
 |
 |
 |
 |
 |
Revisão
|
|
| Biomateriais para formulações de base nanotecnológica visando terapia genética ocular Biomaterials for nanotechnology-based formulations targeting ocular gene therapy |
|
Giovanni K. ZorziI,#, Roselena S. SchuhI,#, Angela M. de CamposII; Edison L. S. CarvalhoIII; Marilise B. RottIV; Helder F. TeixeiraI,*,#
IFaculdade de Farmácia, Universidade Federal do Rio Grande do Sul, 90610-000 Porto Alegre - RS, Brasil Recebido em 17/05/2016 *e-mail: helder.teixeira@ufrgs.br Gene therapy is a promising tool for the treatment of ocular diseases. Nevertheless, there are some import limitations, especially related to the ocular route and the characteristics of nucleic acids that must be overcome to ensure the success of the therapy. In this context, nanotechnology-based carriers such as nanoemulsions, nanoparticles, dendrimers and liposomes have risen as a promising vehicle for the protection of genetic material for vectoring the target cells. The success of such approach is closely related to the judicious selection of the biomaterial, where chemical structure and intermolecular interactions play a major role. So, this manuscript reviews some of the most important biomaterials, lipid and polymeric origin, employed in the manufacture of nanotechnologic carriers for ocular gene therapy involving interfering RNA (siRNA), antisense oligonucleotide (AS-ON) and plasmid DNA (pDNA). INTRODUÇÃO A busca por novos fármacos e ferramentas para o tratamento de doenças constitui um desafio constante no âmbito farmacêutico. Atualmente, a terapia genética ocupa um local de destaque entre as modalidades mais promissoras. A possibilidade de induzir a expressão de uma proteína (inserindo um gene funcional) ou de suprimir uma expressão aberrante cria alternativas interessantes que no futuro podem revolucionar a prática clínica.1 Devido as suas características particulares, a via ocular frequentemente é a primeira cujas novas tecnologias são testadas, pois o olho é um órgão relativamente isolado e imunologicamente privilegiado. Nesse sentido, medicamentos que têm como princípio a terapia genética foram primeiramente aprovados por agências regulatórias para a via ocular, como é o caso do oligonucleotídeo antissentido Vitravene®2 e do aptâmero Macugen®.3 Em seu conjunto, o globo ocular é uma estrutura de baixa permeabilidade, possuindo uma série de barreiras denominadas genericamente barreiras hemato-oculares, que impedem a passagem de elementos sanguíneos ao interior dos tecidos.4 Ainda é possível destacar uma função protetora, já que a parte anterior do olho conta com barreiras específicas formadas pelo filme lacrimal e da córnea, cuja transparência é de capital importância para a manutenção da visão e, por consequência, possui uma estrutura e mecanismos bioquímicos para assegurar sua impermeabilidade.5,6 Dessa maneira encontra-se uma série de limitações para o tratamento da superfície ocular, que impõe limitações à passagem efetiva das moléculas de interesse. A Figura 1 ilustra a estrutura do globo ocular, detalhando a córnea. A presença de diferentes camadas com diferentes propriedades no que refere à lipofilia/hidrofilia limita a absorção de fármacos pela superfície ocular. A via tópica é ainda a principal via de administração ocular, principalmente por não ser invasiva como a via intraocular (segunda forma de administração mais relevante para o olho).
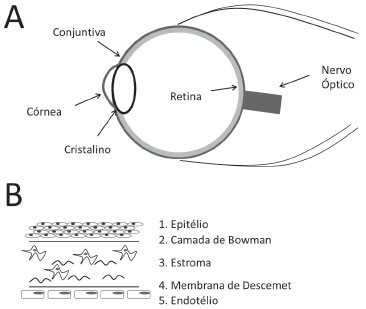 Figura 1. A. Principais estruturas do globo ocular. B. Corte transversal da córnea ilustrando as diferentes camadas: (1) células epiteliais, (2) camada de Bowman, (3) Estroma, (4) Membrana de Descemet, e (5) células endoteliais
O gene terapêutico pode ser introduzido num indivíduo de duas formas: in vivo ou ex vivo. Na técnica in vivo introduz-se o gene diretamente no organismo, enquanto que na técnica ex vivo, as células são removidas do indivíduo, modificadas in vitro e após reintroduzidas. A terapia gênica ocular pode ser realizada por várias vias, dependendo da célula-alvo a ser tratada. Assim, para a avaliação tanto in vitro quanto in vivo da eficácia e segurança de novas formulações, diferentes modelos são empregados, conforme a Tabela 1. A estrutura intraocular mais relevante é o fundo do olho, sendo as células epiteliais pigmentadas da retina (RPE ou ARPE-19) as mais utilizadas, enquanto que para a superfície ocular, as células do epitélio corneal e conjuntival (HCE e IOBA-NHC) são as mais utilizadas para formulações de uso tópico. Para modelos in vivo, são utilizados preferencialmente modelos animais como camundongos, ratos e coelhos.

Nesse contexto, esta revisão relaciona os principais biomateriais de origem lipídica e polimérica empregados na elaboração de carreadores nanotecnológicos para a terapia genética ocular. Tipos de terapia genética No que se refere à manipulação genética propriamente dita, é possível utilizar a terapia genética como estratégia terapêutica de duas maneiras: inserindo um gene mediante um plasmídeo para induzir a expressão de uma proteína ou através da inibição da expressão pela degradação de seu mRNA (RNA mensageiro), consequentemente impedindo a tradução da dita proteína; tais como oligonucleotídeo antissentido ou sequências pequenas de RNA de interferência. Uma exceção a esse mecanismo seriam os aptâmeros, que não regulam necessariamente a expressão gênica, mas em sua maioria atuam na proteína já formada. Em continuação segue uma breve descrição das estratégias mais empregadas para a terapia genética ocular.
VETORES NÃO VIRAIS COM BASE NANOTECNOLÓGICA PARA A VIA OCULAR O futuro da terapia genética como estratégia terapêutica depende da solução de suas limitações. O primeiro obstáculo está relacionado com o conhecimento sobre a doença e se a expressão de determinada proteína desempenha um papel na fisiopatologia dessa doença. Em outras palavras, é dizer se a expressão ou supressão de dita proteína resulta em uma resposta clínica benéfica. Tal informação pode ser obtida mais facilmente para enfermidades monogênicas, mas constitui um verdadeiro desafio para doenças multifatoriais ou que dependem de mais de um gene. A segunda dificuldade se refere à busca de um sistema que seja ao mesmo tempo capaz de proteger o material genético e transportá-lo de maneira eficaz até o seu local de ação, sem representar riscos para a saúde. Nesse sentido, diferentes veículos estão sendo estudados como ferramentas que permitem a transferência de material genético exógeno à célula. Fundamentalmente, é possível diferenciar dois tipos de sistemas transportadores: vetores virais (adenovírus, lentivírus, etc) e vetores não virais ou sintéticos (complexos, micelas, lipossomas, nanopartículas, entre outros). Do ponto de vista químico, os vetores não virais são os mais interessantes especialmente devido a sua segurança, permitindo diferentes estratégias e modificações, e serão o foco da presente revisão. Com o passar dos anos, os carreadores não virais de base nanotecnológica têm ganhado atenção devido à capacidade de superação das limitações tanto da terapia genética como da administração de fármacos pela via ocular. Naturalmente, a eficácia de um sistema em exercer atividade biológica com baixa toxicidade não depende somente do tipo de sistema desenvolvido, mas também do biomaterial empregado em seu preparo. Nesse sentido, alguns sistemas lipídicos (lipossomas e nanoemulsões) e poliméricos (nanopartículas poliméricas e dendrímeros) foram propostos para a terapia genética da superfície ocular. As características mais importantes dos sistemas carreadores são a capacidade de proteger o material genético, internalização celular específica, estabilidade, cinética de liberação compatível com a via de administração, fácil obtenção e preço acessível. Já os biomateriais empregados na construção desses vetores, devem possuir biocompatibilidade, biodegradabilidade, estabilidade química, devem ser de fácil obtenção e ter preço acessível Dentre os sistemas lipídicos, destacam-se os lipossomas e as nanoemulsões. Lipossomas são vesículas lipídicas biocompatíveis e biodegradáveis compostas principalmente por fosfolipídios. Possuem um diâmetro variável entre 25 a 10.000 nm e suas propriedades físico-químicas variam de acordo com o método de preparo e a composição. Com relação ao número de bicamadas, os lipossomas podem ser divididos em: vesículas unilamelares pequenas (small unilamellar vesicle - SUV), vesículas unilamelares grandes (large unilamellar vesicle - LUV), vesículas unilamelares gigantes (giant unilamellar vesicle - GUV) e vesículas multilamelares (multilamellar vesicle - MLV). Os principais constituintes dos lipossomas são fosfolipídios, entretanto outros componentes podem ser empregados, sejam de natureza lipídica ou polimérica, para fins de obtenção das propriedades físico-químicas desejadas. O material genético pode ficar adsorvido na superfície do lipossoma ou no interior de sua cavidade, sendo a taxa de encapsulação dependente do método de preparo.20,21 Dessa maneira os lipossomas são capazes de incrementar a estabilidade dos ácidos nucleicos, diminuindo a sua hidrólise e consequentemente prolongando a sua capacidade de ação.22 Nanoemulsões consistem em uma dispersão líquido-líquido de dois líquidos imiscíveis entre si e estabilizados por um surfactante, ou sistema surfactante, adequado. São sistemas que mesmo não sendo termodinamicamente estáveis apresentam elevada estabilidade cinética.23 Essa estabilidade pode ser convenientemente modificada com uma seleção criteriosa de diferentes misturas de óleos e surfactantes. Nesse sentido o equilíbrio hidrófilo-lipófilo (EHL) mostra-se como uma conveniente abordagem para o preparo das nanoemulsões, tal como para emulsões clássicas.24 Entretanto, essa estratégia nem sempre é bem-sucedida para tensoativos catiônicos e aniônicos, além de não ser capaz de prever a quantidade de tensoativo necessária nem a contribuição de outros constituintes da formulação. Dependendo da proporção dos constituintes, podem formar tanto uma emulsão água-em-óleo (A/O) quanto uma emulsão óleo-em-água (O/A), sendo esta última a mais relevante para a terapia genética da via ocular.25 Assim, devido à elevada lipofilia da fase interna de nanoemulsões O/A, a associação do material genético dá-se geralmente pela adsorção na superfície da gotícula. Da mesma maneira que para os lipossomas, faz-se necessário o emprego de moléculas de carga positiva, sendo esse papel geralmente desempenhado por lipídios catiônicos. Recentemente foi descrita uma abordagem para associação de material genético no interior da nanoemulsão previamente complexado com um lipídio catiônico, abrindo portanto novas possibilidades para a utilização desses sistemas.26 Assim, os constituintes básicos de uma nanoemulsão ocular objetivando a terapia genética seriam óleo, surfactante e lipídio catiônico. Nanopartículas são estruturas geralmente compostas por polímeros, lipídios ou combinações de ambos. Dependendo dos constituintes e do método de preparo, o material genético pode estar associado no interior da estrutura ou adsorvido em sua superfície.27 Quando esses sistemas contêm um polímero (geralmente insolúvel em água), o termo nanopartícula pode se referir tanto à estrutura com núcleo completamente matricial (nanoesfera) quanto à com núcleo oleoso (nanocápsula). Essa última é mais utilizada para fármacos lipofílicos, onde o mesmo encontra-se principalmente dissolvido no núcleo oleoso. Nanopartículas obtidas a partir de polímeros hidrofílicos são preparadas principalmente por geleificação iônica, coacervação ou nanoprecipitação, permitindo uma associação de material hidrofílico, como o material genético, no interior da partícula, o que oferece uma maior proteção contra a degradação.4 Nanopartículas contendo polímeros hidrofóbicos pré-formados invariavelmente necessitam de solvente orgânico, utilizado para a solubilização do polímero, e podem eventualmente desnaturar o ácido nucleico utilizado.28 Nanopartículas lipídicas sólidas são estruturas formadas por lipídios que são sólidos em temperatura ambiente e temperatura corporal.29,30 Existem basicamente dois métodos de preparo: homogeneização à alta pressão e o método de microemulsão. Dependendo do método de preparo pode ser a necessária a presença de um surfactante ou co-surfactante.31 É uma técnica que envolve o uso de energia, sendo que o preparo de estruturas associando material genético em uma só etapa pode ser inviável devido à baixa estabilidade dos ácidos nucleicos. Assim, normalmente o material genético é adsorvido a nanopartículas pré-formadas, de maneira similar ao que ocorre com as nanoemulsões. Entretanto, as nanopartículas lipídicas sólidas podem apresentar alguns problemas relativos a sua estabilidade.32 Essa situação pode ser parcialmente corrigida com o uso de misturas de lipídios, geralmente sólidos e líquidos, de modo a criar um arranjo desordenado. Esses sistemas são denominados carreadores lipídicos nanoestruturados. Tal como ocorre para as nanoemulsões e nanopartículas poliméricas, a associação no interior da nanoestrutura ocorre basicamente com moléculas lipofílicas. Assim, para terapia genética utilizando a via ocular não existe uma diferença no mecanismo entre as estruturas formadas, exceto possíveis diferenças na estabilidade ou tolerância das formulações. Essas diferenças, entretanto, devem ser avaliadas caso a caso. Dendrímeros são moléculas poliméricas sintéticas, de variável peso molecular, que possuem um núcleo central do qual partem ramificações de maneira ordenada e simétrica. As repetidas ramificações dão origem a uma série de camadas concêntricas, chamadas de "gerações". A sua estrutura única permite que possuam uma série de características interessantes como tamanho bem definido, baixa polidispersão, e a capacidade de serem facilmente funcionalizados em sua síntese.33-35 A associação de dendrímeros com material genético dá-se por interações eletrostáticas, principalmente com grupos amino terminais carregados positivamente. Esse é justamente um dos principais limitantes, pois essas interações podem ser facilmente deslocadas devido à força iônica dos fluidos oculares.36,37 Como os principais determinantes das propriedades dos dendrímeros, temos a sua geração (relacionada com o peso molecular) e o grupo terminal que afetarão parâmetros como solubilidade, eficácia, bioadesão e farmacocinética.38-42 A literatura de dendrímeros para a terapia genética ocular ainda é relativamente recente, contando com um número limitado de trabalhos. Dessa maneira, ainda é cedo para predizer o futuro desses sistemas, mesmo já existindo reagentes de transfecção comerciais baseados em dendrímeros (SuperFect®, Qiagen®). Na Figura 2 pode-se observar a estrutura dos principais sistemas carreadores para terapia genética ocular. A seguir será realizada uma breve descrição de cada sistema.
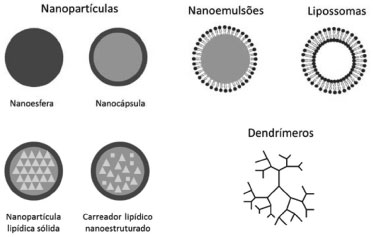 Figura 2. Principais sistemas de base nanotecnológica empregados na terapia genética ocular
BIOMATERIAS EMPREGADOS NO PREPARO DE SISTEMAS DE BASE NANOTECNOLÓGICA PARA TERAPIA GENÉTICA OCULAR Lipídios Os lipídios são biomateriais utilizados em formulações para liberação ocular de ácidos nucleicos devido a sua biocompatibilidade com a camada lipídica mais externa do olho e por sua biodegradabilidade, decorrente de sua composição similar às membranas biológicas.43 Algumas propriedades dos lipídios irão impactar diretamente na estabilidade da formulação e, consequentemente, na extensão de sua atividade. Como exemplo tem-se a temperatura de transição de fases e a temperatura de fusão. Alguns lipídios sólidos à temperatura ambiente necessitam ser fundidos para o preparo das nanoestruturas, como é o caso das nanopartículas lipídicas sólidas e carreadores lipídicos nanoestruturados. A temperatura de transição de fase observada em fosfolipídios, na qual a membrana passa de uma fase gel em que a cadeia de hidrocarbonetos do fosfolipídio se encontra ordenada, para uma fase de cristal líquido em que as moléculas se encontram em uma fase mais livre e desordenada, caracteriza o aspecto da formulação. Misturas de diferentes lipídios solúveis entre si, e de diferentes temperaturas de transição, terão diferentes aspectos e propriedades, já que formulações compostas por lipídios diferentes podem exibir diferentes níveis de fluidez.44 Dentre as vantagens da lipofecção estão a capacidade de carreamento de sequências longas de ácidos nucleicos quando comparada à limitação de tamanho dos vetores virais, a proteção contra a degradação dos ácidos nucleicos encapsulados frente a enzimas, o fato de possuírem baixa imunogenicidade, não oferecerem risco na sua manufatura e serem de fácil produção em larga escala.43 Já as desvantagens incluem uma baixa seletividade ao alvo e baixa eficiência de transfecção quando comparado a vetores virais; limitações que atualmente podem ser transpostas pela funcionalização de superfície visando à endocitose mediada por receptores celulares ou, ainda, através da inibição da digestão lisossômica ou do direcionamento do vetor ao núcleo.43 Dentre os lipídios mais comumente utilizados em formulações para a via ocular estão os fosfolipídios, o colesterol, os lipídios catiônicos, os óleos e tensoativos, e os lipídios sólidos. É importante frisar que o grau de pureza desses lipídios deve ser elevado, a fim de evitar possíveis irritações oculares causadas por contaminantes. Fosfolipídios O material mais comum empregado na confecção de lipossomas é a lecitina/fosfatidilcolina. A lecitina é obtida de diferentes fontes, geralmente da soja e gema de ovo, sendo composta por uma mistura de diferentes fosfolipídios, principalmente a fosfatidilcolina (FC). A fosfatidilcolina é um lipídio neutro em pH fisiológico, porém estruturas formadas tendem a apresentar uma carga superficial negativa devido à presença em pequenas quantidades de outros fosfolipídios carregados negativamente e ácidos graxos livres. Entre os benefícios do uso da lecitina/FC estão a baixa ocorrência de eventos adversos, aumento da estabilidade e modificação do perfil farmacocinético/farmacodinâmico do fármaco.45-50 A lecitina/FC é comumente empregada em formulações que visam uma carga superficial positiva, uma vez que a bicamada negativa formada pela lecitina servirá de ancoradouro para os lipídios catiônicos adicionados à formulação, o que conferirá uma carga residual positiva ao sistema. A incorporação do fosfolipídio neutro DOPE (1,2-dioleoil-3-fosfatidiletanolamina) tem sido frequentemente descrita devido a sua habilidade de escape lisossômico. O mecanismo exato ainda não é completamente entendido, porém o fato de ser um fosfolipídio pH-sensível o torna capaz de desestabilizar as membranas lisossômicas após a internalização celular via endocitose.51 A DOPE é empregada em combinação com outros lipídios, especialmente catiônicos, como agente de transfecção na terapia gênica.52-55 Em um estudo pré-clínico de terapia gênica para glaucoma, as nanopartículas de surfactante com DOPE como lipídio assessor obtiveram a maior taxa de transfecção em células RGC-5 (células ganglionares de retina de rato) e camundongos in vivo, e ainda mostraram-se biocompatíveis.56 Colesterol O colesterol é outro lipídio geralmente empregado em formulações de natureza lipídica, em especial lipossomas. Esse lipídio possui a capacidade de reduzir a fluidez e permeabilidade da bicamada como resultado da supressão da transição sol-gel, assim enrijecendo a membrana lipossomal.45 Como resultado, pode-se obter a redução da toxicidade associada ao fármaco,57 aumento do tempo de residência49 e melhora na estabilidade da formulação,51 fazendo do colesterol um excipiente amplamente utilizado na tecnologia de lipossomas. Lipossomas contendo PEG (polietilenoglicol), colesterol e lecitina de soja demonstraram ser promissores na vetorização intravítrea de oligonucleotídeos, aumentando o tempo de residência, prolongando a liberação do oligonucleotídeo e protegendo-o da degradação.45 Lipídios catiônicos Para a terapia gênica não-viral é praticamente imprescindível a presença de um material catiônico para associar o material genético de interesse, visto que os ácidos nucleicos possuem grupamentos fosfato carregados negativamente que interagem com os lipídios carregados positivamente através de interações eletrostáticas, possibilitando a vetorização do material genético pelos carreadores nanoestruturados catiônicos.58 Lipídios como TMAG (N-(alfa-trimetilamônioacetil)-didodecil-D-glutamato),53,59-61 DC-colesterol (3-beta[N-(N',N'-dimetilaminoetano)-carbamoil] colesterol),53,60,61 DOTAP (1,2-dioleil-3-trimetilamônio- propano),54,55 DOTMA (1,2-dioleiloxi-3-trimetilamônio-cloropropano) e DDBA (dimetil- dioctadecilamônio) permitem o desenho de vesículas capazes de associar eficazmente uma série de moléculas como oligonucleotídeos, DNA plasmídico, siRNA e ribozimas para o tratamento de enfermidade oculares. Atualmente, é fato que a captação de nanoestruturas na superfície ocular é geralmente governada pela sua carga superficial seguindo a seguinte relação: positivos > negativos > neutros.62 Nanoemulsões compostas por DOTAP e lecitina de ovo contendo oligonucleotídeos antissentido específicos para VEGF (fator de crescimento vascular endotelial) foram testados em células humanas do endotélio venoso de cordão umbilical63 e in vivo em ratos e camundongos64 e demonstraram boa transfecção e baixa toxicidade. Lipossomas com DOTMA/colesterol demonstraram alta eficiência de transfecção e expressão gênica no tecido ocular de coelhos em testes realizados in vivo.65 Óleos e tensoativos Óleos são misturas de lipídios líquidos em temperatura ambiente, de composição variada. Diferentes óleos são utilizados no desenvolvimento de formulações oculares. Para formulações clássicas, a escolha do tipo de óleo está intrinsecamente relacionada à solubilidade do fármaco. Entretanto, para formulações que contêm óleo e que objetivam a terapia genética, a associação do material genético dá-se normalmente por adsorção na superfície da nanoestrutura, e não no interior do núcleo oleoso, devido a polaridades opostas. Assim, a escolha do óleo está mais relacionada com a estabilidade da formulação, principalmente devido às interações óleo-surfactante. Os triglicerídeos de cadeia longa (TCL) são óleos de origem vegetal, como óleo de rícino, oliva e soja e possuem ácidos graxos com mais de 12 carbonos estruturais. Os triglicerídeos de cadeia média (TCM) são frações de ácidos graxos reesterificados com glicerina derivados do óleo de coco, em especial ácido caprílico e cáprico. Porém, misturas de TCL e TCM podem ser necessárias para obtenção da solubilização ideal do fármaco na fase oleosa.66 Frequentemente as formulações nanoemulsionadas oculares contêm TCM como componente principal do núcleo oleoso.63,64 Lipídios sólidos São considerados lipídios sólidos aqueles que se encontram em estado sólido em temperatura ambiente (25 ºC) e temperatura corporal (37 ºC). Os lipídios podem ser acilglicerois (como palmitoestearato de glicerila e monoestearato de glicerila), ceras (como a cera de carnaúba e palmitato de cetila), ácidos graxos (como o ácido esteárico) e álcoois graxos (como álcool cetílico e álcool estearílico). Uma vantagem desses lipídios reside na baixa toxicidade em concentrações entre 5% e 30%.67 A mistura de lipídios sólidos promove uma matriz cristalina menos perfeita, resultando em maior capacidade de incorporação dos ácidos nucleicos quando comparada à utilização de um lipídio puro. As nanopartículas lipídicas sólidas utilizam os lipídios sólidos com o auxílio de tensoativos como Span® e Tween® para promover a tensão interfacial e evitar a agregação entre as partículas. Del Pozo-Rodriguez e colaboradores utilizaram nanopartículas lipídicas sólidas para estudar a transfecção em uma linhagem celular de epitélio de retina humana (ARPE-19) e descobriram que a internalização mediada por clatrina direciona as nanopartículas aos lisossomos.68 Em outro estudo foi demonstrada a capacidade de transfecção in vivo em camundongos e a expressão gênica pôde ser demonstrada após uma semana da injeção intravítrea.69 Outros lipídios É conhecido o fato de que o recobrimento da superfície de um lipossoma com polímeros hidrofílicos, como cadeias de PEG, aumenta a estabilidade e o tempo de meia vida em circulação. Lipídios PEGuilados atuam como uma barreira contra o estabelecimento de interações eletrostáticas e podem reduzir o efeito da força iônica do fluido lacrimal na estabilidade das vesículas, bem como aumentar o tempo de residência das mesmas. Por essa razão, o emprego de lipossomas estabilizados estericamente (denominados stealth) é de grande interesse para a via ocular. O emprego de 1,2-diestearoil-sn-glicero-3 fosfoetanolamina-N-polietilenoeglicol (DSPE-PEG) no desenvolvimento de lipossomas já foi proposto para a veiculação ocular de diversas moléculas lábeis como: peptídeos,46,48 oligonucleotídeos45 e plasmídeos.53 A PEGuilação de lipossomas permite mascarar a carga de superfície reduzindo a agregação de proteínas nas vesículas, o que resulta em maior internalização dos oligonucleotídeos na retina/coroide e maior estabilidade das formulações quando comparadas às não PEGuiladas.45,70,71 A estrutura de alguns dos principais lipídios utilizados na terapia genética ocular pode ser observada na Figura 3.
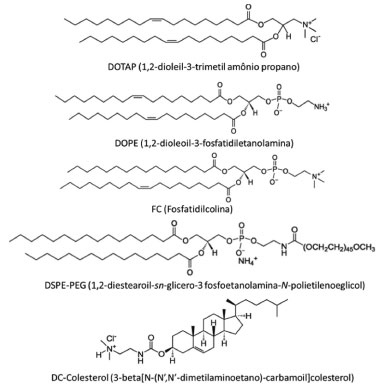 Figura 3. Estrutura de alguns dos principais lipídios empregados no desenvolvimento de formulações de base lipídica para a terapia genética na superfície ocular
Polímeros O outro grupo molecular mais significativo de biomateriais são os polímeros. Para promover interações eletrostáticas com os ácidos nucleicos, poliplexos são formados por polímeros catiônicos com numerosos grupamentos amina, lisina ou arginina protonadas em pH fisiológico. Se apenas uma fração dos grupos são protonados em pH fisiológico, como os dendrímeros de polietileneimina (PEI), eles são capazes de tamponar os endossomas e permitir a liberação do material genético por endocitose dentro da célula.14 Dendrímeros são também capazes de proteger o DNA de DNAses plasmáticas.14 Devido a sua natureza muitas vezes heterogênea, uma caracterização básica deve ser realizada nos excipientes, principalmente no que diz respeito à massa molar, para homo e heteropolímeros, e grau de substituição, quando existente, para heteropolímeros. Por exemplo, para a quitosana os diferentes graus de acetilação fazem com que as cadeias se comportem que maneira diferente, devido à repulsão dos grupamentos desacetilados carregados positivamente no interior da cadeia. Polímeros catiônicos Quitosana A quitosana é um heteropolímero linear, derivado principalmente da desacetilação da quitina em meio básico, composta de repetidas unidades de N-glicosamina e N-acetilglicosamina. Diferentes atividades biológicas já foram descritas para a quitosana, como antimicrobiana e estimulante do processo cicatricial.72,73 Entretanto, no que compete à terapia genética, é a habilidade de interagir com o material genético que torna atrativo o uso da quitosana. Um dos maiores limitantes do emprego da quitosana, não só em formulações para a terapia gênica ocular, mas para outras vias e aplicações, é a dificuldade na caracterização da matéria-prima. Como um heteropolímero, a quitosana pode variar quanto à proporção das diferentes unidades monoméricas, geralmente expressa pelo grau de acetilação (percentual molar de grupos acetilados), quanto ao seu peso molecular, também expresso em termos de graus de polimerização e quanto ao padrão de polimerização. Parâmetros como solubilidade,74 viscosidade75 e biocompatibilidade76 parecem diminuir com o grau de acetilação, enquanto cristalinidade77 e biodegradabilidade78 aumentam com o grau de acetilação. De maneira similar, não há uma relação clara entre o grau de acetilação e a atividade biológica.79-81 Uma das principais aplicações da quitosana na nanotecnologia é a formação de nanopartículas por geleificação iônica.82 Para tanto, o polímero é misturado com agentes reticulantes iônicos (geralmente sais com alta densidade eletrônica como tripolifosfato ou sulfato de sódio), havendo uma transição sol-gel do polímero, conjuntamente com uma nanoprecipitação controlada.83 Ao contrário de sistemas PEGuilados, onde há uma maior penetração das nanopartículas, para o caso de nanopartículas com quitosana parece haver um maior depósito na superfície ocular.84 Entretanto, lipossomas com nanopartículas de quitosana encapsuladas foram capazes de ser internalizados por células de conjuntiva humana (IOBA-NHC).85 Tal aplicação não comprometeu a viabilidade celular, nem a tolerância após administração in vivo em coelhos, bem como em cortes histológicos de córnea e conjuntiva.86 Nanopartículas híbridas de quitosana com ácido hialurônico foram capazes de transfectar in vitro células HCE e IOBA-NHC, utilizando diferentes modelos de pDNA como pEGFP (proteína verde fluorescente), pSEAP (fosfatase alcalina) e pLac-Z (β-galactosidase).87,88 O grau de polimerização (i.e. peso molecular) parece influenciar na transfecção, sendo oligômeros (<10-12 kDa) mais eficazes em transfectar células de córnea HCE.89,90 Essas partículas também demonstraram perfil de toxicidade aceitável, atribuído principalmente à adição de ácido hialurônico. Quanto a sua toxicidade, a quitosana parece ser bem tolerada em aplicações tópicas,85,86,89 podendo ocorrer o risco de inflamação após administração intraocular de nanopartículas.91 Ademais, por possuir um pKa de 6,2 as formulações com quitosana podem apresentar certa instabilidade em pH fisiológico (pH 7,4), existindo uma tendência à agregação.92 Protamina Protamina é uma denominação genérica de proteínas nucleares de baixo peso molecular, ricas em arginina. Devido a esse fato, a protamina está carregada positivamente em pH fisiológico (pH 7,4), sendo capaz de interagir com ácido nucleicos, protegendo-os de degradação por nucleases.93 De fato, as protaminas substituem as histonas (outra classe de proteínas nucleares ricas em arginina) na fase haploide da espermatogênese. Seis ou mais resíduos consecutivos de arginina já são capazes de condensar o DNA, permitindo a passagem desse complexo do citoplasma ao núcleo93 sendo capaz de incrementar a transcrição no interior do mesmo94, ou associar siRNA.95 Complexos de protamina com DNA plasmidial possuem tempo de meia vida reduzido, de modo que somente cerca de 20% do material genético é encontrado na circulação após 60 minutos.96 Nesse sentido, é mais comum a associação de sulfato de protamina com outros nanossistemas pré-formados, como lipossomas97 e nanopartículas lipídicas sólidas.98 O uso de lipossomas pré-formados para a associação de complexos de material genético com protamina compostos por DOTAP e/ou DOPE, DSPE-PEG e colesterol já foram previamente descritos por diversos autores. Toropainen e colaboradores compararam a capacidade de transfecção de lipossomas compostos por DOTAP e DOPE contendo ou não sulfato de protamina para transfecção de pDNA em células de epitélio corneal (HCE) e não foi observada diferença na capacidade de transfecção. Entretanto, formulações similares de lipossomas preparados com DOTAP e sulfato de protamina foram superiores aos lipossomas sem protamina no que se refere à proteção contra degradação enzimática e transfecção in vivo após administração intravenosa.96,99 No que se refere a siRNA, lipossomas PEGuilados contendo DOTAP e colesterol associados a complexos de protamina e siRNA/ácido hialurônico apresentaram alta taxa de associação (> 95%).100 A injeção intravítrea dos lipossomas resultou em decréscimo significativo na expressão do receptor do fator de crescimento vascular endotelial de tipo 1 (VEGFR1).100 Rajala e colaboradores obtiveram uma expressão da proteína 65 de células RPE (Rpe65) e da proteína verde fluorescente (GFP) por 1 semana e 3 meses, respectivamente, com uma formulação de lipossomas de DOTAP, DOPE, colesterol e peptídeo internalizador associados a complexos de protamina e pDNA.101 Nanopartículas lipídicas sólidas pré-formadas contendo DOTAP foram propostas para associar complexos de protamina e pDNA,102 e protamina com sulfato de dextrano e pDNA.103 O uso de protamina parece impactar algumas das principais características físico-químicas das partículas, como tamanho e potencial de superfície. Entretanto, a capacidade de transfecção do pDNA é de cerca de 30-44% em células de retina (ARPE-19) em comparação com os 5-7% relativos às mesmas nanopartículas lipídicas sólidas sem protamina. A presença de sulfato de dextrana parece incrementar a eficácia da formulação, provavelmente devido a uma diminuição da toxicidade intrínseca dos elementos carregados positivamente. A transfecção após administração intravítrea parece variar de acordo como local da injeção, sendo observável após 72h. A grande maioria das células transfectadas pertence a gânglios da retina, sendo quase negligenciável a transfecção em células do epitélio da retina.103 O uso de protamina parece alterar o mecanismo de internalização de nanopartículas lipídicas sólidas contendo o lipídio catiônico DOTAP em células ARPE-19, através de um mecanismo envolvendo principalmente caveolina/rafts de lipídios para internalização por clatrina.102 Uma alta densidade de aminoácidos básicos como lisina ou arginina (como é o caso da protamina) favorece o transporte até o núcleo celular através do complexo de poros nuclear.104,105 Polietilenimina Polietilenimina (PEI) é um polímero linear ou ramificado, considerado um padrão-ouro de sistemas não virais para transfecção in vitro.106,107 Inúmeros fatores estão relacionados à eficiência da PEI, como célula-alvo, peso molecular, ramificações, razão N/P entre os grupos amina da polietilenimina (N) e os grupos fosfato do ácido nucleico (P), e tamanho do complexo.108 A atividade superior da PEI frente a outros reagentes de transfecção é atribuída ao escape lisossomal, comumente chamado "esponja-de-prótons": devido à alta densidade de grupos amino de elevado pKa, o pH ácido da vesícula lisossomal protona esses grupamentos, levando a uma entrada de água para solvatação que colapsa a estrutura do lisossomo.109 Entretanto, muitos autores ainda questionam a veracidade desse mecanismo.110 O principal limitante da aplicação da PEI é a sua toxicidade,111,112 sendo a estrutura ramificada mais tóxica que a linear.113 Nanopartículas de PEI 22kDa associando pDNA (razão N/P 15) foram utilizadas para transfecção in vitro de fibroblastos corneais equinos.114 Esse sistema foi capaz de impedir a diferenciação de fibroblastos relacionada ao TGF-β (fator de transformação do crescimento beta), apresentando toxicidade aceitável. Não obstante, para a terapia genética ocular, a PEI é geralmente associada a outros materiais de modo a diminuir a sua toxicidade ou vetorizar a sua interação com determinados grupos celulares. PEI de 2kDa conjugada a nanopartículas de ouro (razão N/P de 180) aplicada na córnea de coelhos foi capaz de transfectar pDNA por até 7 dias, sem apresentar sintomas de inflamação, vermelhidão ou edema. Entretanto, morte celular moderada e outras reações imunes foram observadas.115 Para a redução da toxicidade intrínseca da PEI, foram utilizadas nanopartículas de PEI/sulfato de condroitina para transfecção in vitro de células de retina ARPE-19.116 As partículas isentas de sulfato de condroitina diminuíram a agregação e a alta toxicidade de partículas constituídas somente de PEI e pDNA, tornando-as adequadas para administração intraocular. Poliamidoamina Dendrímeros de poliamidoamina (PAMAM) são os mais comuns para o carreamento de fármacos ou ácidos nucleicos.117, 118 A farmacocinética desses dendrímeros de geração 2 a geração 4 com terminações amino, carboxilato ou hidroxila foram estudadas por Vandamme e colaboradores, sendo observada uma dependência do peso molecular (i.e. geração) no tempo de residência.42 A comparação entre reagentes comerciais baseados em dendrímeros em relação aos baseados em lipossomas contendo lipídios catiônicos mostrou um efeito superior dos dendrímeros na transfecção de células RPE, sendo a concentração e a razão de cargas os principais moduladores da resposta biológica.119 Dendrímeros de PAMAM ativados foram capazes de transfectar células do endotélio de córnea in vitro. Essa transfecção foi transitória, atingindo seu ápice em 4 dias após a incubação e reduzindo significativamente no oitavo dia.120 Entretanto, a utilização de PAMAM não modificado é quase sempre limitada pela toxicidade intrínseca da molécula, sendo comuns, portanto, as modificações terminais.34 Estudos de dendrímeros PAMAM de gerações 3, 5 e 4 indicaram que a toxicidade pode chegar a níveis de desenvolvimento embrionário, como observado para peixe-zebra.121 Tais modificações são capazes de alterar algumas propriedades físico-químicas, bem como algumas das respostas biológicas associadas aos sistemas, conforme verificado para as gerações 4 e 5.122 A acetilação e a PEGuilação afetam a ligação ao DNA, citotoxicidade e transfecção in vitro. A acetilação terminal é capaz de eliminar quase completamente essa toxicidade. Entretanto, a capacidade de transfecção reduz significativamente, provavelmente devido a uma menor compactação do pDNA. A PEGuilação pode incrementar a estabilidade dos complexos, mas tal efeito é dependente da razão de cargas entre o dendrímeros e o pDNA, sendo que razões superiores a 5 são mais propensas ao aumento da estabilidade.122 No caso de modificações hidróxi-terminais a transfecção de células RPE não foi alterada para dendrímeros de geração 4 que sofreram posterior PEGuilação, mesmo que a estabilidade coloidal tenha sido significativamente incrementada.123 Uma alternativa proposta ao uso de dendrímeors PAMAM são dendrímeros de poli-L-lisina. Esses dendrímeros foram capazes de associar AS-ON anti-VEGF, reduzindo a expressão gênica tanto in vitro (células RPE) quanto in vivo em modelo de neovascularização da coroide em ratos, inibindo dessa maneira neovascularização coroide relacionada à degeneração macular por até dois meses.124,125 Proteínas cationizadas Proteínas cationizadas como albumina, atelocolágeno e gelatina têm sido utilizadas como materiais alternativos para terapia genética. A albumina sérica humana (HSA) é a proteína plasmática mais abundante, possuindo um peso molecular de 66,4 kDa e relativa estabilidade no ambiente (60 ºC e pH 4-9), além de baixa imunogenicidade e toxicidade negligenciável.126,127 O atelocolágeno é o colágeno, principal componente da matriz extracelular, tratado com pepsina. A retirada de ateloproteínas diminui a antigenicidade do atelocolágeno, em relação ao colágeno.128 Já a gelatina é o polímero obtido pela hidrólise do colágeno, com estrutura típica Ala-Gly-Pro-Arg-Gly-Glu-4Hyp-Gly-Pro, composta majoritariamente por glicina, prolina e 4-hidroxiprolina.129 Entretanto, esses polímeros puris naturabilis não possuem suficientes grupos positivos para interagir com o material genético de maneira satisfatória. Dessa maneira, a cationização dos mesmos, pela adição de amina de baixo peso molecular é uma estratégia corriqueira para a elevação do ponto isoelétrico dessas proteínas. Geralmente essa reação se dá por uma reação do tipo carbodiimina, onde os resíduos de ácido carboxílico das proteínas reagem com aminas de baixo peso molecular (como etilenodiamina e espermina), levando à formação de uma ligação do tipo amida com liberação de um derivado isoaciluréia.129 Esses polímeros, uma vez cationizados, são geralmente utilizados para a formação de complexos, hidrogéis e, para a via ocular, como nanopartículas preparadas por nanoprecipitação/geleificação iônica.130,131 Entre as características do polímero que determinam as suas propriedades como materiais para a terapia genética ocular estão peso molecular, número de grupamentos amino e forma da proteína.127,132 O peso molecular influencia diretamente na toxicidade, onde pesos moleculares mais elevados estão associados a uma maior toxicidade em células HCE.113,133 De maneira similar, um elevado número de grupos amino incrementa a toxicidade, porém também a eficácia de transfecção. A estrutura da proteína (linear/globular, cadeia rígida/flexível) está relacionada com a capacidade de formação de partículas e a consequente proteção do material genético. Nanopartículas de sulfato de condroitina e gelatina cationizada com espermina foram capazes de transfectar pDNA codificando a proteína MUC5AC, uma mucina deficiente em casos de olho seco, em duas espécies de animais distintas (camundongos e coelhos).134,135 Mesmo não existindo um tropismo específico para um determinado tipo celular, a expressão in vivo parece dar-se preferencialmente na conjuntiva.134 Polímeros aniônicos Ácido hialurônico O ácido hialurônico é um polissacarídeo linear composto de unidades repetidas de D-ácido glucurônico e N-acetil-D-glicosamina, sendo o único glicosaminoglicano não sulfatado. É um composto relativamente seguro, sendo utilizado na medicina estética e medicina regenerativa, e frequentemente proposto como biomaterial para os mais diversos sistemas de base nanotecnológica. Esse material pode ser utilizado no recobrimento de nanoestruturas mais lipofílicas como nanopartículas poliméricas,136 nanopartículas lipídicas sólidas137 ou nanoemulsões,138 bem como constituinte da matriz de nanopartículas, inclusive como único polímero formador.139 A propriedade mais atrativa do ácido hialurônico no que compete ao desenvolvimento de sistemas carreadores de fármacos é a sua capacidade de interagir especificamente com receptores de internalização CD44 e receptor de endocitose de ácido hialurônico (HARE). A transfecção in vitro de células HCE e IOBA-NHC pareceu ser governada por CD44 quando nanopartículas de quitosana e ácido hialurônico foram utilizadas. O bloqueio do CD44 com o uso de anticorpos resultou em dramática redução na transfecção de pDNA dessas nanopartículas híbridas.89 O sistema de internalização após interação com o CD44 é através de caveolina, onde diferentes cavéolas se fundem dando lugar a um caveossomo.140 Ademais, esses sistemas mostraram ser bem tolerados, não sendo observada nenhuma alteração morfológica, tanto na funcionalidade do olho quanto na produção e drenagem das lágrimas após a sua administração em coelhos.141 De maneira similar a outros polímeros, a atividade do ácido hialurônico parece ser dependente do seu peso molecular. Nanopartículas poliméricas preparadas por geleificação iônica mostraram que os sistemas formados por ácido hialurônico com cadeias menores do que 10 kDa transfectaram mais pDNA in vitro (células HCE e IOBA-NHC).89 Nanopartículas de PEI com 25kDa complexadas com pDNA, de cerca de 180 nm, tiveram seu tamanho substancialmente aumentado pelo uso de ácido hialurônico de baixo peso molecular (<10kDa), variando de cerca de 400 nm a mais de 1 µm.142 A blindagem com ácido hialurônico reduziu o potencial zeta de + 30 mV a 0 mV. Mesmo com propriedades físico-química desfavoráveis no que compete à estabilidade coloidal (levando em consideração estabilização preponderantemente eletrostática), baixa interação com a membrana celular devido à baixa carga positiva e o tamanho elevado (superior ao possível para internalização por clatrina ou caveolina), a internalização dessas partículas foi expressiva em células HCE, devido à alta densidade de receptores CD44 nesta linha celular. De acordo com Lesley e colaboradores, o tamanho mínimo para a ligação de oligômeros de ácido hialurônico com seu receptor CD44 é de 6 - 18 subunidades (mínimo de 1,1 kDa).143 Outros polímeros O sulfato de condroitina é um glicosaminoglicano sulfatado, amplamente distribuído pelo corpo e um dos principais constituintes do tecido conjuntivo. É um heteropolímero linear composto por unidades alternadas de N-acetilglicosamina e ácido glicurônico e tal como o ácido hialurônico, é capaz de interagir com receptores de endocitose (HARE e CD44), que podem ser encontrados na estrutura ocular.144-146 O sulfato de dextrana é um polímero ramificado composto basicamente de unidades de glicose unidas por ligação do tipo α, com grau de sulfatação variável. Ambos os polímeros são utilizados principalmente para diminuir a toxicidade intrínseca dos compostos carregados positivamente, como ocorre para nanopartículas de gelatina cationizada com espermina.147 A incorporação de sulfato de dextrano e sulfato de condroitina reduziu o IC50 dessas nanopartículas de 2 a 3 vezes (79 e 107 µg cm-2), respectivamente, para células de córnea HCE, sem comprometer a capacidade de transfectar pDNA.147 Na Figura 4 estão as estruturas químicas de alguns dos principais polímeros catiônicos e aniônicos utilizados no preparo de nanossistemas para a terapia genética ocular.
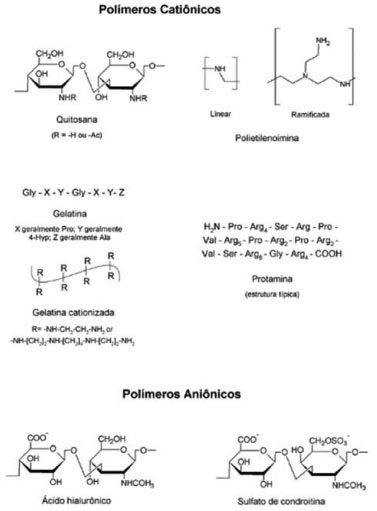 Figura 4. Estrutura de alguns dos principais polímeros catiônicos e aniônicos empregados em sistemas de base nanotecnológica para terapia genética ocular
CONSIDERAÇÕES FINAIS Mesmo que as bases moleculares da terapia genética estejam bem evoluídas, ainda o principal limitante dessa nova abordagem terapêutica reside na ausência de um sistema capaz de proteger o material genético ao mesmo tempo em que permita a sua entrada na célula desejada, até o seu alvo intracelular, seja ele o núcleo ou o citoplasma. A via ocular surge como ideal para que novas tecnologias sejam testadas devido às características particulares, principalmente devido ao seu relativo isolamento, permitindo um tratamento local e com pouca interferência imunológica. De acordo com a plataforma de ensaios clínicos do The Journal of Gene Medicine (wiley.co.uk/genmed/clinical), cerca de 70% dos ensaios clínicos são realizados com sistemas virais e cerca de 18% com ácidos nucleicos sem nenhum carreador. Assim, a quantidade de sistemas aprovados representa meros 0,1% dos sistemas que entram em fase clínica (não contando os inúmeros estudos terminados em fase pré-clínica). Frente a esse relativo insucesso, voltam-se as atenções para sistemas não virais capazes de triunfar ante o fracasso dos sistemas virais e ausência de carreadores. Naturalmente que a terapia genética não viral tem o seu sucesso estreitamente relacionado com a estrutura molecular dos biomateriais utilizados para o preparo desses sistemas, principalmente os de base nanotecnológica. As estratégias atuais são basicamente divididas entre sistemas lipídicos ou poliméricos, sendo relativamente restrita a gama de excipientes utilizados, muito em respeito à toxicidade intrínseca, irritação ocular e baixa eficácia. Lipídios ou polímeros catiônicos como DOTAP ou PEI, respectivamente, já foram exaustivamente testados e mesmo sendo padrões-ouro para transfecção in vitro, são inadequados para aplicações in vivo. Uma tendência atual parece ser a utilização de material endógeno ou com um percentual de sua estrutura baseado em um material de origem natural. A estratégia de utilizar materiais puramente sintéticos, até o momento, não apresentou os resultados esperados. Por essa razão, o uso de materiais como ácido hialurônico ou protamina, associados ou não a esses materiais sintéticos têm ocupado um lugar de destaque atualmente. No que diz respeito à natureza do nanossistema, não parece haver evidências fortes o suficiente para dizer que um sistema é superior ao outro. Os lipossomas são os mais conhecidos, contando com algumas formulações já aprovadas para fármacos, enquanto as nanopa, rtículas têm um crescimento exponencial no que se refere a publicações na área de terapia genética. O que é certo é que a ciência ainda não encontrou o sistema ideal. Assim, a pesquisa por novos sistemas para a terapia genética, seja ocular ou não, invariavelmente deve focar no estudo de novos biomateriais.
AGRADECIMENTOS Os autores gostariam de agradecer ao CNPq e à CAPES pelo apoio financeiro. G.Z. agradece ao programa Ciência sem Fronteiras (BJT 028/2012).
REFERÊNCIAS 1. Organization, W.-W. H. Genomics and World Health - Report of the Advisory Committee on Health Research; 2002. 2. Crooke, S. T.; Antisense Nucleic Acid Drug Dev. 1998, 8, 6. 3. Apte, R. S.; Expert Opin. Pharmacother. 2008, 9, 499. 4. Gaudana, R.; Jwala, J.; Boddu, S. H. S.; Mitra, A. K.; Pharm. Res. 2008, 26, 1197. 5. Hao, J.; Li, S. K.; Kao, W. W. Y.; Liu, C. Y.; Brain Res. Bull. 2010, 81, 256. 6. Borderie, V.; Touzeau, O.; Bourcier, T.; Laroche, L.; EMC-Ophtalmologie 2005, 2, 103. 7. Araki-Sasaki, K.; Ohashi, Y.; Sasabe, T.; Hayashi, K.; Watanabe, H.; Tano, Y.; Handa, H.; Invest. Ophthalmol. Visual Sci. 1995, 36, 614. 8. Diebold, Y.; Calonge, M.; Enriquez de Salamanca, A.; Callejo, S.; Corrales, R. M.; Saez, V.; Siemasko, K. F.; Stern, M. E.; Invest. Ophthalmol. Visual Sci. 2003, 44, 4263. 9. Dunn, K. C.; Aotaki-Keen, A. E.; Putkey, F. R.; Hjelmeland, L. M.; Exp. Eye Res. 1996, 62, 155. 10. Weir, A. B.; Collins, M.; Assessing Ocular Toxicology in Laboratory Animals. Springer: Heidelberg, 2013. 11. Middaugh, C. R.; Evans, R. K.; Montgomery, D. L.; Casimiro, D. R.; J. Pharm. Sci. 1998, 87, 130. 12. Patil, S. D.; Rhodes, D. G.; Burgess, D. J.; The AAPS Journal 2005, 7, E61. 13. Fattal, E.; Bochot, A.; Adv. Drug Delivery Rev. 2006, 58, 1203. 14. Bloquel, C.; Bourges, J. L.; Touchard, E.; Berdugo, M.; BenEzra, D.; Behar-Cohen, F.; Adv. Drug Delivery Rev. 2006, 58, 1224. 15. Elbashir, S. M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T.; Nature 2001, 411, 494. 16. Aagaard, L.; Rossi, J. J.; Adv. Drug Delivery Rev. 2007, 59, 75. 17. Ulrich, H.; Trujillo, C. A.; Nery, A. A.; Alves, J. M.; Majumder, P.; Resende, R. R.; Martins, A. H.; Comb. Chem. High Throughput Screening 2066, 9, 619. 18. Ni, X.; Castanares, M.; Mukherjee, A.; Lupold, S. E.; Curr. Med. Chem. 2011, 18, 4206. 19. Kanwar, J. R.; Shankaranarayanan, J. S.; Gurudevan, S.; Kanwar, R. K.; Drug Discovery Today 2014, 19, 1309. 20. Choi, M. J.; Maibach, H. I.; Skin Pharmacol. Physiol. 2005, 18, 209. 21. Kaur, I. P.; Garg, A.; Singla, A. K.; Aggarwal, D.; Int. J. Pharm. 2004, 269, 1. 22. Fenske, D. B.; Chonn, A.; Cullis, P. R.; Toxicol. Pathol. 2008, 36, 21. 23. Gutiérrez, J. M.; González, C.; Maestro, A.; Solè, I.; Pey, C. M.; Nolla, J.; Curr. Opin. Colloid Interface Sci. 2008, 13, 245. 24. Bouchemal, K.; Briançon, S.; Perrier, E.; Fessi, H.; Int. J. Pharm. 2004, 280, 241. 25. Tamilvanan, S.; Prog. Lipid Res. 2004, 43, 489 26. Fraga, M.; Bruxel, F.; Diel, D.; de Carvalho, T. G.; Perez, C. A.; Magalhães-Paniago, R.; Malachias, Â.; Oliveira, M. C.; Matte, U.; Teixeira, H. F.; J. Controlled Release 2015, 209, 37. 27. Sahoo, S. K.; Dilnawaz, F.; Krishnakumar, K.; Drug Discovery Today 2008, 13, 144. 28. Nagarwal, R. C.; Kant, S.; Singh, P. N.; Maiti, P.; Pandit, J. K.; J. Controlled Release 2009, 136, 2 29. Souto, E. B.; Almeida, A. J.; Muller, R. H.; J. Biomed. Nanotechnol. 2007, 3, 317. 30. Souto, E. B.; Mueller, R. H.; Int. J. Cosmet. Sci. 2008, 30, 157. 31. Muchow, M.; Maincent, P.; Muller, R. H.; Drug Dev. Ind. Pharm. 2008, 34, 1394. 32. Pardeike, J.; Hommoss, A.; Muller, R. H.; Int. J. Pharm. 2009, 366, 170. 33. Dufes, C.; Uchegbu, I. F.; Schatzlein, A. G.; Adv. Drug Delivery Rev. 2005, 57, 2177. 34. Chaplot, S. P.; Rupenthal, I. D.; J. Pharm. Pharmacol. 2014, 66, 542. 35. Raviña, M.; Paolicelli, P.; Seijo, B.; Sanchez, A.; Mini-Rev. Med. Chem. 2010, 10, 73. 36. Bhadra, D.; Bhadra, S.; Jain, S.; Jain, N. K.; Int. J. Pharm. 2003, 257, 111. 37. Ooya, T.; Lee, J.; Park, K.; J. Controlled Release 2003, 93, 121. 38. Durairaj, C.; Kadam, R. S.; Chandler, J. W.; Hutcherson, S. L.; Kompella, U. B.; Invest. Ophthalmol. Visual Sci. 2010, 51, 5804. 39. Holden, C. A.; Tyagi, P.; Thakur, A.; Kadam, R.; Jadhav, G.; Kompella, U. B.; Yang, H.; Nanomedicine: Nanotechnology, Biology and Medicine 2012, 8, 776. 40. Iezzi, R.; Guru, B. R.; Glybina, I. V.; Mishra, M. K.; Kennedy, A.; Kannan, R. M.; Biomaterials 2012, 33, 979. 41. Spataro, G.; Malecaze, F.; Turrin, C.-O.; Soler, V.; Duhayon, C.; Elena, P.-P.; Majoral, J.-P.; Caminade, A.-M.; Eur. J. Med. Chem. 2010, 45, 326. 42. Vandamme, T. F.; Brobeck, L.; J. Controlled Release 2005, 102, 23. 43. Bochot, A. l.; Fattal, E.; Boutet, V. r.; Deverre, J. R.; Jeanny, J. C.; Chacun, H. l. n.; Couvreur, P.; Invest. Ophthalmol. Visual Sci. 2002, 43, 253. 44. Frézard, F.; Schettini, D. A.; Rocha, O. G. F.; Demicheli, C.; Quim. Nova 2005, 28, 511. 45. Bochot, A.; Fattal, E.; Boutet, V.; Deverre, J. R.; Jeanny, J. C.; Chacun, H.; Couvreur, P.; Invest. Ophthalmol. Visual Sci. 2002, 43, 253. 46. Camelo, S.; Lajavardi, L.; Bochot, A.; Goldenberg, B.; Naud, M. C.; Fattal, E.; Behar-Cohen, F.; de Kozak, Y.; Mol. Vision 2007, 13, 2263. 47. Hathout, R. M.; Mansour, S.; Mortada, N. D.; Guinedi, A. S.; AAPS PharmSciTech 2007, 8, 1. 48. Lajavardi, L.; Bochot, A.; Camelo, S.; Goldenberg, B.; Naud, M. C.; Behar-Cohen, F.; Fattal, E.; de Kozak, Y.; Invest. Ophthalmol. Visual Sci. 2007, 48, 3230. 49. Shen, Y.; Tu, J.; The AAPS Journal 2007, 9, 371. 50. Schuh, R. S.; Bruxel, F.; Teixeira, H. F.; Quim. Nova 2014, 37, 1193. 51. Kawakami, S.; Harada, A.; Sakanaka, K.; Nishida, K.; Nakamura, J.; Sakaeda, T.; Ichikawa, N.; Nakashima, M.; Sasaki, H.; Int. J. Pharm. 2004, 8, 255. 52. Mannermaa, E.; Rönkkö, S.; Ruponen, M.; Reinisalo, M.; Urtti, A.; Curr. Eye Res. 2005, 30, 345. 53. Matsuo, T.; Masuda, Y.; Yasuda, T.; Matsuo, N.; Biochem. Biophys. Res. Commun. 1996, 219, 947. 54. Peeters, L.; Sanders, N. N.; Braeckmans, K.; Boussery, K.; de Voorde, J. V.; De Smedt, S. C.; Demeester, J.; Invest. Ophthalmol. Visual Sci. 2005, 46, 3553. 55. Toropainen, E.; Hornof, M.; Kaarniranta, K.; Johansson, P.; Urtti, A.; J. Gene Med. 2007, 9, 208. 56. Alqawlaq, S.; Sivak, J. M.; Huzil, J. T.; Ivanova, M. V.; Flanagan, J. G.; Beazely, M. A.; Foldvari, M.; Nanomedicine: Nanotechnology, Biology and Medicine 2014, 10, 1637. 57. Tremblay, C.; Barza, M.; Szoka, F.; Lahav, M.; Baum, J.; Invest. Ophthalmol. Visual Sci. 1985, 26, 711. 58. Martini, É.; Carvalho, E.; Teixeira, H.; Leão, F.; Oliveira, M. C. d.; Quim. Nova 2007, 30, 930. 59. Arima, H.; Aramaki, Y.; Tsuchiya, S.; J. Pharm. Sci. 1997, 86, 438. 60. Masuda, I.; Matsuo, T.; Yasuda, T.; Matsuo, N.; Invest. Ophthalmol. Visual Sci. 1996, 37, 1914. 61. Zhao, D. D.; Watarai, S.; Lee, J. T.; Kouchi, S.; Ohmori, H.; Yasuda, T.; Acta Med. Okayama 1997, 51, 149. 62. Rabinovich-Guilatt, L.; Couvreur, P.; Lambert, G.; Dubernet, C.; J. Drug Targeting 2004, 12, 623. 63. Hagigit, T.; Abdulrazik, M.; Orucov, F.; Valamanesh, F.; Lambert, M.; Lambert, G.; Behar-Cohen, F.; Benita, S.; J. Controlled Release 2010, 145, 297. 64. Hagigit, T.; Abdulrazik, M.; Valamanesh, F.; Behar-Cohen, F.; Benita, S.; J. Controlled Release 2012, 160, 225. 65. Kawakami, S.; Harada, A.; Sakanaka, K.; Nishida, K.; Nakamura, J.; Sakaeda, T.; Ichikawa, N.; Nakashima, M.; Sasaki, H.; Int. J. Pharm. 2004, 278, 255. 66. Bruxel, F.; Laux, M.; Wild, L. B.; Fraga, M.; Koester, L. S.; Teixeira, H. F.; Quim. Nova 2012, 35, 1827. 67. Souto, E. B.; Muller, R. H.; Die Pharmazie 2006, 61, 431. 68. del Pozo-Rodríguez, A.; Delgado, D.; Solinís, M. A.; Gascón, A. R.; Pedraz, J. L.; Int. J. Pharm. 2008, 360, 177. 69. del Pozo-Rodríguez, A.; Delgado, D.; Solinís, M. Á.; Pedraz, J. L.; Echevarría, E.; Rodríguez, J. M.; Gascón, A. R.; Int. J. Pharm. 2010, 385, 157. 70. Geng, S.; Yang, B.; Wang, G.; Qin, G.; Wada, S.; Wang, J. Y.; Nanotechnology 2014, 25, 275103. 71. Lajunen, T.; Hisazumi, K.; Kanazawa, T.; Okada, H.; Seta, Y.; Yliperttula, M.; Urtti, A.; Takashima, Y.; Eur. J. Pharm. Sci. 2014, 62, 23. 72. Pires, A. L. R.; Bierhalz, A. C. K.; Moraes, Â. M.; Quim. Nova 2015, 38, 957. 73. Laranjeira, M. C. M.; Fávere, V. T. d.; Quim. Nova 2009, 32, 672. 74. Cho, Y.-W.; Jang, J.; Park, C. R.; Ko, S.-W.; Biomacromolecules 2000, 1, 609. 75. Kofuji, K.; Qian, C.-J.; Nishimura, M.; Sugiyama, I.; Murata, Y.; Kawashima, S.; Eur. Polym. J. 2005, 41, 2784. 76. Chatelet, C.; Damour, O.; Domard, A.; Biomaterials 2001, 22, 261. 77. Aranaz, I.; Mengibar, M.; Harris, R.; Panos, I.; Miralles, B.; Acosta, N.; Galed, G.; Heras, A.; Curr. Chem. Biol. 2009, 3, 203. 78. Zhang, H.; Neau, S. H.; Biomaterials 2001, 22, 1653. 79. Dash, M.; Chiellini, F.; Ottenbrite, R. M.; Chiellini, E.; Prog. Polym. Sci. 2011,36, 981. 80. Huang, Y.; Lapitsky, Y.; Langmuir 2011, 27, 10392. 81. Park, K.; Kim, J.-H.; Nam, Y. S.; Lee, S.; Nam, H. Y.; Kim, K.; Park, J. H.; Kim, I.-S.; Choi, K.; Kim, S. Y.; Kwon, I. C.; J. Controlled Release 2007, 122, 305. 82. Paolicelli, P.; de la Fuente, M.; Sanchez, A.; Seijo, B.; Alonso, M. J.; Expert Opin. Drug Delivery 2009, 6, 239. 83. Calvo, P.; Remunan-Lopez, C.; VilaJato, J. L.; Alonso, M. J.; Pharm. Res. 1997, 14, 1431. 84. de Campos, A. M.; Sanchez, A.; Gref, R.; Calvo, P.; Alonso, M. J.; Eur. J. Pharm. Sci. 2003, 20, 73. 85. Diebold, Y.; Jarrin, M.; Saez, V.; Carvalho, E. L. S.; Orea, M.; Calonge, M.; Seijo, B.; Alonso, M. J.; Biomaterials 2007, 28, 1553. 86. Carvalho, E. L. S.; Grenha, A.; Remuñan-Lopez, C.; Alonso, M. J.; Seijo, B.; Methods Enzymol. 2009, 465, 289. 87. de la Fuente, M.; Seijo, B.; Alonso, M. J.; Gene Ther. 2008, 15, 668. 88. de la Fuente, M.; Seijo, B.; Alonso, M. J.; Nanotechnology 2008, 19, 075105 89. de la Fuente, M.; Seijo, B.; Alonso, M. J.; Invest. Ophthalmol. Visual Sci. 2008, 49, 2016. 90. Janes, K. A.; Alonso, M. J.; J. Appl. Polym. Sci. 2003, 88, 2769. 91. Prow, T. W.; Bhutto, I.; Kim, S. Y.; Grebe, R.; Merges, C.; McLeod, D. S.; Uno, K.; Mennon, M.; Rodriguez, L.; Leong, K.; Lutty, G. A.; Nanomedicine: Nanotechnology, Biology and Medicine 2008, 4, 340. 92. de la Fuente, M.; Seijo, B.; Alonso, M. J.; Macromol. Biosci. 2008, 8, 441. 93. Biegeleisen, K.; J. Theor. Biol. 2006, 241, 533. 94. Masuda, T.; Akita, H.; Harashima, H.; FEBS Lett. 2005, 579, 2143. 95. Cui, Z.; Kang, J.; Hu, D.; Zhou, J.; Wang, Y.; Mol. Cells 2014, 37, 613. 96. Li, S.; Huang, L.; Gene Ther. 1997, 4, 891. 97. Wang, Y.; Rajala, A.; Rajala, R. V.; J. Funct. Biomater. 2015, 6, 379. 98. del Pozo-Rodríguez, A.; Delgado, D.; Gascón, A. R.; Solinís, M. Á.; J. Ocul. Pharmacol. Ther. 2013, 29, 173. 99. Li, S.; Rizzo, M. A.; Bhattacharya, S.; Huang, L.; Gene Ther. 1998, 5, 930. 100. Liu, H. A.; Liu, Y. L.; Ma, Z. Z.; Wang, J. C.; Zhang, Q.; Invest. Ophthalmol. Visual Sci. 2011, 52, 4789. 101. Rajala, A.; Wang, Y.; Zhu, Y.; Ranjo-Bishop, M.; Ma, J.-X.; Mao, C.; Rajala, R. V. S.; Nano Lett. 2014, 14, 5257. 102. Delgado, D.; del Pozo-Rodríguez, A.; Solinís, M. Á.; Rodríguez-Gascón, A.; Eur. J. Pharm. Biopharm. 2011, 79, 495. 103. Delgado, D.; del Pozo-Rodríguez, A.; Solinís, M. Á.; Avilés-Triqueros, M.; Weber, B. H. F.; Fernández, E.; R. Gascón, A.; Hum. Gene Ther. 2012, 23, 345. 104. Khalil, I. A.; Kogure, K.; Futaki, S.; Harashima, H.; Int. J. Pharm. 2008, 354, 39. 105. Veldhoen, S.; Laufer, S. D.; Restle, T.; Int. J. Mol. Sci. 2008, 9, 1276. 106. Boussif, O.; Lezoualc'h, F.; Zanta, M. A.; Mergny, M. D.; Scherman, D.; Demeneix, B.; Behr, J. P.; Proc. Natl. Acad. Sci. U. S. A. 1995, 92, 7297. 107. Mohan, R. R.; Tovey, J. C.; Sharma, A.; Tandon, A.; Prog. Retinal Eye Res. 2012, 31, 43. 108. Thomas, M.; Ge, Q.; Lu, J. J.; Chen, J.; Klibanov, A. M.; Pharm. Res. 2005, 22, 373. 109. Gunther, M.; Lipka, J.; Malek, A.; Gutsch, D.; Kreyling, W.; Aigner, A.; Eur. J. Pharm. Biopharm. 2011, 77, 438. 110. Benjaminsen, R. V.; Mattebjerg, M. A.; Henriksen, J. R.; Moghimi, S. M.; Andresen, T. L.; Mol. Ther. 2013, 21, 149. 111. Basarkar, A.; Singh, J.; Int. J. Nanomed. 2007, 2, 353. 112. Tiyaboonchai, W.; Woiszwillo, J.; Middaugh, C. R.; Eur. J. Pharm. Sci. 2003, 19, 191. 113. Fischer, D.; Li, Y.; Ahlemeyer, B.; Krieglstein, J.; Kissel, T.; Biomaterials 2003, 24, 1121. 114. Donnelly, K. S.; Giuliano, E. A.; Sharma, A.; Tandon, A.; Rodier, J. T.; Mohan, R. R.; Vet. Ophthalmol. 2014, 17, 162. 115. Sharma, A.; Tandon, A.; Tovey, J. C.; Gupta, R.; Robertson, J. D.; Fortune, J. A.; Klibanov, A. M.; Cowden, J. W.; Rieger, F. G.; Mohan, R. R.; Nanomedicine: Nanotechnology, Biology and Medicine 2011, 7, 505. 116. Kurosaki, T.; Uematsu, M.; Shimoda, K.; Suzuma, K.; Nakai, M.; Nakamura, T.; Kitahara, T.; Kitaoka, T.; Sasaki, H.; Biol. Pharm. Bull. 2013, 36, 96. 117. Kambhampati, S. P.; Kannan, R. M.; J. Ocul. Pharmacol. Ther. 2013, 29, 151. 118. Yavuz, B.; Pehlivan, S. B.; Ünlü, N.; Sci. World J. 2013, 2013, 13. 119. Chaum, E.; Hatton, M. P.; Stein, G.; J. Cell. Biochem. 1999, 76, 153. 120. Hudde, T.; Rayner, S. A.; Comer, R. M.; Weber, M.; Isaacs, J. D.; Waldmann, H.; Larkin, D. F.; George, A. J.; Gene Ther. 1999, 6, 939. 121. King Heiden, T. C.; Dengler, E.; Kao, W. J.; Heideman, W.; Peterson, R. E.; Toxicol. Appl. Pharmacol. 2007, 225, 70. 122. Fant, K.; Esbjörner, E. K.; Jenkins, A.; Grossel, M. C.; Lincoln, P.; Nordén, B.; Mol. Pharmaceutics 2010, 7, 1734. 123. Mastorakos, P.; Kambhampati, S. P.; Mishra, M. K.; Wu, T.; Song, E.; Hanes, J.; Kannan, R. M.; Nanoscale 2015, 7, 3845. 124. Marano, R. J.; Toth, I.; Wimmer, N.; Brankov, M.; Rakoczy, P. E.; Gene Ther. 2005, 12, 1544. 125. Marano, R. J.; Wimmer, N.; Kearns, P. S.; Thomas, B. G.; Toth, I.; Brankov, M.; Rakoczy, P. E.; Exp. Eye Res. 2004, 79, 525. 126. Howard, K. A.; Adv. Drug Delivery Rev. 2009, 61, 710. 127. Elzoghby, A. O.; J. Controlled Release 2013, 172, 1075. 128. Minakuchi, Y.; Takeshita, F.; Kosaka, N.; Sasaki, H.; Yamamoto, Y.; Kouno, M.; Honma, K.; Nagahara, S.; Hanai, K.; Sano, A.; Kato, T.; Terada, T.; Ochiya, T.; Nucleic Acids Res. 2004, 32, e109. 129. Zorzi, G. K.; Seijo, B.; Sanchez, A., Endogenous Polymers as Biomaterials for Nanoparticulate Gene Therapy. In Handbook of Polymers for Pharmaceutical Technologies, John Wiley & Sons, Inc.: 2015; pp 237. 130. López-Cebral, R.; Martín-Pastor, M.; Párraga, J. E.; Zorzi, G. K.; Seijo, B.; Sánchez, A.; Med. Chem. 2011, 7, 145. 131. Elzoghby, A. O.; J. Controlled Release 2013, 172, 1075. 132. Zorzi, G. K.; Párraga, J. E.; Seijo, B.; Sanchez, A.; Colloids Surf., B 2015, 135, 533. 133. Fischer, D.; Bieber, T.; Brüsselbach, S.; Elsässer, H. P.; Kissel, T.; Int. J. Pharm. 2001, 225, 97. 134. Contreras-Ruiz, L.; Zorzi, G. K.; Hileeto, D.; Lopez-Garcia, A.; Calonge, M.; Seijo, B.; Sanchez, A.; Diebold, Y.; Gene Ther. 2013, 20, 467. 135. Konat Zorzi, G.; Contreras-Ruiz, L.; Párraga, J. E.; López-García, A.; Romero Bello, R.; Diebold, Y.; Seijo, B.; Sánchez, A.; Mol. Pharmaceutics 2011, 8, 1783. 136. Yenice, I.; Mocan, M. C.; Palaska, E.; Bochot, A.; Bilensoy, E.; Vural, I.; Irkec, M.; Hincal, A. A.; Exp. Eye Res. 2008, 87, 162. 137. Apaolaza, P. S.; Delgado, D.; Pozo-Rodríguez, A. d.; Gascón, A. R.; Solinís, M. Á.; Int. J. Pharm. 2014, 465, 413. 138. Kong, M.; Park, H. J.; Carbohydr. Polym. 2011, 83, 1303. 139. Parraga, J. E.; Zorzi, G. K.; Diebold, Y.; Seijo, B.; Sanchez, A.; Int. J. Pharm. 2014, 477, 12. 140. Contreras-Ruiz, L.; de la Fuente, M.; Lopez-Garcia, A.; Fernández, I.; Seijo, B.; Sánchez, A.; Calonge, M.; Diebold, Y.; Mol. Vision 2011, 17, 279. 141. Contreras-Ruiz, L.; de la Fuente, M.; Garcia-Vazquez, C.; Saez, V.; Seijo, B.; Alonso, M. J.; Calonge, M.; Diebold, Y.; Cornea 2010, 29, 550. 142. Hornof, M.; de la Fuente, M.; Hallikainen, M.; Tammi, R. H.; Urtti, A.; J. Gene Med. 2008, 10, 70. 143. Lesley, J.; Hascall, V. C.; Tammi, M.; Hyman, R.; J. Biol. Chem. 2000, 275, 26967. 144. Kawashima, H.; Li, Y. F.; Watanabe, N.; Hirose, J.; Hirose, M.; Miyasaka, M.; Int. Immunol. 1999, 11, 393. 145. Kawashima, H.; Miyasaka, M.; Trends Glycosci. Glycotechnol. 2000, 12, 283. 146. Weigel, J. A.; Raymond, R. C.; Weigel, P. H.; Biochem. Biophys. Res. Commun. 2002, 294, 918. 147. Zorzi, G. K.; Párraga, J. E.; Seijo, B.; Sánchez, A.; Macromol. Biosci. 2011, 11, 905.
#Programa de Pós-Graduação em Ciências Farmacêuticas |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access