
 |
 |
 |
 |
 |
Artigo
| On-line solid phase extraction coupled with liquid chromatography tandem mass spectrometry for the determination of codeine in human plasma |
|
Alessandro de C. CruzI,*; Eunice M. SuenagaII; Eduardo AbibIII; José PedrazzoliI
IDepartamento de Farmacologia, Faculdade de Ciências Médicas, Universidade de Campinas, 13086010 Campinas - SP, Brasil Recebido em 02/03/2016 *e-mail: alesscruz@yahoo.com.br A specific LC-MS/MS method was developed and validated for automated determination of codeine in human plasma, using on-line solid phase extraction (SPE) system coupled with positive ion electrospray ionization tandem mass spectrometry. The method allowed plasma direct injection onto cartridge without sample pre-treatment. Total analysis time per run was 3 min, allowing high-throughput for codeine determination. SPE on-line along a monolithic column (Chromolith Performance RP-18e, 100 mm x 4.6 mm) demonstrated to be highly effective in terms of backpressure, separation speed and peak asymmetry. Calibration curves range was linear 5.0-200 ng mL-1. Method showed an excellent intra-day and inter-day precision ranged from 2.34 to 7.25% (CV%) as well as great intra-day and inter-day accuracy, ranging from 97.64 to 110% (RE%). SPE-LC-MS/MS method provided selectivity, accuracy, precision, fastness and high-throughput to assess codeine pharmacokinetics in human plasma samples. INTRODUCTION Codeine, 7,8-didehydro-4,5-epoxy-3-methoxy-17-methylmorphinan-6-ol (methylmorphine) is a phenanthrene-derivative opiate agonist used in mild to moderate pain relief (Figure 1). Several studies have demonstrated that it is still a drug of choice for treating acute and chronic pain.1-5Although, many bioanalytical methods have been developed for determination of codeine by LC-MS/MS and CG-MS, such as toxicological and forensic aplicattions,6-13 most of these methods are based on off-line liquid-liquid or solid phase extraction (SPE). These procedures require complex steps and laborious repetitive work for sample clean-up, which may not meet the requirement for high-throughput, robustness and speed in bioanalysis and pharmacokinetic studies.14 Hence, more reliable, accurate and fast analytical methods are required.
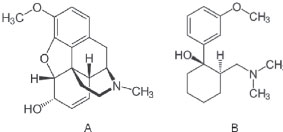 Figure 1. Molecular structure of codeine (A) and IS (B)
On-line SPE has been used as an alternative to traditional extraction, offering significantly higher assay efficiency due to chromatographic run time synchronized to sample extraction, resulting in notably decrease analysis time.15 Accordingly, monolithic silica columns improve conventional chromatographic techniques (C8 and C18), allowing high-flow on-line extraction and higher throughput on sample analysis.16 This paper describes an improved, rapid, selective and sensitive on-line SPE-LC-MS/MS method for the determination of codeine in human plasma using tramadol as an internal standard (IS) and its application to a bioequivalence study.
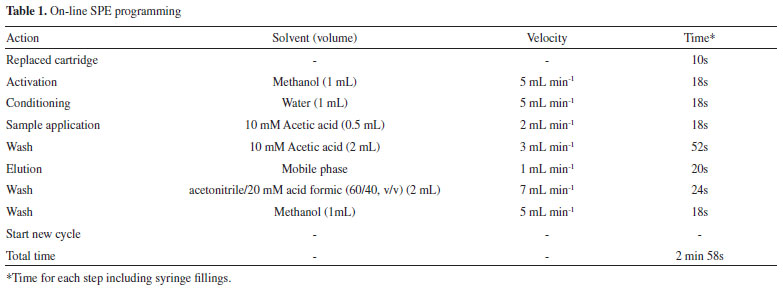
EXPERIMENTAL Chemicals Codeine phosphate was obtained from U.S. Pharmacopeia (Rockville, MD) and tramadol (IS) was obtained from Biosintetica Farmaceutica LTDA. (Sao Paulo, Brazil). HPLC grade acetonitrile, methanol and acetic acid was purchased from J.T. Baker (Phillipsburg, NJ, USA). Water was purified using a Milli-Q Gradient System from Millipore (Sao Paulo, Brazil). Instrumentation On-line SPE was performed with a Prospekt-2 automated SPE system (Spark Holland, Emmen, Netherlands) including an autosampler (Endurance), a high pressure dispenser (HPD) and an automated cartridge exchanger module (ACE). SPE cartridges used were HySphere C18, HD, 7 mm (Spark Holland, Emmen, Netherlands). HPLC system used was Shimadzu LC-10ADVP pump (Kyoto, Japan). LC separation was run through a Chromolith Performance RP-18e (100 mm x 4.6 mm) column (Merck, Germany) with a SecurityGuard C-18 guard column (4 mm x 3 mm) from Phenomenex (Torrance, CA, USA). Mass spectrometry was performed in a Quattro LC triple-quadrupole (Waters, Milford, MA, USA) equipped with an API electrospray source operating in positive mode. Standards and quality control samples preparation Drug stock solutions were prepared separately at a concentration of 10 µg mL-1 in acetonitrile/water (1:1 v/v). Working solution of tramadol (IS) was prepared in the same solvent at concentration of 70 ng mL-1. Codeine stock solution aliquots were used to spike blank human plasma in order to obtain calibration standards of 5.0, 10, 20, 50, 100, 200 ng mL-1. Aliquots of 200 ng mL-1 samples were used to spike blank human plasma in order to obtain three levels of quality control (QCs) at 15, 80, and 160 ng mL-1 (low, medium and high). Aliquots of 450 µL from calibration standards and quality controls were spiked with 50 µL IS work solution. Following these samples were rapidly mixed and centrifuged at 14000 rpm for 3 min and aliquots of 300 µL were transferred to 96-well plates. Liquid chromatography, SPE and mass spectrometry conditions The mobile phase was a mixture of acetonitrile/acetic acid 10 mmol L-1 pH 3.5 (50:50, v/v) at a flow rate of 1 mL min-1. Analytical column was operated at room temperature (25 ºC) and total run time was set for 3.0 min. Autosampler temperature was maintained at 4.0 ºC and was set to make 100 µL sample injection. The SPE sequence was: cartridges were solvated with 1 mL methanol (at 5.0 mL min-1) and equilibrated with 1 mL water (at 5.0 mL min-1) to obtain suitable conditions for analyte adsorption. Subsequently, 100 µL of spiked plasma was loaded onto cartridge using 0.5 mL of 10 mmol L-1 acetic acid. Then, the cartridge was washed with 4.0 mL of 10 mmol L-1 acetic acid. After completion of SPE, cartridge was switched in-line with the LC-MS/MS mobile phase to elute the analyte onto the analytical column. The column effluent was split to 200 µL min-1 before it entered the mass spectrometer ESI source. Two channels of positive ion multiple reaction monitoring (MRM) modes were used to detect codeine and IS. The most abundant product ions were: m/z 215.2 from the parent ion m/z 300.0 for codeine and m/z 58.4 from the parent ion m/z 264.3 for IS, respectively. Source temperature was optimized at 120 ºC, desolvation temperature was 300 ºC, and desolvation gas flow was 550 L h-1. The capillary voltage was set at 3.0 kV, while cone energy for codeine and IS were 40 and 20 V, respectively. Collision energy was optimized for codeine (25 V) and IS (15 V). The multiplier was set at 750 V and cone gas flow was 110 L h-1. Validation study The method was validated for selectivity, linearity, precision, accuracy, extract recovery and stability based upon ANVISA bioanalytical method guideline.17 Selectivity was determined analyzing six different blank human plasma (including six distinct lots of normal, lipemic and hemolyzed) spiked with codeine and IS (70 ng mL-1). To evaluate linearity, calibration curves with six codeine concentration levels (5.0-200 ng mL-1) were prepared and analyzed in duplicates three consecutive days. Standard curves were obtained by weighted least-squares regression (weighting = 1/x) of measured peak area ratios analyte/IS versus analyte concentrations added to plasma. Precision and accuracy were evaluated by analyzing six replicates at 15, 80, 160 ng mL-1 (low, medium and high QC samples, respectively) of codeine and were analyzed on three different days to determine intra- and inter-day variation. Precision was estimated from coefficient of variation (CV%) and accuracy was measured as % of recovery. Quantification limit was estimated based on signal intensity and noise for the m/z analyte trace (S/N > 10) with precision and accuracy less than or equal 20%. Codeine stability in plasma was studied under a variety of storage and handling conditions using the low, medium and high QC samples. Short-term temperature stability was assessed through QC sample analysis, kept at ambient temperature for 6 h. Sample stability in autosampler was conducted analyzing QC samples kept under the autosampler condition (4 ºC) for 24 h. Freeze-thaw stability (-20 ºC in plasma) was checked through three cycles. Long-term stability was performed at -20 ºC in plasma for 30 days. Recovery was calculated by comparing mean analyte peak areas extracted from plasma samples versus mean peak areas obtained from solutions prepared in mobile phase in the same concentration. Matrix effect was evaluated by ion suppression degree due to matrix components. Continuous infusion postcolumn of codeine and IS (1 µg mL-1) were performed through a syringe pump, while a blank human plasma sample was extracted and chromatographed in the conditions of on-line SPE. Ion suppression degree was monitored at elution time of codeine and IS by continuously acquiring MRM mode.
RESULTS AND DISCUSSION HPLC-MS/MS condition optimization Mass spectrometric parameters were optimized to achieve maximum scan and product abundance from compounds analyzed. Full scan codeine and IS product ion mass spectra are displayed in Figure 2. Monolithic columns improve high-throughput due to higher separation efficiency at high flow velocities when compared to conventional LC columns.16 Svensson et al.9 determined codeine retention time of approximately 5 min using solid phase extraction (SPE) and conventional C18 column chromatography. Most forensic application methods determined codeine with long chromatographic run time.6-13 Therefore, mobile phase optimization is important to improve peak shape and sensitivity detection as well as codeine and tramadol chromatographic run time reduction (IS).
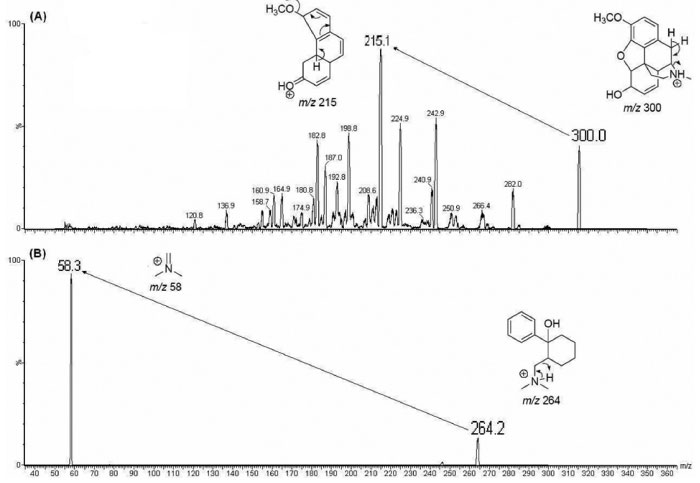 Figure 2. Precursor and product ion spectra of codeine (A) and IS (B)
Best chromatographic condition was achieved using a Chromolith Performance RP-18e (100 mm x 4.6 mm) column maintained at ambient temperature (25º C). Mobile phase was a mixture of acetonitrile/acetic acid 10 mmol L-1 pH 3.5 (50:50, v/v) at a flow rate of 1 mL min-1. Codeine and IS were eluted at 1.37 and 1.53 min, respectively and total analysis run time was 3 min synchronized to sample extraction, allowing high-throughput. No interference from drug-free human plasma was observed, and method selectivity in presence of endogenous plasma compounds was satisfactory. Figure 3 shows MRM chromatograms of blank plasma, plasma spiked with codeine, plasma spiked with IS and QC.
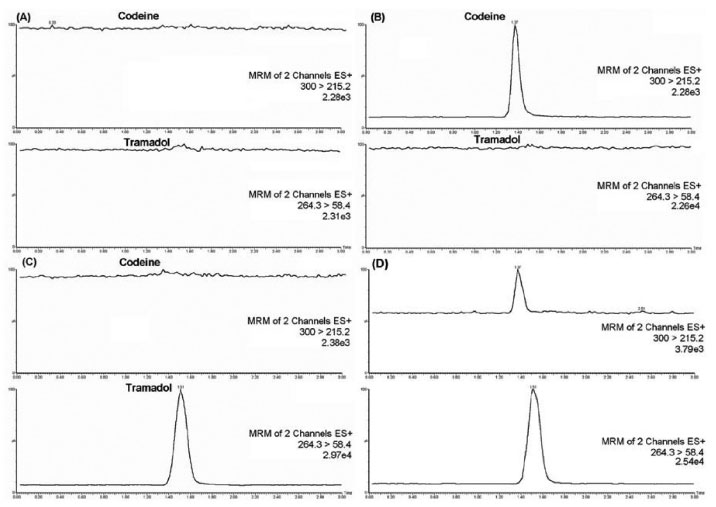 Figure 3. Representative MRM chromatograms of codeine and IS in human plasma: blank plasma sample (A), plasma spiked with codeine 50 ng mL-1 (B), plasma spiked with IS 70 ng mL-1 (C) and QC sample LQ (D)
On-line SPE process On-line SPE technique offers speed, high precision and labor decrease for sample clean-up.16 Methods based on liquid-liquid or solid-phase off-line extractions for determination of codeine require laborious repetitive work and time.15 Furthermore, organic solvents employed in this technique expose analyst and environment to biohazard.14 In recent publication, Verplaetse and Henion18 determined a series of opioids by automated dried blood spot desorption coupled on-line SPE-LC-MS/MS method. Although showed excellent precision, accuracy and reduced analysis time, this technique was not applied a large number of samples or even complete codeine pharmacokinetic study. Other few methods were performed based upon on-line SPE to determined codeine. However, such methods were performed in other matrices and sample extraction steps were semi-automated.6,18-22 Liao et al.23 developed a codeine determination method with chromatographic run time of 2 min and applied it successfully to a bioequivalence study. In spite of results achieved in relation to recovery of about 80%, the method based on liquid-liquid extraction consists of several steps to clean samples, including 15 min mixing, 10 min centrifugation, plus solvent evaporation time at 40 ºC. Prospeckt 2 system offers a wide range of cartridge sorbents and versatility to change a cartridge automatically.16 Hysphere C18 HD SPE stationary phase was chosen for sample preparation purpose, allowing higher load capacity and more uniform analyte extraction reproducibility.24-28 After mobile phase desorbed analytes and transferred to analytical column, cartridge was washed with 2 mL of mixture acetonitrile/acid formic 20 mmol L-1 pH 3.2 (60/40, v/v) and methanol (1 mL). Therefore, no carry-over was observed when a blank solution was injected into cartridge after injecting the highest calibration point. Cartridge was then physically moved with a robotic arm, leaving the extraction clamp set to new sample. Total cycle time of 3 min allows 480 samples to be analyzed per day.
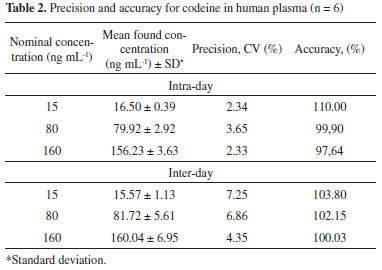
Method validation Matrix effect was evaluated by post-column analyte infusion experiments based on method described by Bonfiglio et al.29 Four different blank plasma sample (including normal, lipemic and hemolysed) were extracted under on-line SPE conditions and chromatographed. No significant ion suppression effect was observed in codeine and IS retention time. The recovery of codeine was evaluated with four different concentrations 5, 15, 80, 160 ng mL-1 by comparing the peak area ratios (peak area of the solution/peak area of the plasma sample extracted). Mean extraction recoveries of codeine were 66.62, 62.82, 60.51 and 75.14%, respectively. Tramadol (IS) average recovery was 74.57% ranging from 69.89-77.07%. Regarding codeine recovery values some studies18-21 also reached similar results, indicating that low recovery is a characteristic of these compounds when submitted to SPE conditions. Linearity was evaluated using codeine calibration curves from 5.0 to 200 ng mL-1. A good linear relationship between peak area ratios and concentrations was established. The mean regression equation and determination coefficient (r2) from duplicate calibration curves on three non-consecutive days were: y = 0.013215(±0.001475)x + 0.018946(±0.013191) and r2 = 0.998573(±0.001093). Lowest concentration calibration standard (5 ng mL-1) was taken as quantification limit. Precision and accuracy for this method were controlled by calculating intra- and inter-day variation at three concentrations (15, 80, 160 ng mL-1) of QC samples in six replicates. Intra-day mean accuracy ranged from -2.36 to 10%, and intra-day precision ranged from 2.33 to 3.65% (CV). Inter-day accuracy ranged from 0.03 to 3.80%, and inter-day precision ranged from 4.35 to 7.25, as shown in Table 2. Although, the toxicological and forensic methods6-13 described here are sufficiently precise and accurate for the measurement of codeine in human plasma, this current method showed excellent results, with intra and inter-day accuracy and precision less than 10% and within the ranges specified by ANVISA guideline.17 Other automated and pharmacokinetics determination of the codeine18-23 also failed to achieve intra-day precision less than 5% as was obtained in this study. These results demonstrate that this method has been effectively optimized, with less sample manipulation and higher accuracy and precision as described by Xu et al.16
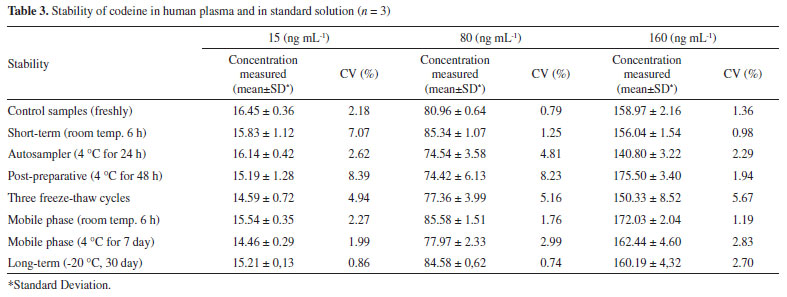
Codeine stability in human plasma under different storage conditions is summarized in Table 3. There was no significant degradation at any tested concentration and all CV(%) values between post-storage and initial QC samples were within ± 15%. Codeine was stable under all conditions described and no stability-related problems were encountered during routine sample analysis.
Method application This validated on-line SPE-LC-MS/MS method was successfully applied to pharmacokinetic study of codeine tablet in 31 healthy volunteers following oral administration of 30 mg codeine. High-throughput sample analyses are very important in studies that require a large number of samples. In the present study, 1700 samples were analyzed, including clinical samples, calibration curves and QC samples. There was no significant change related to backpressure, peak asymmetry and retention time reproducibility. Figure 4 shows a representative plasma concentration-time profile for codeine.
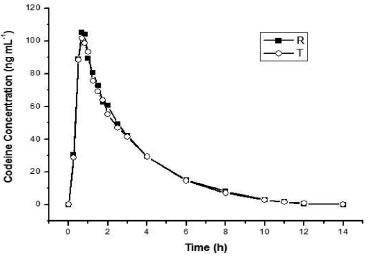 Figure 4. Representative plasma concentration-time profile for codeine after 30 mg single oral dose (R: reference formulation and T: test formulation)
CONCLUSIONS An automated LC-MS/MS method has been developed and validated for codeine determination in human plasma utilizing on-line solid phase extraction. This method awards compensation over those previously reported6-13,23 in terms of time per sample clean-up, improved separation speed by monolithic column and high-throughput due to fast chromatographic run time synchronized to sample extraction (3.0 min). On-line SPE-LC-MS/MS method allowed a rapid assay development and sample processing. Method was validated the concentration range 5.0-200 ng mL-1. Assay performance results indicate that the method is sufficiently precise and accurate for routine determination of the codeine in human plasma.
REFERENCES 1. Colini, B. G.; Cobianchi, M. R.; Minerva Med. 2012, 103, 177. 2. Mattia, C.; Ferrari, A.; Minerva Med. 2014, 105, 501. 3. Kircher, J.; Drendel, A. L.; Newton, A.S.; Plint, A.C.; Vandermeer, B.; Dulai, S.; Ali, S.; Clin. Pediatr. (Philadelphia) 2014, 53, 1326. 4. Mahic, M.; Fredheim, O. M.; Borchgrevink, P. C.; Skurtveit, S.; Eur. J. Pain 2014, 20, 632. 5. Mofizul, I. M.; McRae, I. S.; Mazumdar, S.; Taplin, S.; McKetin, R.; Intern. Med. J. 2015, 25, 6. Fritch, D.; Blum, K.; Nonnemacher, S.; Haggerty, B. J.; Sullivan, M. P.; Cone, E. J.; J. Anal. Toxicol. 2009, 33, 569. 7. Zheng, N.; Jiang, H.; Zeng, J.; Bioanalysis 2014, 6, 2441. 8. Oiestad, E. L.; Johansen, U.; Christophersen, A. S.; Clin. Chem. 2007, 53, 300. 9. Svensson, J. O.; Andersson, M.; Gustavsson, E.; Beck, O.; J. Anal. Toxicol. 2007, 31, 81. 10. Al-Asmari, A. I.; Anderson, R. A.; J. Anal. Toxicol. 2007, 31, 394. 11. Hegstad, S.; Khiabani, H. Z.; Kristoffersen, L.; Kunoe, N.; Lobmaier, P. P.; Christophersen, A. S.; J. Anal. Toxicol. 2008, 32, 364. 12. Huang, D.; Liu, Huang, C. M.; Chien, C.; Rapid Commun. Mass Spectrom. 2009, 23, 957. 13. Coles, R.; Kushnir, M. M.; Nelson, G. J.; McMillin, G. A.; Urry, F. M.; J. Anal. Toxicol. 2007, 31, 1. 14. Namera, A.; Saito, T.; Bioanalysis 2013, 8, 915. 15. Mullett, W. M.; J. Biochem. Biophys. Methods 2007, 70, 263. 16. Xu, R. N.; Fan, L.; Rieser, M. J.; El-Shourbagy, T. A.; J. Pharm. Biomed. Anal. 2007, 44, 342. 17. Agência Nacional de Vigilância Sanitária (ANVISA); Resolução Nº 899, de 29 de maio de 2003. Guia para validação de métodos analíticos e bioanalíticos. Diário oficial da República Federativa do Brasil, Poder Executivo, Brasília, DF, 02 de junho de 2003. Seções 2-4, 13 p. (Métodos Bioanalíticos). 18. Verplaetse, R.; Henion, J.; Drug Test. Anal. 2016, 1, 30. 19. Ramírez Fernández Mdel, M.; Van Durme, F.; Wille, S. M.; di Fazio, V.; Kummer, N.; Samyn, N.; J. Anal. Toxicol. 2014, 38, 280. 20. Lerch, O.; Temme, O.; Daldrup, T.; Anal. Bioanal. Chem. 2014, 406, 4443. 21. Fontanals, N.; Marcé, R. M.; Borrull, F.; J. Chromatogr. A 2011, 1218, 5975. 22. Musshoff, F.; Trafkowski, J.; Kuepper, U.; Madea, B.; J. Mass Spectrom. 2006, 41, 633. 23. Liao, Q.; Deng, Y.; Xie, Z.; Zhang, L.; J. Sep. Sci. 2009, 32, 202. 24. Alnouti, Y.; Srinivasan, K.; Waddell, D.; Bi, H.; Kavetskaia, O.; Gusev, A. I.; J. Chromatogr. A 2005, 1080, 99. 25. Schellen, A.; Ooms, B.; Lagemaat, D.; Vreeken, R.; Dongen, W. D.; J. Chromatogr. B 2003, 788, 251. 26. Niederlander, H. A.; Koster, E. H.; Hilhorst, M. J.; Metting, H. J.; Eilders, M.; Ooms, B.; de Jong, G. J.; J. Chromatogr. B 2006, 834, 98. 27. Souverian, S.; Rudaz, S.; Veuthey, J. L.; J. Chromatogr. B 2004, 801, 141. 28. Rodriguez-Mozaz, S.; de Alda, M. J. L.; Barceló, D.; J. Chromatogr. A 2007, 1152, 97. 29. Bonfiglio, R.; King, R. C.; Olah, T. V.; Merkle, K.; Rapid Commun. Mass Spectrom. 1999, 13, 1175. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access