
 |
 |
 |
 |
 |
Artigo
|
|
| Diseño y caracterización teórica de materiales fotoactivos eficientes basados en derivados de naftopirrol y de naftotiofeno para celdas solares orgánicas Theoretical characterization and design of efficient photoactive materials based on naphthopyrrole and naphthothiophene derivatives aimed towards organic solar cells |
|
Walter J. Cuadro*; Adolfo E. Ensuncho; Juana R. Robles
Departamento de Química, Universidad de Córdoba. Montería, Colombia Recebido em 06/02/2016 *e-mail: w1534@hotmail.com In this research we have designed electron donors D-π-A type containing two different π fragments to obtain naphthopyrrole (D-NPR-A) and naphthotiophene (D-NTP-A) derivatives, proposed for the use in organic bulk hetero-junction (BHJ) solar cells (OSCs). These derivatives were characterized by DFT and TD-DFT calculations. For all the electron donors the anchorage fragment was 2-methylenemalononitrile, while the chromophore fragment was spanned between diphenylamine, triphenylamine, thiophene. Properties affecting open-circuit photovoltage (VOC) and short-circuit photocurrent (JSC) from D-π-A type derivatives, such as geometric structure, frontier-molecular orbital energies, exciton driving force energy, natural bond orbital analysis, absorption spectra and light harvesting efficiency. Energy from HOMO and LUMO orbitals was discussed. Theoretical calculations from TD-DFT within Coulumb attenuation method CAM-B3LYP were able to predict excited state properties. The electron donors D-π-A type exhibit photoelectric conversion efficiency above 10%, being the naphthopyrrole derivatives (D-NPR-A) along with the [6,6]-phenyl-C61-butyric acid methyl ester (PC61BM) the complexes with higher photoelectric properties, these complexes are proposed as photoactive materials in the construction of organic bulk hetero-junction solar cells. INTRODUCCION Con la creciente demanda mundial de energía combinada con el agotamiento de los recursos derivados del petróleo y los riesgos de calentamiento global se ilustra la necesidad urgente de una transición hacia fuentes alternativas de energía renovable.1 Siendo las celdas solares orgánicas (OSCs) libres de metales una potencial alternativa "verde" para la generación de energía eléctrica, ya que estas son comercialmente atractivas por su bajo costo de fabricación, semitransparentes, bajo peso, flexibilidad física, fácil integración, producción continua utilizando herramientas de impresión y su desempeño relativamente bueno en presencia de luz solar difusa, además este tipo de celdas son sostenibles y amigables con el ambiente.1-5 Recientemente, las celdas solares orgánicas (OSC) han surgido como una nueva tecnología emergente para competir con las celdas solares tradicionales basadas en Silicio.2,6,7 Para llevar esta tecnología a una gran escala y hacerla comercialmente viable es necesario alcanzar una eficiencia de conversión fotoeléctrica (PCE) superior al 10% y garantizar un tiempo de vida útil de 10 años (mayor estabilidad fotoquímica).8-11 La capa fotoactiva de las OSCs se constituye por dos componentes, uno electrodonador (D) y otro electroaceptor (A), ensamblados generalmente en una estructura bi-capa (tipo wafer) o en forma de mezcla, conocida como heterounión de volumen (BHJ).1,12,13 Las OSCs del tipo BHJ han incrementado en un factor de 10 la PCE respecto a los dispositivos convencionales en forma de bi-capa.2 Diferentes estudios muestran que los componentes electroaceptores con mejores propiedades semiconductoras son los derivados de fullereno, por ser excelentes captadores de electrones, amigables con el ambiente e incrementar la estabilidad de la capa fotoactiva, por tanto, mejoran las propiedades fotovoltaicas de las OSCs del tipo BHJ.2,13-20 Siendo el éster metílico del ácido [6,6]-fenil-C61-butírico (PC61BM) uno de los más promisorios. Por otro lado tenemos a la molécula electrodonadora que está conformada por un fragmento cromóforo, un fragmento enlazador π-conjugado y un fragmento aceptor de anclaje, denotado como sistema electrodonador [D-π-A] y esta configuración permite una mayor captura de luz del espectro solar.6,8,9,14,21-24 Una gran variedad de cromóforos se han estudiado como trifenilmetanos, azulenos, indolinas, perilenos, cumarinas, cianinas, hemicianinas, carotenoides, fenotiazinas, fenoxazinas, etc.6,7,21,23,25-27 Respecto a los enlazadores o puentes π-conjugados generalmente se utilizan grupos heterocíclicos, por su mayor estabilidad ambiental y como fragmentos aceptores de anclaje se tienen funciones de ácido carboxílico, de ácido malónico ó de ácido cianoacrílico.6,9,21-24,26-31 En el desarrollo de las OSCs los grupos cromóforos trifenilamina, difenilamina y derivados de tiofeno exhiben excelentes propiedades ópticas y fotovoltaicas, como también los derivados del ácido malónico en el fragmento aceptor.8,9,18,23,27 Paralelamente a los nuevos resultados experimentales sobre estos materiales orgánicos fotoactivos, las investigaciones teóricas han comenzado a constituir una importante fuente de información que complementa los estudios experimentales, contribuyendo así en el diseño racional y en la predicción de propiedades ópticas, electrónicas y fotovoltaicas en los sistemas captadores de luz a nivel molecular y a su vez reduce el grado de empirismo en estas investigaciones.1,14,26,28 En el marco de la química computacional se han publicado numerosos estudios utilizando diferentes metodologías como la Teoría del Funcional de la Densidad (DFT) y la Teoría del Funcional de la Densidad Dependiente del Tiempo (TD-DFT) para diseñar, evaluar y predecir propiedades fotovoltaicas de los materiales fotoactivos utilizados en OSCs.1,6,8,9,14,21-23,27,31-35 Por tal razón, muchos investigadores sugieren que los métodos de química cuántica podrían ilustrarnos moléculas electrodonadoras eficientes y proporcionar una ayuda visual para comprender como la estructura molecular y sus propiedades afectan el rendimiento del dispositivo fotovoltaico, minimizando costos y ahorrando esfuerzos de síntesis.9 En 2006, Scharber et al.,13 propone un modelo empírico que describe como estimar la PCE de las celdas solares de BHJ, y para ello requiere el conocimiento de los orbitales frontera HOMO-LUMO de los componentes electrodonador y electroaceptor del material fotoactivo. El modelo Scharber ha tenido una gran acogida por los investigadores por su versatilidad y además predice claramente algunas propiedades fotovoltaicas tales como, energía del gap (ΔEH-L), densidad de fotocorriente de corto circuito (JSC), fotovoltaje de circuito abierto (VOC) y la PCE cuando se utiliza en conjunto con la metodología DFT.2,17,20,33,34 Sin embargo, Bérubé et al.,10 observaron una sobrestimación en las propiedades electrónicas y fotovoltaicas de algunos componentes fotoactivos, que fueron calculadas con la DFT en estudios teóricos comparado con los datos experimentales. Donde la estimación de estas propiedades fue ajustada linealmente con los datos y así se obtuvo una desviación estándar menor a 0,2 eV en la predicción de la propiedad después de la respectiva correlación. En el presente estudio, nosotros investigamos teóricamente el éster metílico del ácido [6,6]-fenil-C61-butírico (PC61BM) como componente electroaceptor y cinco componentes electrodonadores del tipo D-π-A utilizando la DFT y la TD-DFT, señalando los siguientes derivados Da-NPR-A, Db-NPR-A, Dc-NPR-A, Da-NTP-A y Db-NTP-A que pueden ser considerados moléculas D-π-A, donde los grupos trifenilamina (Da), difenilamina (Db) y tiofeno (Dc) son utilizados como fragmento cromóforo (R2); el nafto[2,3-c]pirrol (NPR) y el nafto[2,3-c]tiofeno (NTP) son utilizados como fragmento π enlazador; y el 2-metilen-malononitrilo (A) es utilizado como fragmento aceptor de anclaje (R1) (Ver Figura 1).
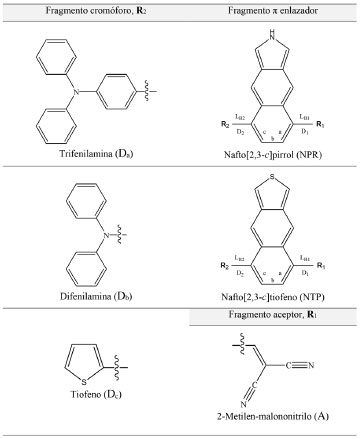 Figura 1. Fragmentos para la construcción de los derivados D-π-A de los componentes electrodonadores D-NPR-A y D-NTP-A
METODOLOGIA Antecedentes teóricos El modelo Scharber,2,13 es un modelo empírico para predecir PCE de las celdas solares orgánicas (OSCs) con una arquitectura de heterounión de volumen (BHJ), donde se estiman teóricamente las propiedades fotovoltaicas de estos dispositivos a partir de la energía de los orbitales frontera HOMO - LUMO. La eficiencia de conversión fotoeléctrica (PCE) de una celda fotovoltaica bajo irradiación solar (por ejemplo AM 1.5G) puede ser determinada por la densidad de fotocorriente de corto circuito (JSC) y el fotovoltaje de circuito abierto (VOC). La PCE puede ser expresada por la siguiente ecuación:8  donde FF es el factor de forma o de relleno eléctrico, el cual es proporcional a la potencia máxima de la celda solar y Pinc es la potencia de luz incidente. La relación entre JSC, VOC, FF y las estructuras electrónicas de los componentes fotoactivos es todavía desconocida. Generalmente, la densidad de fotocorriente de corto circuito JSC puede ser determinada como:36 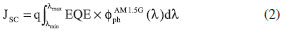 donde, q es la carga elemental, EQE es la eficiencia cuántica externa y El fotovoltaje de circuito abierto VOC puede ser determinado por: 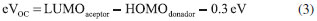 donde e es la carga elemental, LUMOaceptor y HOMOdonador es la energía del orbital frontera del PC61BM y los electrodonadores tipo D-π-A respectivamente y el valor de 0.3 eV es un factor empírico asociado a las interfaces de los componentes que constituyen el material fotoactivo.2,10,13 Para estudiar un poco más fondo el comportamiento del material fotoactivo de las OSCs de BHJ, nosotros investigamos la energía de la fuerza motriz del excitón (ΔE) y la eficiencia de captura de luz (LHE). Métodos computacionales Todos los cálculos en este estudio fueron desarrollados con el paquete de Software Gaussian 03.37 La teoría del funcional de la densidad (DFT) con el funcional de intercambio de tres parámetros de Becke,38 y el funcional de correlación Lee-Yang-Parr (B3LYP),39 y el conjunto base 6-31G(d) fue empleado para investigar la optimización geométrica del estado fundamental (S0) en fase gas de la molécula electroaceptora y las moléculas electrodonadoras del tipo D-π-A. Para la estimación de la energía de los orbitales frontera HOMO - LUMO de los componentes fotoactivos se tuvieron en cuenta las ecuaciones ajustadas propuestas por Bérubé et al.10 Las cuales pueden ser expresadas como: 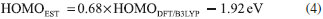 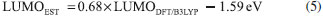 El análisis del orbital natural de enlace (NBO) se llevo a cabo mediante un análisis poblacional de cargas (NPA), utilizando el programa NBO 3.1 incluido en el paquete de software de Gaussian a través de cálculos a punto simple al mismo nivel de teoría.6,21-23,31 En trabajos recientes, se ha observado que los estados de transferencia de carga en sistemas D-π-A se describen mejor cuando se utiliza un alto porcentaje de intercambio HF en el funcional.6,8,22,27 Por lo tanto, para la evaluación de las propiedades del estado excitado se implementaron cálculos a punto simple utilizando un funcional con interacciones de largo alcance TD-CAM-B3LYP.40
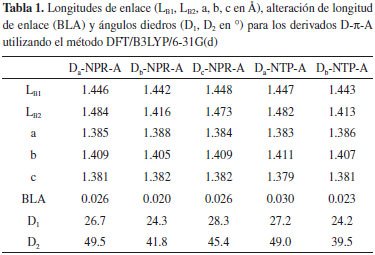
RESULTADOS Y DISCUSION Propiedades geométricas Para comprender la transferencia de carga intramolecular y las propiedades de empaquetamiento π-π intermolecular en moléculas orgánicas π-conjugadas es necesario identificar los factores estructurales que son influenciados por la arquitectura molecular. En particular, las propiedades de transferencia de carga intramolecular pueden ser analizadas en función de las longitudes de enlace (LB1 y LB2), la alteración de longitud de enlace (BLA) y los ángulos diedros (D1 y D2) (Figura 1) calculados entre el fragmento R1/R2 y el fragmento π-enlazador de las moléculas D-π-A se encuentran listados en la Tabla 1.
Las estructuras optimizadas de los electrodonadores D-NPR-A, D-NTP-A y el electroaceptor PC61BM se muestran en la Figura 2, obtenidos con el nivel de teoría B3LYP/6-31G(d) en el vacío.
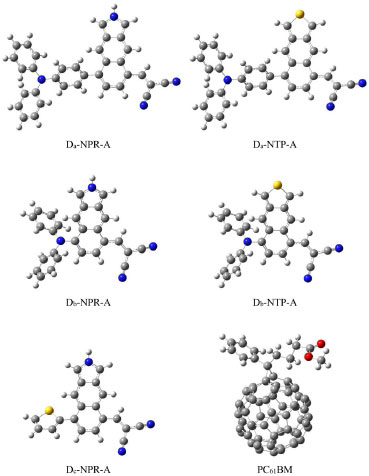 Figura 2. Estructuras optimizadas de los derivados D-NPR-A, D-NTP-A y PC61BM
El fragmento difenilamina (Db) de las moléculas D-NPR-A y D-NTP-A, se muestra como un grupo donador de electrones relativamente fuerte comparado con los otros fragmentos, por lo cual, el átomo de N con su par de electrones libres puede tomar parte importante en los procesos de transferencia de carga del fragmento cromóforo al aceptor en toda la molécula.8,9 Y esto se observa con longitudes de enlaces más cortos LB2 (1.416 - 1.413 Å) entre los fragmentos Db y los fragmentos π-enlazadores de los derivados D-π-A (Tabla 1). El fragmento aceptor 2-Metilen-malononitrilo (A) es el mismo para todos los derivados D-π-A y de igual forma el fragmento Db influye en la disminución de la longitud de enlace LB1 (1.443 - 1.442 Å) (Tabla 1) entre el fragmento π-enlazador y el fragmento aceptor de anclaje. En este análisis puede considerarse que el fragmento Db mejora las propiedades de transferencia de carga intramolecular comparado con los fragmentos Da y Dc. Para estimar con precisión el grado de conjugación del fragmento π-enlazador empleamos el parámetro de alteración de longitud de enlace (BLA),9,17 utilizando las longitudes de enlace del anillo aromático descritas en la Figura 1. El BLA puede ser expresado como:  Los resultados muestran que las moléculas Db-NPR-A y Db-NTP-A poseen los más bajos BLA (0.020 - 0.023) (Tabla 1), lo cual significa que los electrones π tienen buena deslocalización y redistribución, mejorando la transferencia y transporte de carga intramolecular y además garantiza un desplazamiento distintivo hacia el rojo como respuesta espectral.9 En la Tabla 1, se observan los ángulos diedros (D1 y D2) (Figura 1) de los derivados D-π-A, los cuales se encuentran en un intervalo de 24 ° a 49 °. Dando lugar, a los efectos estéricos entre los diferentes fragmentos que constituyen la molécula electrodonadora y por tanto, estos sistemas moleculares exhiben una leve conformación no planar (Figura 2), lo cual puede influir en cierta medida en la transferencia de carga intramolecular y el empaquetamiento π-π en el estado sólido.8,9 Y estos resultados muestran que las moléculas Db-NPR-A y Db-NTP-A poseen los ángulos diedros más bajos (24.3 °/41.8 °) y (24.2 °/39.5 °) respectivamente. Propiedades electrónicas En las OSCs, la energía de los orbitales frontera HOMO - LUMO de los componentes fotoactivos tiene una estrecha relación con las propiedades fotovoltaicas.10 Por lo tanto, empleamos las ecuaciones (4) y (5) propuestas por Bérubé et al.,10 para la estimación de la energía de los orbitales moleculares HOMOEST - LUMOEST respectivamente, ya que su determinación es un paso crucial en el diseño de nuevos materiales fotovoltaicos y los resultados se encuentran registrados en la Tabla 2.
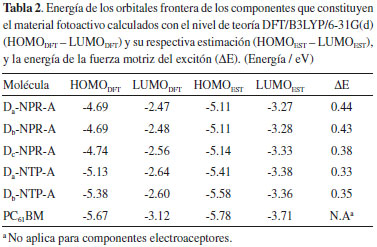
En la Tabla 2, se observa que la energía del orbital HOMOEST de los derivados D-NPR-A posee un valor promedio de -5.12 eV y en los derivados D-NTP-A el promedio es de -5.49 eV, exhibiendo una mayor conjugación π en los primeros derivados debido a la mayor contribución electrónica del átomo de N del anillo pirrólico en el fragmento π-enlazador y esto se manifiesta con valores más bajos en la energía del gap (ΔEH-L) y consecuentemente, mejorando las propiedades fotovoltaicas.8 En lo que respecta a la energía del orbital LUMOEST los valores son muy similares en todos los derivados D-π-A, en promedio -3.32 eV. Wu et al.,7 reportaron la energía de los orbitales HOMO-LUMO del electroaceptor PC61BM (-6.10 eV / -3.70 respectivamente) obtenidos de forma experimental, los cuales tienen mucha correlación con los datos teóricos registrados en la Tabla 2 (-5.78 eV / -3.71 eV respectivamente). Energía de la fuerza motriz del excitón Un parámetro muy importante que ayuda a predecir el grado de transferencia de carga eficiente entre los componentes del material fotoactivo de las OSCs, es la estimación de la energía de la fuerza motriz del excitón (ΔE), la cual se puede calcular como una diferencia de energía de orbitales LUMOs entre la molécula electrodonadora y el electroaceptor (PC61BM).32,34 La ΔE puede ser expresada como:9 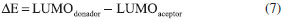 Los resultados muestran que la energía (ΔE) de todos los derivados D-π-A poseen un valor superior a 0.3 eV (ver Tabla 2), por lo cual se puede garantizar una división eficiente de los excitones y se asegura una eficiente transferencia de electrones entre el componente electrodonador y el electroaceptor, y a demás se minimizan las pérdidas de energía por recombinación de los portadores de carga.8,33 También se observa que los derivados D-NPR-A poseen los valores más altos de ΔE mejorando así este parámetro y esto se debe a la alta densidad electrónica que le imprime el átomo de N del anillo pirrólico a la molécula como un todo. Poblaciones electrónicas Para analizar la distribución de carga y el mecanismo de transferencia electrónica de los derivados D-π-A, el análisis del orbital natural de enlace (NBO) se llevo a cabo a partir de las estructuras optimizadas en el estado fundamental (S0) obtenidas con el nivel de teoría DFT/B3LYP/6-31G(d). Las cargas naturales calculadas que poblaron los fragmentos cromóforos, los π enlazadores y el aceptor de anclaje de los derivados electrodonadores se registran en la Tabla 3.
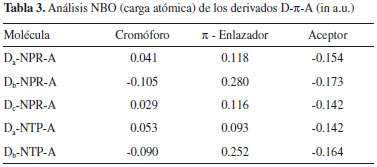
Los datos sugieren que la carga en el fragmento aceptor son bastante similares en los derivados D-π-A, lo cual indica que el 2-Metilen-malononitrilo (A) es un excelente grupo extractor de electrones. Los resultados muestran que el fragmento cromóforo difenilamina (Db) induce a una acumulación de carga negativa (Tabla 3) y esto se debe a la elevada electronegatividad del átomo de N. Esto significa que el fragmento Db puede actuar como un extractor de electrones y por tanto, facilitar los procesos de transferencia de carga intramolecular. En lo que respecta a los fragmentos π-enlazadores tienen una doble función, una es servir como donador de electrones y la otra es mejorar la conjugación electrónica.6,21,23,27 Propiedades ópticas Para los materiales fotoactivos de las OSCs, las moléculas electrodonadoras juegan un papel importante en la absorción de luz solar porque el electroaceptor PC61BM tiene una absorción débil en la región visible y el infrarrojo cercano. Para investigar las propiedades de absorción de estos electrodonadores, tales como energías de excitación, osciladores de fuerza para excitaciones electrónicas, composición de las transiciones verticales y espectros de absorción UV/VIS nosotros utilizamos TDDFT con el funcional CAM-B3LYP.8,27 Los correspondientes parámetros espectroscópicos de los derivados D-π-A fueron resumidos en la Tabla 4 y las curvas de absorción simuladas son presentadas en la Figura 3.
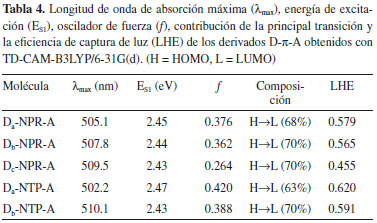
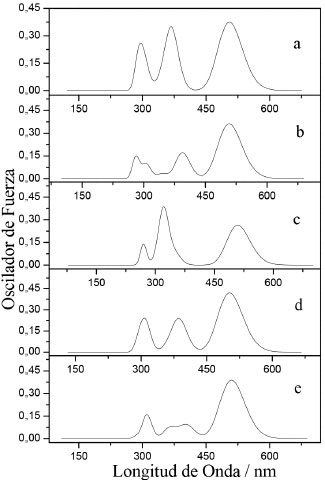 Figura 3. Espectros de absorción simulados de los derivados D-π-A. (a) Da-NPR-A, (b) Db-NPR-A, (c) Dc-NPR-A, (d) Da-NTP-A y (e) Db-NTP-A
Los espectros muestran un perfil similar para todos los derivados, los cuales presentan una banda principal intensa entre los 450 nm y 600 nm. Esta banda corresponde a la transición dominante de electrones del HOMO al LUMO, es decir, del orbital molecular π (fragmentos cromóforos - π-enlazador) al orbital π* (fragmento aceptor) y este proceso puede ser adscrito a la transferencia de carga intramolecular. Los picos de absorción máximo calculados (λmax) también son atribuidos a la transición HOMO - LUMO, los cuales están alrededor de los 505 nm (Tabla 4). En general, la posición y el ancho de la banda principal en el espectro son dos parámetros que pueden estar relacionados con la eficiencia del material electrodonador, ya que amplitudes mayores en regiones de baja energía (visible - infrarrojo cercano) favorecen los procesos de captación de luz solar.9,27 En la Tabla 4, nosotros observamos que las energías de excitación (ES1) son relativamente bajas en todos los derivados D-π-A, lo cual indica que podrían capturar más luz a longitudes de onda más altas y a su vez incrementar la PCE de la celda solar.27 Además de las propiedades de absorción, la correspondiente eficiencia de captura de luz (LHE) de estos derivados D-π-A fue estimada, la cual es bastante aproximada con la magnitud del oscilador de fuerza (f) asociado a la longitud de onda (λmax). La LHE puede ser expresada como:41  La LHE es un factor importante en las OSCs, ya este valor da una impresión intuitiva de la eficiencia del componente electrodonador en el proceso de absorción de fotones. De acuerdo a la Ecuación 8, los valores altos de f se reflejan en una mayor LHE. Se muestra en la Tabla 4, los valores de la LHE de los derivados D-π-A que se encuentran en el rango 0.450 - 0.620. Esto significa que todos los electrodonadores poseen una sensibilidad similar frente a la luz solar. Pero además se observa una mayor contribución por parte de los fragmentos trifenilamina (Da) y difenilamina (Db) a la LHE.9,27 Propiedades fotovoltaicas Las OSCs basadas en una estructura de BHJ generalmente representan una mezcla de un electrodonador π conjugado con un electroaceptor derivado del fullereno.2,7-9,14,18 Por tanto, examinamos el comportamiento fotovoltaico de los derivados D-π-A mezclados con el PC61BM, el cual es un electroaceptor ampliamente utilizado en dispositivos relacionados con celdas solares. Los principales parámetros fotovoltaicos se relacionan en la Tabla 5.
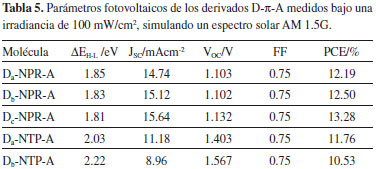
Las propiedades fotovoltaicas estimadas de los derivados D-NPR-A y D-NTP-A registradas en la Tabla 5 fueron determinadas teóricamente y hasta el momento no se han reportado valores experimentales. El valor de la energía del gap (ΔEH-L) puede ser determinada como la diferencia entre la energía del orbital LUMO y el orbital HOMO del componente electrodonador D-π-A.9 Basados en la Ecuación (2), en la energía ΔEH-L y el espectro de irradiancia solar AM 1.5G (ASTM G173),42 se estimo la densidad de fotocorriente de corto circuito (JSC) para todos los derivados D-π-A, asumiendo una eficiencia cuántica externa (EQE) del 80%.2 Los resultados muestran que los derivados D-NPR-A poseen los valores más bajos en la energía ΔEH-L y debido a esto se incrementa sustancialmente la JSC y la eficiencia de conversión fotoeléctrica (PCE) (Ver Tabla 5). Para calcular el fotovoltaje de circuito abierto (VOC) de los derivados D-π-A se utilizó la Ecuación (3), y se puede observar que los derivados del naftotiofeno (D-NTP-A) poseen los valores más altos de VOC, lo cual indica una mejora en los procesos de transferencia de electrones desde la molécula electrodonadora excitada a la banda de conducción del electroaceptor (PC61BM),14 pero esto no es suficiente para reflejar una alta PCE (Tabla 5). En la estimación de la eficiencia de fotoconversión de los materiales fotoactivos se utilizo la Ecuación (1), donde se asume un factor de forma (FF) del 75%,2 y se puede observa en la Tabla 5, que los derivados del naftopirrol (D-NPR-A) poseen los valores más altos de fotoconversión (PCE). Sin desconocer que los aspectos morfológicos de la capa fotoactiva juegan un papel determinante en la eficiencia real de las OSCs de BHJ.2
CONCLUSIONES Hemos diseñado una serie de electrodonadores D-π-A que contienen diferentes fragmentos cromóforos que fueron combinados con dos fragmentos π enlazadores para obtener los derivados del naftopirrol (D-NPR-A) y los del naftotiofeno (D-NTP-A). Las propiedades geométricas, electrónicas, ópticas y fotovoltaicas de los sistemas diseñados fueron investigadas mediante cálculos DFT con los niveles de teoría B3LYP/6-31G(d) y TD-CAM-B3LYP/6-31G(d). De acuerdo al análisis estructural el fragmento cromóforo difenilamina (Db), se muestra como un grupo donador de electrones relativamente fuerte comparado con los fragmentos trifenilamina (Da) y tiofeno (Dc), reflejando así una alta conjugación y redistribución de los electrones π, mejorando la transferencia y transporte de carga intramolecular. Pero en general todos los derivados D-π-A exhibieron una leve conformación no planar. La estimación de la energía de los orbitales moleculares de frontera HOMOEST-LUMOEST mostraron una importante correlación con los datos experimentales disponibles. La energía de la fuerza motriz del excitón (ΔE) de todos los derivados D-π-A poseen un valor superior a 0.3 eV, lo cual puede garantizar una división eficiente de los excitones. Los espectros de absorción UV/VIS simulados muestran un perfil similar para todos los electrodonadores, los cuales presentan una banda principal intensa entre los 450 nm y 600 nm. En general, los derivados del naftopirrol (D-NPR-A) poseen las mejores propiedades fotovoltaicas comparado con los derivados del naftotiofeno (D-NTP-A), donde se observaron valores bajos en la energía del gap (ΔEH-L) (1.81 - 1.83 eV) y valores altos en la densidad de fotocorriente de corto circuito (JSC) (14.74 - 15.64 mA cm-2) y en la eficiencia de conversión fotoeléctrica (PCE) (12.19 - 13.28%), por tanto, estos derivados pueden ser potencialmente utilizados en celdas solares orgánicas (OSCs) de heterounión de volumen (BHJ) y además se evidencia la gran importancia del estudio teórico de los orbitales moleculares frontera en la predicción de propiedades fotovoltaicas y así contribuir al diseño racional de materiales fotoactivos.
AGRADECIMIENTOS Los autores de este artículo muestran su agradecimiento al Centro de Investigaciones de la Universidad de Córdoba (CIUC), por el soporte económico brindado para el desarrollo del estudio en el marco del proyecto de investigación código 1.2.08.109 (numeral FCB-03-09).
REFERENCIAS 1. Agnihotri, N.; J. Photochem. Photobiol. C 2014, 18, 18. DOI: http://dx.doi.org/10.1016/j.jphotochemrev.2013.10.004 2. Scharber, M.; Sariciftci, N.; Prog. Polym. Sci. 2013, 38, 1929. DOI: http://dx.doi.org/10.1016/j.progpolymsci.2013.05.001 PMID: 24302787 3. Wild, J.; Meijerink, A.; Rath, J. K.; Sark, W. G. J. H. M.; Schropp, R. E. I.; Energy Environ. Sci. 2011, 4, 4835. DOI: http://dx.doi.org/10.1039/c1ee01659h 4. Lu, N.; Li, L.; Sun, P.; Liu, M.; Chem. Phys. Lett. 2014, 614, 27. DOI: http://dx.doi.org/10.1016/j.cplett.2014.08.070 5. Sharma, G.; Keshtov, M.; Khokhlov, A.; Tasis, D.; Galiotis, C.; Org. Electron. 2014, 15, 348. DOI: http://dx.doi.org/10.1016/j.orgel.2013.11.027 6. Fu, J-J.; Duan, Y-A.; Zhang, J-Z.; Guo, M-S., Liao, Y.; Comput. Theor. Chem. 2014, 1045, 145. DOI: http://dx.doi.org/10.1016/j.comptc.2014.07.008 7. Wu, Z.; Fan, B.; Xue, F.; Adachi, C.; Ouyang, J.; Sol. Energ. Mat. Sol. C. 2010, 94, 2230. DOI: http://dx.doi.org/10.1016/j.solmat.2010.07.017 8. Tai, C-K.; Hsieh, C-A.; Hsiao, K-L.; Wang, B-C.; Wei, Y.; Org. Electron. 2015, 16, 54. DOI: http://dx.doi.org/10.1016/j.orgel.2014.10.031 9. Wang, D.; Zhang, X.; Ding, W.; Zhao, X.; Geng, Z.; Comput. Theor. Chem. 2014, 1029, 68. DOI: http://dx.doi.org/10.1016/j.comptc.2013.12.011 10. Bérubé, N.; Gosselin, V.; Gaudreau, J.; Côté, M.; J. Phys. Chem. C 2013, 117, 7964. DOI: http://dx.doi.org/10.1021/jp309800f 11. Lizin, S.; Van, S.; De Schepper, E.; Vranken, L.; Sol. Energy Mater. Sol. Cells 2012, 103, 1. DOI: http://dx.doi.org/10.1016/j.solmat.2012.04.001 12. Coakley, K.; McGehee, M.; Chem. Mater. 2004, 16, 4533. DOI: http://dx.doi.org/10.1021/cm049654n 13. Scharber, M.; Mühlbacher, D.; Koppe, M.; Denk, P.; Waldauf, C.; Heeger, A.; Brabec, C.; Adv. Mater. 2006, 18, 789. DOI: http://dx.doi.org/10.1002/adma.200501717 14. Bourass, M.; Touimi, A.; Hamidi, M.; Benzakour, M.; Mcharfi, M.; Sfaira, M.; Serein-Spirau, F.; Lère-Porte, J.; Sotiropoulos, J.; Bouzzine, S.; Bouachrine, M.; J. Saudi Chem. Soc. (2013), http://dx.doi.org/10.1016/j.jscs.2013.01.003. DOI: http://dx.doi.org/10.1016/j.jscs.2013.01.003 15. Shalabi, A.; Abdel, S.; Assem, M.; Soliman, K.; Org. Electron. 2012, 13, 2063. DOI: http://dx.doi.org/10.1016/j.orgel.2012.06.028 16. Hoke, E.; Sachs-Quintana, I.; Lloyd, M.; Kauvar, I.; Mateker, W.; Nardes, A.; Peters, C.; Kopidakis, N.; McGehee, M.; Adv. Energy Mater. 2012, 2, 1351. DOI: http://dx.doi.org/10.1002/aenm.201200169 17. Azazi, A.; Mabrouk, A.; Alimi, K.; Comput. Theor. Chem. 2011, 978, 7. DOI: http://dx.doi.org/10.1016/j.comptc.2011.08.020 18. Wang, Y.; Zheng, B.; Tamai, Y.; Ohkita, H.; Benten, H.; Ito, S.; J. Electrochem. Soc. 2014, 161, D3093. DOI: http://dx.doi.org/10.1149/2.0561410jes 19. Zimmermann, Y-S.; Schäffer, A.; Hugi, C.; Fent, K.; Corvini, P.; Lenz, M.; Environ. Int. 2012, 49, 128. DOI: http://dx.doi.org/10.1016/j.envint.2012.08.015 PMID: 23022661 20. Hachmann, J.; Olivares-Amaya, R.; Jinich, A.; Appleton, A.; Blood-Forsythe, M.; Seress, L.; Román-Delgado, C.; Trepte, K.; Atahan-Evrenk, S.; Er, S.; Shrestha, S.; Mondal, R.; Sokolov, A.; Bao, Z.; Aspuru-Guzik, A.; Energy Environ. Sci. 2014, 7, 698. DOI: http://dx.doi.org/10.1039/C3EE42756K 21. Zhang, C-R.; Liu, L.; Liu, Z-J.; Shen, Y-L.; Sun, Y-T.; Wu, Y-Z.; Chen, Y-H.; Yuan, L-H.; Wang, W.; Chen, H-S.; J. Mol. Graph. Model. 2012, 38, 419. DOI: http://dx.doi.org/10.1016/j.jmgm.2012.09.004 PMID: 23117291 22. Fitri, A.; Benjelloun, A.; Benzakour, M.; Mcharfi, M.; Hamidi, M.; Bouachrine, M.; Spectrochim. Acta A 2014, 124, 646. DOI: http://dx.doi.org/10.1016/j.saa.2014.01.052 23. Tai, C-K.; Chen, Y-J.; Chang, H-W.; Yeh, P-L.; Wang, B-C.; Comput. Theor. Chem. 2011, 971, 42. DOI: http://dx.doi.org/10.1016/j.comptc.2011.05.036 24. Jo, H-J.; Nam, J-E.; Kim, D-H.; Kim, H.; Kang, J-K.; Dyes Pigments 2014, 102, 285. DOI: http://dx.doi.org/10.1016/j.dyepig.2013.10.032 25. Koyama, Y.; Kakitani, Y.; Nagae, H.; Molecules 2012, 17, 2188. DOI: http://dx.doi.org/10.3390/molecules17022188 PMID: 22357317 26. Reddy, M.; Vinayak, B.; Suresh, T.; Niveditha, S.; Bhanuprakash, K.; Singh, S.; Islam, A.; Han, L.; Chandrasekharam, M.; Synth. Met. 2014, 195, 208. DOI: http://dx.doi.org/10.1016/j.synthmet.2014.06.009 27. Fitri, A.; Benjelloun, A.; Benzakour, M.; Mcharfi, M.; Hamidi, M.; Bouachrine, M.; Spectrochim. Acta A 2014, 132, 232. DOI: http://dx.doi.org/10.1016/j.saa.2014.04.164 28. Yao, Z.; Wu, H.; Ren, Y.; Guo, Y.; Wang, P.; Energy Environ. Sci. 2015, 8, 1438. DOI: http://dx.doi.org/10.1039/C4EE03934C 29. Al-Sehemi, A.; Irfan, A.; Asiri, A.; Ammar, Y.; J. Mol. Struct. 2012, 1019, 130. DOI: http://dx.doi.org/10.1016/j.molstruc.2012.02.035 30. Jeon, B. C.; Kim, M. S.; Cho, M. J.; Choi, D. H.; Ahn, K-S.; Kim, J. H.; Synth. Met. 2014, 188, 130. DOI: http://dx.doi.org/10.1016/j.synthmet.2013.12.006 31. Mohr, T.; Aroulmoji, V.; Ravindran, R.; Müller, M.; Ranjitha, S.; Rajarajan, G.; Anbarasan, P.; Spectrochem. Acta A 2015, 135, 1066. DOI: http://dx.doi.org/10.1016/j.saa.2014.07.094 32. Balanay, M.; Kim, D-H.; Comput. Theor. Chem. 2015, 1055, 15. DOI: http://dx.doi.org/10.1016/j.comptc.2014.12.016 33. Yu, M.; Zhang, L.; Peng, Q.; Zhao, H.; Gao, J.; Comput. Theor. Chem. 2015, 1055, 88. DOI: http://dx.doi.org/10.1016/j.comptc.2014.12.027 34. Liu, X.; He, R.; Shen, W.; Li, M.; J. Power Sources 2014, 245, 217. DOI: http://dx.doi.org/10.1016/j.jpowsour.2014.04.043 35. Zhang, C-R.; Han, L-H.; Zhe, J-W.; Jin, N-Z.; Shen, Y-L.; Yuan, L-H.; Wu, Y-Z.; Liu, Z-J.; J. Nanometer. 2013, 2013, 1. DOI: http://dx.doi.org/10.1155/2013/596313 36. Nayak, P.; Garcia-Belmonte, G.; Kahm, A.; Bisquert, J.; Cahen, D.; Energy Environ. Sci. 2012, 5, 6022. DOI: http://dx.doi.org/10.1039/c2ee03178g 37. Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Montgomery, Jr., J. A.; Vreven, T.; Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G. A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Gonzalez, C.; Pople, J. A.; Gaussian 03, Gaussian, Inc., Wallingford CT, 2004. 38. Becke, A. D.; Phys. Rev. B 1988, 38, 3098. DOI: http://dx.doi.org/10.1103/PhysRevA.38.3098 39. Lee, C.; Yang, W.; Parr, R. G.; Phys. Rev. B 1988, 37, 785. DOI: http://dx.doi.org/10.1103/PhysRevB.37.131 40. Yanai, T.; Tew, D.; Handy, N.; Chem. Phys. Lett. 2004, 393, 51. DOI: http://dx.doi.org/10.1016/j.cplett.2004.06.011 41. Tai, C-K.; Hsieh, C-A.; Hsiao, K-L.; Wang, B-C.; Wei, Y.; Org. Electron. 2015, 16, 54. DOI: http://dx.doi.org/10.1016/j.orgel.2014.10.031 42. http://rredc.nrel.gov/solar/spectra/am1.5/, accesada en Enero 2016. |
 es el flujo de fotones asociado al espectro de irradiancia solar AM 1.5G.
es el flujo de fotones asociado al espectro de irradiancia solar AM 1.5G.On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access