverse heterocycles using such cyclo-release approaches and fluorous tags,23-25 a methodological tool that was mainly developed to simplify the purification procedure in the parallel synthesis strategies.26-29 We herein report an efficient synthetic route to some structurally diverse pyrazolone derivatives from perfluorinated allylic alcohols. We investigated the palladium-catalyzed transformation of allylic alcohols to β-ketoesters and their subsequent cyclization into pyrazolones. Besides the practical benefits of fluorous-phase reactions, heterocyclization-cleavage strategy afford the opportunity to provide a final product with high purity. Our synthetic approach is outlined in scheme 1. Baylis-Hillman adducts and allylic alcohols are of particular interest as starting building blocks in the Heck coupling reaction since the β-hydride elimination of the organopalladium intermediate gives rise, in a regioselective manner, to β-keto ester derivatives, which upon reaction with hydrazines may give pyrazolones in a cyclative cleavage. Each step allows the introduction of diversity and purification steps can be considerably simplified. We expect this method to find extensive application in the fields of combinatorial chemistry, diversity-oriented synthesis and drug discovery.
EXPERIMENTAL General All reagents of high quality were purchased from commercial suppliers, and used without further purification. All reactions requiring anhydrous conditions were performed under an argon atmosphere using oven dried glassware. DMF and THF were distilled from CaH2 and Na/benzophenone, respectively. 1H and 13C NMR were recorded at 300 and 75 MHz, respectively, on a Bruker AM 300 spectrometer or at 270 MHz on a Jeol GSX 270 WB, using CDCl3 (and TMS as internal standard) or DMSO-d6. δ values are given in parts per million (ppm), coupling constants (J values) are given in Hertz (Hz), and multiplicity of signals are reported as follows: s, singlet; d, doublet; t, triplet; q, quadruplet; m, multiplet; bs, broad singlet. Thin layer chromatography was performed using precoated silica gel plate (0.2 mm thickness). All melting points are uncorrected. IR spectra were run on a PerkinElmer Spectrum 16 PC spectrometer; only selected absorbances are quoted. HRMS (EI) measurements were recorded on a Varian MAT 311 mass spectrometer at the Centre Régional de Mesures Physique de l'Ouest. F-SPE was performed using silica gel 60 C8-reversed phase perfluorinated (35-70 µm, Fluka), which was conditioned in a cartridge for under vacuum filtration. Chemistry Synthesis of Baylis-Hillman adducts (1-4) To a 3,3,4,4,5,5,6,6,7,7,8,8,9,9,10,10,10-heptadecafluorodecyl-acrylate (2 g, 3.86 mmol, 1 eq) was added DABCO (490 mg, 3.86 mmol, 1 eq) and 2 eq of aldehyde (R1CHO) in neat conditions. After stirring at room temperature for 48 h, the crude reaction mixture is diluted in CH2Cl2 (30 mL), washed three times with water and dried over anhydrous sodium sulphate. After filtration, the mixture was evaporated in vacuo, and then dissolved in 10 mL of [80/20] MeOH/H2O (v/v). 2 mL of this material was applied to the top of a fluorous silica gel cartridge (3 g) which had been preconditioned with [80/20] MeOH/H2O. The cartridge was eluted with [80/20] MeOH/H2O (2 x 10 mL), then with CH2Cl2 (2 x 10 mL). This operation was repeated 5 times on the same cartridge. The CH2Cl2 fractions were then combined and removal of the solvent in vacuo afforded a mixture of the desired fluorous Baylis-Hillman adduct and the fluorous alcohol. Perfluorinated Baylis-Hillman adduct (1): pale yellow oil (82%). 1H NMR (270 MHz, CDCl3): δH 0.96 (3H, t, H5, JH5-H4 = 7.4 Hz), 1.60-1.78 (2H, m, H4), 2.53 (2H, tt, Hb, JHb-Ha = 6.4 Hz, JHb-F = 18.1 Hz), 2.61 (1H, bs, -OH), 4.33-4.39 (1H, m, H3), 4.49 (2H, t, Ha, JHa-Hb = 6.4 Hz), 5.87 (1H, s, =CH2), 6.28 (1H, s, =CH2). 13C NMR (67.5 MHz, CDCl3): δC 10.1 (C5), 29.2 (C4), 30.6 (Cb, t, JCb-F = 21.8 Hz), 56.7 (Ca, bs), 72.9(C3), 126.0 (=CH2), 142.0 (C2), 166.1 (C1). 19F NMR (470.6 MHz, CDCl3): δF -80.8 (3F), -113.6 (2F), -121.7 (2F), -121.9 (4F), -122.8 (2F), -123.6 (2F), -126.2 (2F). FTIR: 1100 to 1300 (CF), 1629 (C=C methylene), 1719 (CO), 3430 (OH). HRMS (EI): (m/z) calcd for C16H13F17O3 576.0593, found 576.0504. Perfluorinated Baylis-Hillman adduct (2): pale yellow oil (72%). 1H NMR (270 MHz, CDCl3): δH 0.90 (3H, t, H7, JH7-H6 = 6.8 Hz), 1.26-1.73 (6H, m, H4,5,6), 2.42 (1H, bs, -OH), 2.53 (2H, tt, Hb, JHb-Ha = 6.4 Hz, JHb-F = 18.1 Hz), 4.38-4.45 (1H, m, H3), 4.49 (2H, t, Ha, JHa-Hb = 6.4 Hz), 5.87 (1H, s, =CH2), 6.26 (1H, s, =CH2). 13C NMR (67.5 MHz, CDCl3): δC 14.0 (C7), 22.5 (C6), 28.0 (C5), 30.6 (Cb, t, JCb-F = 21.8 Hz), 36.0 (C4), 56.7 (Ca, bs), 71.6 (C3), 125.8 (=CH2), 142.3 (C2), 166.1 (C1). 19F NMR (470.6 MHz, CDCl3): δF -80.9 (3F), -113.6 (2F), -121.7 (2F), -122.0 (4F), -122.8 (2F), -123.6 (2F), -126.2 (2F). FTIR: 1100 to 1300 (CF), 1627 (C=C methylene), 1720 (CO), 3425 (OH). HRMS: (m/z) calcd for C18H17F17O3 604.0906, found 604.0862. Perfluorinated Baylis-Hillman adduct (3): white solid (85%). 1H NMR (270 MHz, CDCl3): δH 2.43 (2H, tt, Hb, JHb-Ha = 6,4 Hz, JHb-F = 18.1 Hz), 2.80 (1H, bs, -OH), 4.42 (2H, t, Ha, JHa-Hb = 6.4 Hz), 5.58 (1H, s, H3), 5.93 (1H, s, =CH2), 6.38 (1H, s, =CH2), 7.30-7.37 (5H, m, Harom). 13C NMR (67.5 MHz, CDCl3): δC 30.4 (Cb, t, JCb-F = 21.8 Hz), 56.7 (Ca, bs), 73.1 (C3), 126.6; 126.9; 128.0; 128.5 (=CH2 and CH arom), 141.0 (CIV), 141.5 (CIV), 165.7 (C1). 19F NMR (470.6 MHz, CDCl3): δF -80.8 (3F), -113.6 (2F), -121.6 (2F), -121.9 (4F), -122.7 (2F), -123.5 (2F), -126.1 (2F). FTIR: 1100 to 1300 (CF), 1602 (C=C arom), 1630 (C=C methylene), 1718 (CO), 3470 (OH). HRMS (EI): (m/z) calcd for C20H13F17O3 624.0593, found 624.0642. Perfluorinated Baylis-Hillman adduct (4): colorless oil (72%). 1H NMR (270 MHz, CDCl3): δH 1.70 (1H, bs, -OH), 1.88-2.10 (2H, m, H4), 2.38-3.00 (4H, m, H5,b), 4.42-4.47 (3H, m, H3,a), 5.87 (1H, s, =CH2), 6.27 (1H, s, =CH2), 7.10-7.30 (5H, m, Harom). 13C NMR (67.5 MHz, CDCl3): δC 30.5 (Cb, t, JCb-F = 21.8 Hz), 32.1 (C4), 37.8 (C5), 56.8 (Ca, bs), 70.8 (C3), 126.0; 126.1; 128.5; 128.6 (=CH2 and CHarom), 141.6 (CIV), 142.1 (CIV), 166.0 (C1). 19F NMR (470.6 MHz, CDCl3): δF -80.8 (3F), -113.6 (2F), -121.7 (2F), -121.9 (4F), -122.7 (2F), -123.5 (2F), -126.2 (2F). FTIR: 1100 to 1300 (CF), 1603 (C=C arom), 1628 (C=C methylene), 1718 (CO), 3420 (OH). HRMS (EI): (m/z) calcd for [M-H2O]+. C22H15F17O2 634.0801, found 634.0829. Synthesis of perfluorinated allylic alcohol 5 To a stirred solution of 5 g of 1H,1H,2H,2H-perfluorooctanol (13.7 mmol) in 90 mL of dry CH2Cl2, under inert atmosphere, were added freshly distilled triethylamine (1.2 eq., 2.3 mL), then slowly acetyl chloride (1.2 eq., 1.2 mL) in 10 mL of dry CH2Cl2. After 18 h at room temperature, the reaction mixture was washed four times with 30 mL of water and this aqueous layer was extracted two times with 30 mL of CH2Cl2. The combined organic layers were dried (Na2SO4) and evaporated under reduced pressure to give the polyfluorooctyl acetate (98%). 1H,1H,2H,2H-perfluorooctyl acetate: colorless oil; FTIR: 1145 to 1234 (CF), 1369 (-OC-), 1751 (-OCO-) cm-1, 1H NMR (270 MHz, CDCl3): δH 2.00 (3H, s, H2); 2.45 (2H, m, H2'); 4.38 (2H, t, H1', J = 6.75 Hz);13C NMR (67.5 MHz, CDCl3) : δC 20.7 (C2), 30.5 (C2'), 56.3 (C1'), 170.6 (C1); 19F NMR (254 MHz, CDCl3): δF -81.3 (3F), -114.2 (2F), -122.4 (2F), -123.4 (2F), -124.1 (2F), -126.7 (2F). HRMS (EI): (m/z) calcd for C19H32O4 324.2301, found 324.2291. TLC: DCM (100%) R = 0.80. To a stirred solution of polyfluorooctyl acetate (2 g, 5.0 mmol) in dry THF (50 mL), under inert atmosphere, was added a solution of LHMDS (5.2 mL, 1.1 eq) at -78 °C and the resulting solution was stirred for 45 minutes. Then acrolein (0.5 mL, 1.5 eq.) and, ten minutes later, TMSCl (0.6 mL, 1.0 eq.) were added and the resulting solution was stirred for 4 hours at -78 °C. The reaction was then quenched by addition of sat. NH4Cl followed by addition of 1 mol L-1 HCl until the pH was acid. The mixture was extracted with diethyl ether. The organic layers were dried and evaporated under reduced pressure. The residue was purified through fluorous flash chromatography to give the ester (70%). 1H,1H,2H,2H-perfluorooctyl-3-hydroxypent-4-enoate (5): yellow oil; FTIR: 1144 to 1236 (CF), 1740 (-OCO-), 3435 (-OH) cm-1.; 1H NMR (270 MHz, CDCl3) δH 2.20-2.60 (4H, m, H2 and H2'), 4.28 (2H, t, H1', JH1'-H2' = 6.54 Hz), 4.50 (1H, m, H3), 5.30 (1H, dt, H5, JH5-H4 = 17.2 Hz, 1.2 Hz ), 5.70-5.95 (1H, ddd, H4, JH4-H5 = 17.1 Hz, 10.6 Hz, 5.7 Hz).; 13C NMR (67.8 MHz, CDCl3): δC 30.5 (C2'), 41.1 (C2), 56.6 (C1'), 68.9 (C3), 115.7 (C5), 138.6 (C4), 171.6 (C1), 19F NMR (254 MHz, CDCl3): δF -81.3 (3F), -114.2 (2F), -122.4 (2F), -123.4 (2F), -124.1 (2F), -126.7 (2F). TLC: DCM/AcOEt (90/10) R = 0.40. Representative procedure for the Heck coupling/isomerization reaction from BH adducts To a stirred solution of Baylis-Hillman adduct 1 (270 mg, 0.47 mmol) in THF (3 mL) was added NaHCO3 (138 mg, 3.5 eq.), NBu4Br (151 mg, 1 eq) and iodoanisole (329 mg, 3.0 eq). Then, the solution was degassed with nitrogen and the catalyst [Pd(OAc)2 (5 mg, 5 mol%)] was added. After the reaction mixture was stirred 18 hours under reflux in an oil bath, the crude mixture was allowed to cool at room temperature, and then diluted in 1 mL of diethyl ether. This suspension was applied over diatomaceous earth and washed with diethyl ether (3 x 10 mL). The filtrate was then washed three times with water and dried over anhydrous sodium sulphate. After filtration, the mixture was evaporated in vacuo, and then dissolved in 1 mL of [80/20] MeOH/H2O. The material was applied to the top of a fluorous silica gel cartridge (3 g) which had been preconditioned with [80/20] MeOH/H2O. The cartridge was eluted with [80/20] MeOH/H2O (2 x 10 mL), then diethyl ether (2 x 10 mL). The combined diethyl ether fractions were dried over anhydrous sodium sulfate. After filtration, solvent evaporation resulted in the desired perflurinated β-keto ester 6 (90%). 1H,1H,2H,2H-perfluorodecyl-2-(4-methoxybenzyl)-3-oxopentanoate (6): clear oil; 1H NMR (270 MHz, CDCl3) δH 0.98 (3H, t, J = 7.0 Hz), 2.20-2.60 (4H, m), 3.10 (2H, d, J = 7.6 Hz), 3.76-3.79 (1H, m), 3.77 (3H, s), 4.37 (2H, t, J = 6.5 Hz), 6.80 (2H, d, J = 8.6 Hz), 7.07 (2H, d, J = 8.6 Hz); 13C NMR (67.8 MHz, CDCl3): δC 7.4, 30.4 (t), 33.5, 36.4, 55.2, 57.1, 60.2, 114.1, 129.8, 158.5, 168.9, 205.0; TLC: DCM/AcOEt (90/10) R = 0.40. HRMS (ESI) (m/z): [M+Na]+ calcd for C23H19O4F17Na 705.0910, found 705.0909. Representative procedure for the Heck coupling/isomerization reaction from intermediate 5 To a stirred solution of ester 5 (0.3 g, 0.65 mmol) in dry DMF (5 mL) was added NaHCO3 (0.11 g, 2.0 eq.), NBu4Br (0.42 g, 2.0 eq.) and iodobenzene (73 µL, 1.0 eq). The reaction mixture is then degassed with nitrogen before adding the catalyst [Pd(OAc)2 (14.6 mg, 10 mol%)]. After the reaction mixture was stirred 18 hours at 50 °C, H2O was added and the resulting mixture was extracted with CH2Cl2. The organic layers were evaporated under reduced pressure and the residue was poured into H2O and extracted with diethyl ether. The organic layers were dried over Na2SO4 and evaporated under reduced pressure. The residue was purified through fluorous flash chromatography to give 7 (65%). 1H,1H,2H,2H-Perfluorooctyl 3-oxo-5-phenylpentanoate (7): brown oil; FTIR (cm-1): 698 and 746 (Arom), 1144 to 1235 (CF), 1751 (ester), 1719 (ketone); 1H NMR (270 MHz, CDCl3) δH 2.38-2.50 (2H, m), 2.83-2.91 (4H, m), 3.43 (2H, s), 4.40 (2H, t, J = 6.4 Hz), 7.15-7.27 (5H, m); 13C NMR (67.8 MHz, CDCl3): δC 29.4, 30,3, 44.6, 49.0, 57.1, 126.3, 128.3, 128.6, 140.3, 166.6, 201.2, 19F NMR (67.8 MHz, CDCl3): δF -81.2 (3F), -114.1 (2F), -122.4 (2F), -123.4 (2F), -124.0 (2F), -126.6 (2F). Representative procedure for the cyclization into 8-17 To a stirred solution of 6 (200 mg, 0.29 mmol) in ethanol (3 mL) was added hydrazine monohydrate (43 µL, 3 eq.). The resulting mixture was stirred under reflux for 3 hours. After cooling to room temperature, the desired product 8 precipitated and was collected after filtration. The precipitate was washed with cold ethanol to give 8 (66%) (in the case that products did not precipitate, they were purified through F-SPE). 3-Ethyl-4-(4-methoxybenzyl)-1H-pyrazol-5(4H)-one (8): white solid, mp: 176 °C; 1H NMR (400 MHz, DMSO-d6) δH 1.01 (3H, t, J = 7.5 Hz), 2.38 (2H, q, J = 7.5 Hz), 3.49 (2H, s), 3.69 (3H, s), 6.80 (2H, d, J = 8.4 Hz), 7.06 (2H, d, J = 8.4 Hz), 10.31 (2H, bs); 13C NMR (100 MHz, DMSO-d6): δC 13.4, 17.9, 26.3, 54.9, 99.5, 113.5, 128.8, 134.0, 142.4, 157.2, 159.6; HRMS (ESI) (m/z): [M+Na]+ calcd for C13H16N2O2Na 255.1109, found 255.1110. 3-Butyl-4-(4-methoxybenzyl)-1H-pyrazol-5(4H)-one (9): white solid, mp: 120 °C; 1H NMR (400 MHz, DMSO-d6) δH 0.79 (3H, t, J = 7.3 Hz), 1.15-1.20 (2H, m), 1.33-1.40 (2H, m), 2.35 (2H, t, J = 7.2 Hz), 3.48 (2H, s), 3.68 (3H, s), 6.79 (2H, d, J = 8.4 Hz), 7.04 (2H, d, J = 8.4 Hz), 10.31 (2H, bs); 13C NMR (100 MHz, DMSO-d6): δC 13.4, 17.9, 26.3, 54.9, 99.5, 113.5, 128.8, 134.0, 142.4, 157.2, 159.6; HRMS (ESI) (m/z): [M+Na]+ calcd for C15H20N2O2Na 283.1422, found 283.1421. 3-Phenyl-4-(4-methoxybenzyl)-1H-pyrazol-5(4H)-one (10): white solid, mp: 212 °C; 1H NMR (400 MHz, DMSO-d6) δH 3.68 (3H, s), 3.73 (2H, s), 6.81 (2H, d, J = 8.7 Hz), 7.05 (2H, d, J = 8.7 Hz), 7.30-7.45 (5H, m), 10.31 (2H, bs); 13C NMR (100 MHz, DMSO-d6): δC 26.6, 55.0, 99.9, 113.7, 126.4, 127.7, 128.7, 128.8, 130.8, 133.3, 140.0, 157.3, 160.5; HRMS (ESI) (m/z): [M+Na]+ calcd for C17H16N2O2Na 303.1109, found 303.1107. 3-(2-Phenylethyl)-4-(4-methoxybenzyl)-1H-pyrazol-5(4H)-one (11): white solid, mp: 173 °C; 1H NMR (400 MHz, DMSO-d6) δH 2.62-2.70 (4H, m), 3.45 (2H, s), 3.68 (3H, s), 6.79 (2H, d, J = 8.5 Hz), 7.07 (2H, d, J = 8.5 Hz), 7.10-7.30 (5H, m), 10.35 (2H, bs); 13C NMR (100 MHz, DMSO-d6): δC 26.3, 26.7, 55.0, 100.2, 113.5, 125.9, 128.2, 128.3, 128.9, 133.9, 140.4, 141.1, 157.2, 159.4; HRMS (ESI) (m/z): [M+H]+ calcd for C19H21N2O2 309.1603, found 309.1598. 3-(2-Phenylethyl)-1H-pyrazol-5(4H)-one (12): white solid, mp: 197 °C; 1H NMR (400 MHz, DMSO-d6) δH 2.70-2.76 (2H, m), 2.82-2.90 (2H, m), 5.23 (1H, s), 7.10-7.30 (5H, m), 11.14 (2H, bs); 13C NMR (100 MHz, DMSO-d6): δC 27.6, 34.5, 88.2, 126.0, 128.3, 141.2, 143.7, 160.8; HRMS (ESI) (m/z): [M+Na]+ calcd for C11H12N2ONa 211.0847, found 211.0848. 1-Methyl-3-(2-phenylethyl)-1H-pyrazol-5(4H)-one (13): white solid, mp: 104 °C; 1H NMR (400 MHz, DMSO-d6) δH 2.60-2.66 (2H, m), 2.79-2.85 (2H, m), 3.40 (3H, s), 5.15 (1H, s), 7.10-7.30 (5H, m), 10.73 (1H, bs); 13C NMR (100 MHz, DMSO-d6): δC 30.3, 32.5, 34.8, 84.7, 125.7, 128.2, 128.3, 141.8, 149.2, 152.3; HRMS (ESI) (m/z): [M+H]+ calcd for C12H15N2O 203.1184, found 203.1184. 3-(2-(4-Fluorophenyl)-ethyl)-1H-pyrazol-5(4H)-one (14): white solid, mp: 206 °C; 1H NMR (400 MHz, DMSO-d6) δH 2.68-2.74 (2H, m), 2.82-2.88 (2H, m), 5.22 (1H, s), 7.05-7.26 (4H, m), 10.34 (2H, bs); 13C NMR (100 MHz, DMSO-d6): δC 27.6, 33.6, 88.2, 115.0 (d, JC-F = 21.0 Hz), 130.1 (d, JC-F = 7.8 Hz), 137.3 (d, JC-F = 3.0 Hz), 143.5, 160.7 (d, JC-F = 241.1 Hz), 160.8; HRMS (ESI) (m/z): [M+Na]+ calcd for C11H11N2OFNa 229.0753, found 229.0753. 1-Methyl-3-(2-(4-fluorophenyl)-ethyl)-4-(4-methoxybenzyl)-1H-pyrazol-5(4H)-one(15): white solid, mp: 150 °C; 1H NMR (400 MHz, DMSO-d6) δH 2.59-2.64 (2H, m), 2.78-2.83 (2H, m), 3.39 (3H, s), 5.13 (1H, s), 7.04-7.26 (4H, m), 10.85 (1H, bs); 13C NMR (100 MHz, DMSO-d6): δC 30.4, 32.5, 33.9, 85.3, 114.9 (d, JC-F = 20.9 Hz), 130.1 (d, JC-F = 7.8 Hz), 137.9 (d, JC-F = 2.9 Hz), 149.1, 153.3, 160.7 (d, JC-F = 241.0 Hz); HRMS (ESI) (m/z): [M+Na]+ calcd for C12H13N2OFNa 243.0910, found 243.0908. 3-(2-(4-Methoxyphenyl)-ethyl)-1H-pyrazol-5(4H)-one (16): white solid, mp: 226 °C; 1H NMR (400 MHz, DMSO-d6) δH 2.65-2.80 (4H, m), 3.70 (3H, s), 5.22 (1H, s), 6.83 (2H, d, J = 8.7 Hz), 7.11 (2H, d, J = 8.7 Hz), 10.83 (2H, bs); 13C NMR (100 MHz, DMSO-d6): δC 27.8, 33.7, 55.0, 88.2, 113.7, 129.2, 133.1, 143.8, 157.6, 160.8; HRMS (ESI) (m/z): [M+Na]+ calcd for C12H14N2O2Na 241.0953, found 241.0953. 1-Methyl-3-(2-(4-Methoxyphenyl)-ethyl)-4-(4-methoxybenzyl)-1H-pyrazol-5(4H)-one (17): white solid, mp: 141 °C; 1H NMR (400 MHz, DMSO-d6) δH 2.55-2.80 (4H, m), 3.39 (3H, s), 3.71 (3H, s), 5.12 (1H, s), 6.83 (2H, d, J = 8.7 Hz), 7.12 (2H, d, J = 8.7 Hz), 10.68 (1H, bs); 13C NMR (100 MHz, DMSO-d6): δC 30.6, 32.5, 33.9, 54.9, 84.7, 113.6, 129.2, 133.7, 149.2, 157.4; HRMS (ESI) (m/z): [M+Na]+ calcd for C13H16N2O2Na 255.1109, found 255.1107.
RESULTS AND DISCUSSION We initially focused on the synthesis of fluorous substrates 1-5 for the Heck reaction. The starting Baylis-Hillman adducts 1-4 were synthesized according to our previous protocol.23 Commercially available fluorous acrylate was coupled to an aliphatic or aromatic aldehyde using DABCO in neat conditions at room temperature for 48 h (disappearance of the starting acrylate was monitored by TLC) to produce the corresponding fluorous-tagged derivatives 1-4 (Scheme 2).
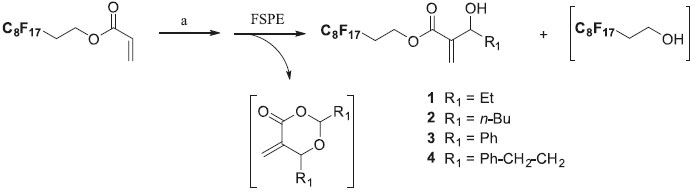 Scheme 2. (a) R1CHO (2 eq.), DABCO (1 eq.), r.t., 48 h, neat (72-85%)
Purification was accomplished using a fluorous silica gel chromatography. The crude reaction mixture was added to the fluorous silica column and the excess reagents and other non fluorous products were eluted following a fluorophobic wash (methanol and water in 80:20 ratio) whereas the fluorous adduct was retained onto the SPE cartridge until elution with a fluorophilic solvent (100% CH2Cl2). This purification method was rapid (10 min per compound) and generated desired compounds 1-4 with a 72-85% yield. In some instances, 1H NMR analyses also revealed the presence of a perfluorinated alcohol, resulting in a hydrolysis of the BH adducts into a dioxanone derivative due to the excess of aldehyde. However, these two byproducts did not induce further purification steps since the dioxanone was removed by the fluorophobic wash, and the fluorous alcohol did not interfere in the following steps. Synthesis of perfluorinated hydroxypentenoate 5 was accomplished conveniently in two steps as outlined in scheme 3.
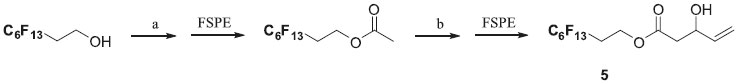 Scheme 3. (a) AcCl (1.2 eq.), TEA, DCM, r.t., 18 h (98%); (b) i. LHMDS (1.1 eq.), THF, -78 °C, 45 min.; ii. Acrolein (1.5 eq.), THF, -78 °C, 10 min.; iii. TMSCl (1.0 eq.), THF, -78 °C, 4 h. (70%)
Polyfluorooctanol was used as a perfluorous tag and gave under standard conditions30 (acetyl chloride and triethylamine in dichloromethane) the corresponding acetate in a very good yield (98%) after F-SPE purification. We next investigated the aldol condensation of this acetate with acrolein. Formation of lithium enolate was attempted with LDA or LHMDS as a base in THF at -78 or -50 °C. During these first attempts ester hydrolysis occurred and was easily confirmed by the presence of perfluorous alcool by 1H NMR analysis. The best result, 70% purified yield of 5 was obtained by treatment of perfluorinated acetate with LHMDS (1.1 eq.), followed by the addition of acrolein then trimethylsilyl chloride (1.0 eq.). Actually, it was shown that the yield could be significantly improved by the addition of trimethylsilyl chloride to stabilize the intermediate aldolate.30 With these perfluorinated allylic alcohols 1-5 in hand, we next focused on the tandem Mizoroki-Heck cross-coupling reaction and double-bond isomerization. This tandem Heck-isomerization reaction is well documented with allylic alcohols31,32 and Baylis-Hillman adducts.33-36 During this reaction, Heck reaction with an adequate palladium catalyst leads to the formation of the β-keto ester required for the end of the sequence. Basavaiah and Muthukumaran34 described a very efficient Heck reaction (using the 'Jeffery protocol')37 to convert Baylis-Hillman adducts into α-benzyl-β-keto esters avoiding the formation of mixture of products.34 Based on these conditions and after a few optimization, the fluorous Baylis-Hillman adduct 1 led, in the presence of 4-iodoanisole (3.0 eq.), NaHCO3 (3.5 eq.), Bu4NBr (1.0 eq.) and Pd(OAc)2 (5 mol%), in refluxing THF to the predominant formation of the β-keto ester 6 with a good yield (90%) after F-SPE purification (Scheme 4). However, 1H NMR analysis of the crude material revealed the presence of small amounts of byproduct A (about 10%).
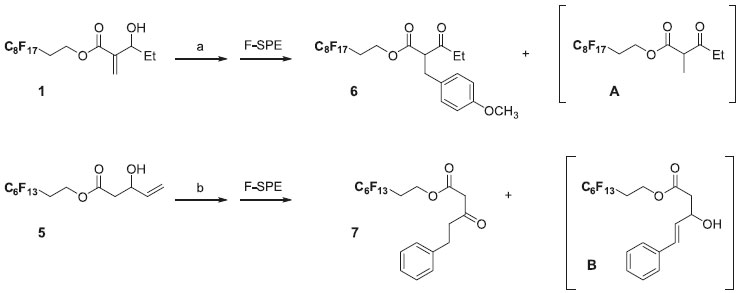 Scheme 4. (a) 4-Iodoanisole (3.0 eq.), NaHCO3 (3.5 eq.), Bu4NBr (1.0 eq.), Pd(OAc)2 (5 mol%), THF, reflux, 18 h (90%); (b) Iodobenzene (1.0 eq.), NaHCO3 (2.0 eq.), Bu4NBr (2.0 eq.), Pd(OAc)2 (10 mol%), DMF, 50 °C, 18 h (65%)
Similarly, fluorous allylic alcohol 5 was submitted to the same reaction conditions in order to introduce a second diversity. The reaction conditions were optimized using 5 and iodobenzene as the coupling partner. The best results were obtained when hydroxypentenoate 5 reacted with 4-fluoro-iodobenzene (1.0 eq.) in DMF at 50 °C for 18 hours in the presence of Pd(OAc)2 (10 mol%), NaHCO3 as base and Bu4NBr (1.0 eq.), thus affording the required derivative 6 in satisfactory isolated yield (65%). After F-SPE purification, presence of small amounts of compound B and prefluorinated alcohol -due to ester hydrolysis- (about 10% in each case) was observed by 1H NMR analysis of the crude material. However, these byproducts did not induce further purification step since they did not interfere in the following step. Once the starting materials were prepared and the Heck coupling and isomerization reactions to obtain β-keto esters validated, we planned to obtain a library of pyrazolones (Tables 1 and 2). The intermediates β-keto esters obtained from Baylis-Hillman adducts were condensed with hydrazine. The reaction was carried out in EtOH under reflux for 3 hours. After cooling to room temperature, the cyclized products 8-11 precipitated out from the reaction medium since they had low solubility in such a solvent, and were collected in a moderate overall yield by filtration.
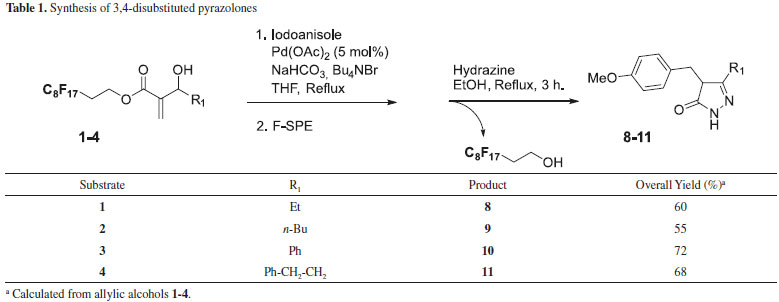
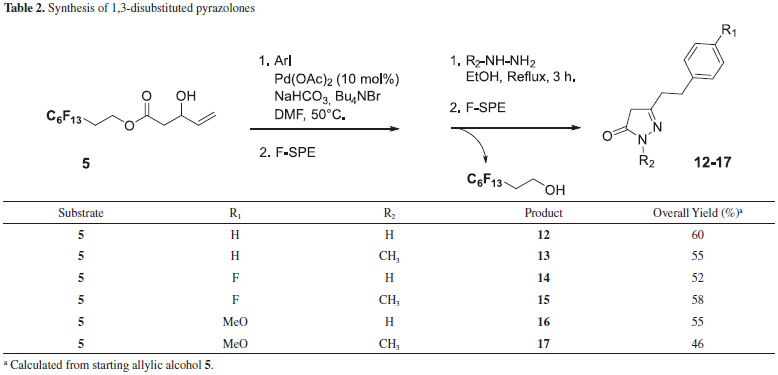
Compounds 12-17 were obtained in a similar manner starting from intermediate 5. The cyclized products did not precipitated out from the reaction medium when using methylhydrazine (compounds 13, 15 and 17). In this case, the reaction media was evaporated to remove solvent and the crude mixture was taken up with dichloromethane and washed with water. The residue was then purified through F-SPE to afford the substituted pyrazolones in the fluorophobic wash and complete removal of fluorous products in the fluorophilic wash. The efficiency of this purification step was good regarding the 1H NMR spectra of the final compounds. All the products were identified by 1H, 13C NMR and mass spectrometry. The 1H-pyrazolin-5-one presents three tautomeric forms due to their keto-enolic tautomerism, the full aromatic enol form (OH tautomer) being the predominent one (Figure 2).
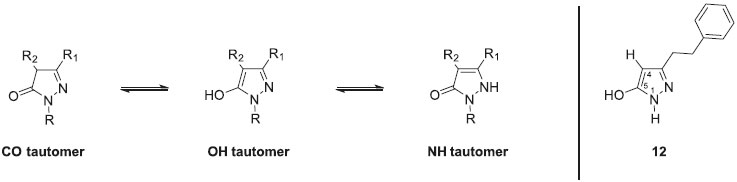 Figure 2. Tautomerism of the pyrazolin-5-one derivatives
Analysis of these compounds by NMR was not so easy in CDCl3 due to their existence as a mixture of three tautomeric forms (CO tautomer being predominent), whereas in DMSO-d6 they appeared as a single OH tautomer form. The OH tautomeric form was confirmed by 1H and 13C NMR spectra analyses. Actually, for 12 as an example, a two protons broad signal between 9 and 11 ppm due to the presence of two exchangeable protons (OH and NH), and a H-4 signal at 5.23 ppm confirmed this tautomeric form. Furthermore, the 13C NMR spectrum of this compound is also in accordance with the OH tautomer by the presence of a C-4 signal at 88.2 ppm. These chemical shifts are in accordance with the literature.38,39
CONCLUSIONS In summary, ten substituted pyrazolone derivatives were easily synthesized from perfluorinated allylic alcohols, via a straightforward two steps reaction involving a Heck-coupling/isomerization reaction and subsequent condensation of hydrazines that allows for a cyclo-release of the final products with a good purity. The combination of this approach and F-SPE purification represents an attractive method that has potential for automation to prepare libraries of compounds for high-throughput screening.
SUPPLEMENTARY MATERIAL 1H and 13C NMR spectra for compounds 8-17 are available at http://quimicanova.sbq.org.br, in pdf file with free access.
ACKNOWLEDGEMENTS The authors thank the Brazilian program Ciência Sem Fronteiras and CNPq for financial support and fellowships. We gratefully acknowledge the CRMPO for mass spectrometry experiments.
REFERENCES 1. Elguero, J. In Comprehensive Heterocyclic ChemistryII; Katritzky, A. R., Rees, C. W., Scriven, E. F. V., eds.; Pergamon Press: Oxford, 1996; Vol. 3, p 1. 2. Yet, L. In Comprehensive Heterocyclic ChemistryII; Katritzky, A. R., Ramsden, C. A., Scriven, E. F. V., Taylor, R. J. K., eds.; Elsevier: Oxford, 2008; Vol. 4, p 1. 3. Fustero, S.; Sanchez-Rosello, M.; Barrio, P.; Simon-Fuentes, A.; Chem. Rev. 2011, 111, 6984. DOI: http://dx.doi.org/10.1021/cr2000459 PMID: 21806021 4. Keri, R. S.; Chand, K.; Ramakrishnappa, T.; Nagaraja, B. M.; Arch. Pharm. Chem. Life Sci. 2015, 348, 299. DOI: http://dx.doi.org/10.1002/ardp.201400452 5. Brogden, R. N.; Drug 1986, 32, 60. DOI: http://dx.doi.org/10.2165/00003495-198600324-00006 6. Kuo, S.-C.; Huang, L.-J.; Nakamura, H.; J. Med. Chem. 1984, 27, 539. DOI: http://dx.doi.org/10.1021/jm00370a020 PMID: 6708056 7. Volz, M.; Kellner, H. M.; J. Clin. Pharmacol. 1980, 10, 299S. DOI: http://dx.doi.org/10.1111/j.1365-2125.1980.tb01813.x 8. Bondock, S.; Rabie, R.; Etman, H. A.; Fadda, A. A.; Eur. J. Med. Chem. 2008, 43, 2122. DOI: http://dx.doi.org/10.1016/j.ejmech.2007.12.009 PMID: 18255196 9. Orrling, K. M.; Jansen, C.; Vu, X. L.; Balmer, V.; Bregy, P.; Shanmugham, A.; England, P.; Bailey, D.; Cos, P.; Maes, L.; Adams, E.; van den Bogart, E.; Chatelain, E.; Ioset, J.-R.; van de Stolpe, A.; Zorg, S.; Veerman, J.; Seebeck, T.; Sterk, G. J.; de Esch, I. J. P.; Leurs, R.; J. Med. Chem. 2012, 55, 8745. DOI: http://dx.doi.org/10.1021/jm301059b PMID: 22963052 10. Sujatha, K.; Shanthi, G.; Selvam, N. P.; Manoharan, S.; Perumal, P. T.; Rajendran, M.; Bioorg. Med. Chem. Lett. 2009, 19, 4501. DOI: http://dx.doi.org/10.1016/j.bmcl.2009.02.113 PMID: 19482473 11. Mahajan, S. S.; Scian, M.; Sripathy, S.; Posakony, J.; Lao, U.; Loe, T. K.; Leko, V.; Thalhofer, A.; Schuler, A. D.; Bedalov, A.; Simon, J. A.; J. Med. Chem. 2014, 57, 3283. DOI: http://dx.doi.org/10.1021/jm4018064 PMID: 24697269 12. Okuyama, S.; Morita, M.; Sawamoto, A.; Terugo, T.; Nakajima, M.; Furukawa, Y.; Pharmaceuticals 2015, 8, 176. DOI: http://dx.doi.org/10.3390/ph8020176 13. Jiao, S.-S.; Yao, X.-Q.; Liu, Y.-H.; Wang, Q.-H.; Zeng, F.; Lu, J.-J.; Liu, J.; Zhu, C.; Shen, L.-L.; Liu, C.-H.; Wang, Y.-R.; Zeng, G.-H.; Parikh, A.; Chen, J.; Liang, C.-R.; Xiang, Y.; Bu, X.-L.; Deng, J.; Li, J.; Xu, J.; Zeng, Y.-Q.; Xu, X.; Xu, H.-W.; Zhong, J.-H.; Zhou, H.-D.; Zhou, X.-F.; Wang, Y.-J. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 5225. DOI: http://dx.doi.org/10.1073/pnas.1422998112 PMID: 25847999 14. Hadi, V.; Koh, Y.-H.; Sanchez, T. W.; Barrios, D.; Neamati, N.; Jung, K. W.; Bioorg. Med. Chem. Lett. 2010, 20, 6854. DOI: http://dx.doi.org/10.1016/j.bmcl.2010.08.057 PMID: 20864343 15. Laleu, B.; Gaggini, F.; Orchard, M.; Fioraso-Cartier, L.; Cagnon, L.; Houngninou-Molango, S.; Gradia, A.; Duboux, G.; Merlot, C.; Heitz, F.; Szyndralewiez, C.; Page, P.; J. Med. Chem. 2010, 53, 7715. DOI: http://dx.doi.org/10.1021/jm100773e PMID: 20942471 16. Knorr, L.; Chem. Ber. 1884, 17, 546. DOI: http://dx.doi.org/10.1002/cber.188401701152 17. Claisen, L.; Lowman, O.; Chem. Ber. 1887, 20, 651. DOI: http://dx.doi.org/10.1002/cber.188702001149 18. Meldrum, A. N.; J. Chem. Soc., Trans. 1908, 93, 598. DOI: http://dx.doi.org/10.1039/CT9089300598 19. Oikawa, Y.; Sugano, K.; Yonemitsu, O.; J. Org. Chem. 1978, 43, 2087. DOI: http://dx.doi.org/10.1021/jo00404a066 20. Basavaiah, D.; Muthukumaran, K.; Tetrahedron 1998, 54, 4943. DOI: http://dx.doi.org/10.1016/S0040-4020(98)00200-2 21. Amaral, P. A.; Petrignet, J.; Gouault, N.; Agustini, T.; Lohézic-Le Dévéhat, F.; Cariou, A.; Grée, R.; Eifler-Lima, V. L.; David, M.; J. Braz. Chem. Soc. 2009, 20, 1687. DOI: http://dx.doi.org/10.1590/S0103-50532009000900018 22. Nicolaou, K. C.; Hanko, R.; Hartwig, W.; Handbook of combinatorial chemistry. Drugs, catalysts, materials; Wiley-VCH: Weinheim, 2002; Vol. 1, p. 1114. 23. Le Lamer, A.-C.; Gouault, N.; David, M.; Boustie, J.; Uriac, P.; J. Comb. Chem. 2006, 8, 643. DOI: http://dx.doi.org/10.1021/cc060046+ PMID: 16961398 24. Horhant, D.; Le Lamer, A.-C.; Boustie, J.; Uriac, P.; Gouault, N.; Tetrahedron Lett. 2007, 48, 6031. DOI: http://dx.doi.org/10.1016/j.tetlet.2007.06.077 25. Le Lamer, A.-C.; Authier, H.; Rouaud, I.; Coste, A.; Boustie, J.; Pipy, B.; Gouault, N.; Bioorg. Med. Chem. Lett. 2014, 24, 3819. DOI: http://dx.doi.org/10.1016/j.bmcl.2014.06.062 PMID: 25027935 26. Studer, A.; Hadida, S.; Ferrito, R.; Kim, S.-Y.; Jeger, P.; Wipf, P.; Curran, D. P.; Science 1997, 275, 823. DOI: http://dx.doi.org/10.1126/science.275.5301.823 PMID: 9012347 27. Zhang, W.; Chem. Rev. 2004, 104, 2531. DOI: http://dx.doi.org/10.1021/cr020738u PMID: 15137799 28. Curran, D. P.; Luo, Z.; J. Am. Chem. Soc. 1999, 121, 9069. DOI: http://dx.doi.org/10.1021/ja991496r 29. Eifler-Lima, V. L.; Amaral, P. A.; Rev. Virtual Quim. 2010, 2, 298. DOI: http://dx.doi.org/10.5935/1984-6835.20100027 30. Eames J.; Khanom H.; Molecules 2004, 9, 266. DOI: http://dx.doi.org/10.3390/90500266 PMID: 18007430 31. Larock, R. C.; Leung, W. Y.; Stolz-Dunn, S.;Tetrahedron Lett. 1989, 30, 6629. DOI: http://dx.doi.org/10.1016/S0040-4039(00)70636-8 32. Bollikonda, S.; Mohanarangam, S.; Jinna, R. R.; Kandirelli, V. K. K.; Makthala, L.; Sen, S.; Chaplin, D. A.; Lloyd, R. C.; Mahoney, T.; Dahanukar, V. H.; Oruganti, S.; Fox, M. E.; J. Org. Chem. 2015, 80, 3891. DOI: http://dx.doi.org/10.1021/acs.joc.5b00197 PMID: 25807000 33. Ferrera, B. R. V.; Pirovani, R. V.; Souza-Filhi, L. G.; Coelho, F.; Tetrahedron 2009, 65, 7712. DOI: http://dx.doi.org/10.1016/j.tet.2009.06.084 34. Basavaiah, D.; Muthukumaran, K.; Tetrahedron 1998, 54, 4943. DOI: http://dx.doi.org/10.1016/S0040-4020(98)00200-2 35. Kulkarni, B. A.; Ganesan, A.; J. Comb. Chem. 1999, 1, 373. DOI: http://dx.doi.org/10.1021/cc990007g 36. Coelho, F.; Veronese, D.; Pavam, C. H.; De Paula, V. I.; Buffon, R.; Tetrahedron 2006, 62, 4563. DOI: http://dx.doi.org/10.1016/j.tet.2006.02.045 37. Jeffery, T. J.; J. Chem. Soc., Chem. Commun. 1984, 1287. DOI: http://dx.doi.org/10.1021/ja00318a006 38. Isaad, J.; Tetrahedron 2013, 69, 2239. DOI: http://dx.doi.org/10.1016/j.tet.2013.01.038 39. Metwally, M. A.; Bondock, S. A.; El-Desouky, S. I.; Abdou, M. M.; Int. J. Modern Org. Chem. 2012, 1, 19. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access