
 |
 |
 |
 |
 |
Assuntos Gerais
|
|
| O prêmio nobel de química em 2016: máquinas moleculares# The nobel prize in chemistry 2016: molecular machines |
|
Karla S. FeuI,*; Francisco F. de AssisI; Siddavatam NagendraI,II; Ronaldo A. PilliI
IInstituto de Química, Universidade Estadual de Campinas, 13084-971 Campinas - SP, Brasil Recebido em 16/11/2016 *e-mail: karlafeu@gmail.com The Nobel Prize in Chemistry 2016 was granted to Jean-Pierre Sauvage, Sir James F. Stoddart and Bernard (Ben) L. Feringa. These scientists were responsible for the creation and development of molecular machines. The development of such systems represents a breakthrough in the field of supramolecular chemistry, nanotechnology and organic synthesis. This article aims to bring simple and objective information to the scientific community about the work of these three researchers. INTRODUÇÃO A Academia Real Sueca de Ciências laureou os cientistas Jean-Pierre Sauvage, Sir James Fraser Stoddart e Bernard L. Feringa com o prêmio Nobel de Química em 2016 "pelo design e síntese das máquinas moleculares". Em nosso dia a dia, estamos acostumados a utilizar diversos tipos de máquinas para nos ajudar em tarefas dos mais variados tipos, algumas tão simples como a preparação de um café e outras extremamente complexas como a linha de montagem de um automóvel. O design e construção de novas maquinarias é algo que está fortemente associado ao desenvolvimento tecnológico e científico e, como resultado, vem trazendo mais conforto e recursos para a nossa sociedade. O conceito de construir máquinas moleculares é bastante recente e a construção efetiva do que se considera máquinas moleculares ocorreu a cerca de 25 anos atrás.1 Uma das maiores influências e inspirações para construção de máquinas moleculares veio do físico Richard Feynman, também laureado com o prêmio Nobel de Física em 1965. Em sua famosa palestra para a reunião anual da Sociedade Americana de Física no ano de 1959, cujo título era "There's plenty of room at the bottom" que, em tradução livre para a língua portuguesa podemos entender como "Ainda há muito espaço lá no fundo", Feynman queria transmitir a idéia que ainda havia muito espaço para ser explorado no interior dos átomos, apontando claramente para um novo caminho para o desenvolvimento da ciência e tecnologia da época e, de forma particular, no que se configurou nos anos seguintes como a área de nanociência e nanotecnologia, com a perspectiva inclusive de construção de máquinas moleculares, apontada por Drexler em seu livro "Engines of Creation".2 Quando se pensa na construção de uma máquina é necessário dispor de várias peças. O funcionamento adequado desta máquina depende de como os seus componentes respondem uns aos outros quando há um estímulo externo de energia, ou seja, quando a máquina é ligada. Muitas das máquinas com as quais estamos acostumados têm seu funcionamento baseado em movimentos de seus componentes internos. Saber como controlar esses movimentos para que a máquina possa desempenhar de forma correta a sua função é algo fundamental para os projetistas e engenheiros. Construir máquinas em nível molecular possui os mesmos desafios (e outros mais!) para os químicos, pois estamos acostumados a construir moléculas através da formação de ligações covalentes entre átomos, de forma que os movimentos do sistema estão baseados na rotação em torno de ligações simples, estiramento das ligações e na deformação dos ângulos de ligação. Outra inspiração dos químicos para a construção das máquinas moleculares é a própria Natureza, como, por exemplo, o motor rotatório da síntese da ATP que converte ADP em ATP, o motor linear miosina-actina capaz de criar as contrações musculares e, também, a linha de produção de proteínas existentes no interior os ribossomos. Vale a pena ressaltar, que o estudos com os ribossomos levaram ao prêmio Nobel de Química, em 2009.1 Entretanto, a construção de máquinas moleculares só foi possível graças ao desenvolvimento de moléculas interligadas sem o uso de ligações covalentes e de moléculas contendo ligações isomerizáveis (ligações duplas) capazes de girar em uma única direção. Contribuições do laboratório de J-P. Sauvage: Catenanos O estudo das moléculas interligadas teve início com a síntese dos catenanos que são arquiteturas moleculares formadas por dois (ou mais) anéis macrocíclicos interligados um ao outro, de forma que, apesar de não haver ligações colaventes entre eles, um não pode se separar do outro (Figura 1). A este tipo de junção molecular se dá o nome de ligação mecânica. O nome dessa classe de compostos é derivado da palavra "catena" que, em Latim, significa "cadeia, corrente".
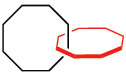 Figura 1. Catenanos
O primeiro relato sobre a síntese de catenanos surgiu no início da década de 60, quando o grupo do professor Wasserman3 publicou um trabalho descrevendo a preparação do composto 3, mediante reação entre 1 e 2 (Esquema 1). A formação do produto 3 está fortemente atrelada à remota probabilidade de que, no momento da ciclização, o composto 2 se encontre no interior de 1, justificando assim o baixo rendimento obtido na época. Neste caso, o composto sintetizado foi um [2]catenano, onde o prefixo "[2]" indica o número de anéis macrocíclicos interligados.
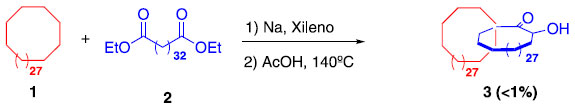 Esquema 1. Síntese do [2]catenano 3 relatada por Wasserman3
Em 1983, o grupo do professor Jean-Pierre Sauvage introduziu um novo conceito denominado de "síntese por molde", que permitiu a obtenção de catenanos de maneira bastante eficiente, dada a sua complexidade.4 Assim como Wasserman, Sauvage fez com que uma molécula acíclica se fechasse ao redor de uma estrutura cíclica pré-existente. A grande diferença é que ele utilizou um íon Cu(I) para colocar as moléculas na posição adequada para que o fechamento pudesse acontecer. O rendimento global da obtenção de 9 foi de 42% (Esquema 2), que pode ser considerado extremamente eficiente, dada a complexidade de formação de 9 e dos precedentes existentes até então. Para obter o composto livre do íon Cu(I), bastou tratar 9 com um ligante com alta afinidade pelo metal, neste caso, o íon cianeto (Esquema 3).4
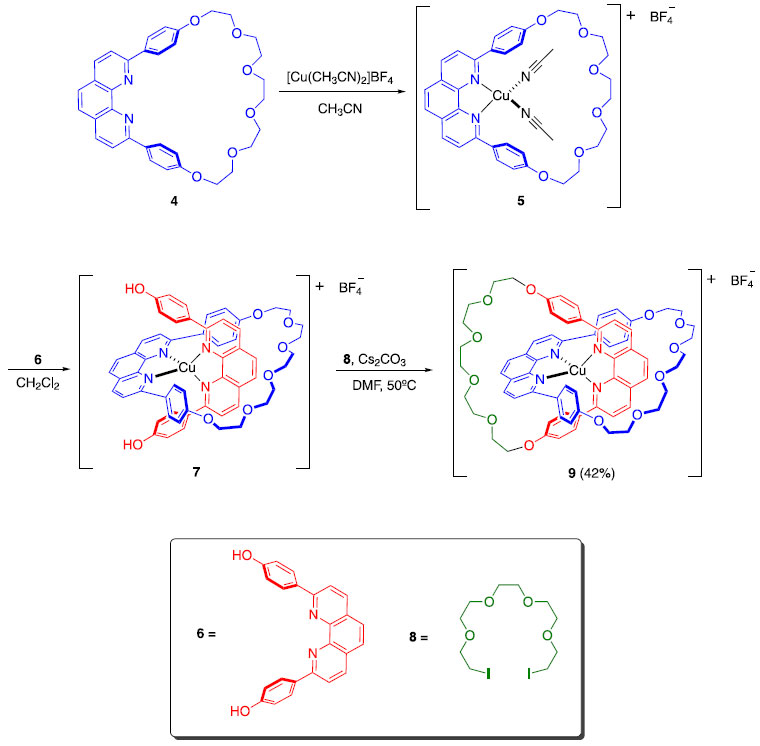 Esquema 2. Síntese do [2]catenano 9 relatada por Sauvage4
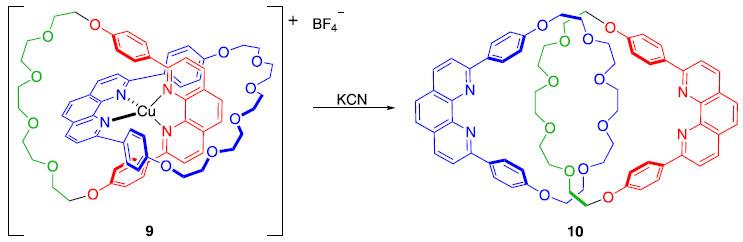 Esquema 3. Obtenção do [2]catenano 10 a partir de 9 pela remoção do íon Cu(I)4
Pode-se dizer que este foi um marco que alavancou vigorosamente o desenvolvimento da química de compostos contendo ligações mecânicas, pois forneceu aos cientistas da época uma maneira eficiente de preparar essas estruturas. O grupo de Sauvage empregou sua metodologia na obtenção de estruturas ainda mais desafiadoras que os [2]catenanos. Um dos exemplos que se pode citar é a síntese de estruturas conhecidas como Trefoil Knot, cuja tradução é "nó de trevo" (Figura 3-A). Esses símbolos fazem parte da iconografia de muitas culturas antigas, como no caso do Triquetra (Figura 3-B), um símbolo encontrado em livros da cultura celta que representa a santa trindade da fé cristã.5,6
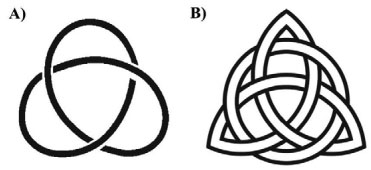 Figura 3. Representação genérica do Trefoil Knot7 e da Triquetra8. Reprinted (adapted) with permission from (Forgan R. S.; Sauvage, J. P.; Stoddart, J. F. Chem. Rev, 2011, 111, 5434-5464 Copyright (2016) American Chemical Society.
Nos primeiros trabalhos relatados pelo grupo de Sauvage, esses compostos foram obtidos em baixo rendimento (3%), servindo como prova de conceito de que era possível obter tais estruturas.9 O grupo de Sauvage investiu esforços para aprimorar sua metodologia de síntese de moléculas interligadas mecanicamente, de forma a permitir que tais estruturas pudessem ser obtidas com alta eficiência, viabilizando assim possíveis aplicações. Cerca de 10 anos depois, o mesmo grupo publicou trabalhos em que mostrava ser possível obter [2]catenanos10 e Trefoil Knots11 em excelentes rendimentos (Esquema 4).· Como é possível notar, a estratégia de "síntese por molde" utilizando íons Cu(I) foi mantida, porém, a principal diferença está na etapa de macrociclização, que foi alcançada utilizando a reação de metátese de fechamento de anel (RCM) em lugar de reações de alquilação. Vale ressaltar que o desenvolvimento desta reação foi responsável pela concessão do Prêmio Nobel de Química, em 2005, aos pesquisadores Yves Chauvin, Robert H. Grubbs e Richard R. Schrock.12
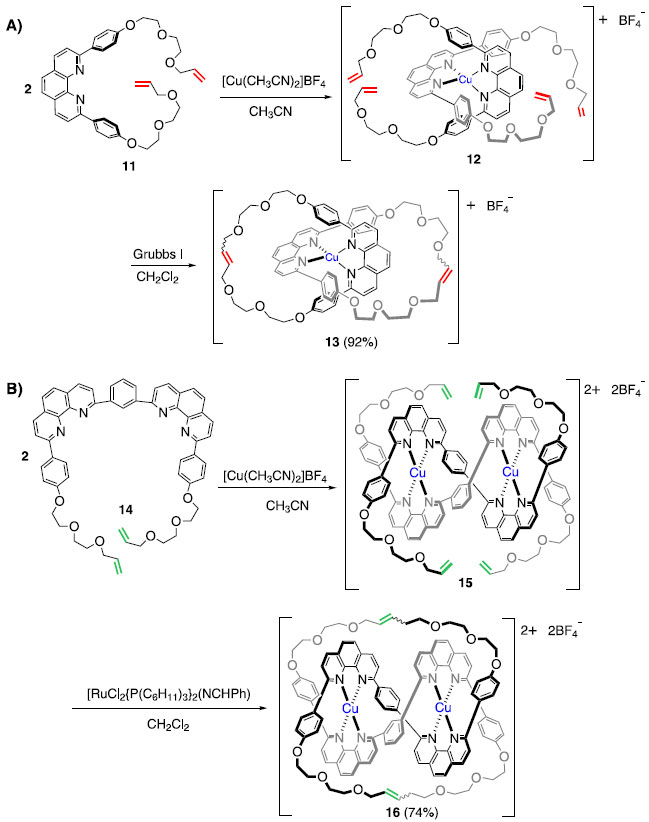 Esquema 4. Síntese aprimorada de [2]catenanos e compostos do tipo Trefoil Knot10,11
Uma das características das máquinas com as quais lidamos no nosso dia a dia é o fato de que, através de um estímulo externo, podemos induzi-las a realizar uma determinada ação/movimento. O grupo de Sauvage desenvolveu alguns sistemas do tipo [2]catenanos capazes de se movimentar devido a estímulos externos como: variação de pH, luz e calor. No caso do sistema mostrado no Esquema 5, a mudança de pH é o que provoca o movimento dos anéis macrocíclicos.13 Quando o meio encontra-se neutro ou básico, a conformação mais estável é aquela na qual os anéis bipiridínicos de um dos macrociclos formam uma espécie de estrutura sanduiche com o anel naftalenodioxi do outro macrociclo. Isso ocorre porque os anéis dos cátions bipiridínicos encontram-se deficientes de densidade eletrônica (presença das cargas positivas), ao passo que o anel naftalenodioxi é um anel rico em densidade eletrônica (efeito mesomérico dos átomos de oxigênio). Sendo assim, existe atração eletrostática entre eles e, ainda mais, ocorrem também interações entre o sistema de orbitais π dos anéis, denominadas de interações π-stacking. Quando o meio se encontra ácido, ocorre a protonação de uma das unidades de fenantrolina, permitindo a formação de ligações de hidrogênio com os pares de elétrons não-ligantes dos átomos de nitrogênio da outra unidade de fenantrolina (Esquema 5). Isso leva à conformação mostrada na estrutura 18.
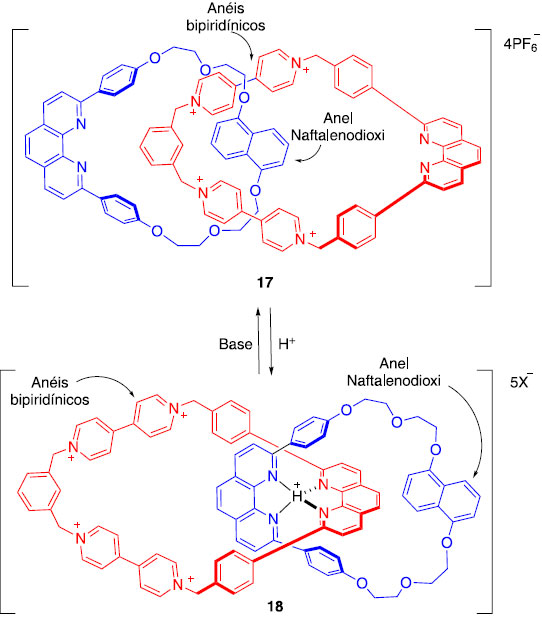 Esquema 5. Movimento dos anéis de um [2]catenano mediante alteração do pH da solução12
Outra maneira de induzir movimentos nesse tipo de sistema é através do uso de luz. Sabe-se que em complexos do tipo [Ru(di-imina)3]2+, a presença de ligantes volumosos leva à uma distorção da geometria octaédrica, facilitando a dissociação do complexo a partir do estado excitado.14 Utilizando uma metodologia semelhante àquela mostrada no Esquema 2, o grupo de Sauvage sintetizou um [2]catenano na forma de um complexo de Ru(II). Mediante a ação de luz ocorre a dissociação do ligante bipiridina e inserção de íons cloreto na esfera de coordenação do íon Ru(II) (Esquema 6).15 Quando a energia é fornecida na forma de calor, o processo se reverte.
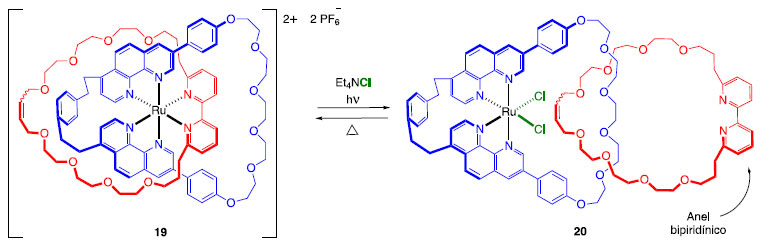 Esquema 6. Movimento dos anéis de um [2]catenano mediante ação de luz14
Contribuições do laboratório de J. F. Stoddart: Rotaxanos As propriedades dinâmicas dos catenanos encorajaram o grupo de Stoddart a criar, em 1991, os rotaxanos que, assim como os catenanos, são estruturas interligadas por ligações mecânicas (Esquema 7).16
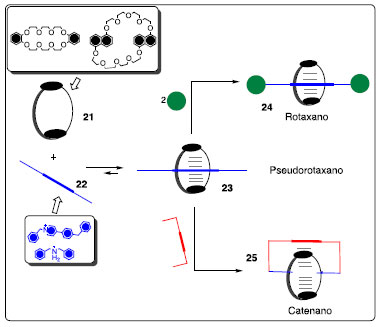 Esquema 7. Síntese por molde dos rotaxano, pseudorotaxano e catenano15
Logo, como podemos observar no Esquema 7, a inserção de uma molécula acíclica 22 dentro da cavidade de um macrocíclico 21 leva a um complexo supramolecular estabilizado por interações intermoleculares (como ligações de hidrogênio), denominado de pseudorotaxano (23), que pode se dissociar em seus componentes 21 e 22 devido ao equilíbrio entre as espécies controlado pela energia livre de complexação. Entretanto, com a introdução de dois grupos volumosos no final do componente acíclico do pseudorotaxano, há formação de um composto que não se dissocia em seus componentes, denominado rotaxano (24). Já os catenanos (25) são oriundos da ligação do pseudorotaxano com outro componente acíclico, o qual também não pode ser dissociado em seus componentes.16 Desta forma, Stoddart desenvolveu os rotaxanos que funcionam como um transportador molecular, ou seja, uma estrutura molecular em que uma plataforma tetracatiônica move-se ao longo de um eixo entre duas estações idênticas nas quais o reconhecimento ocorre por meio de interações moleculares de forma controlada, em movimento de ida e volta.17 Para o caso do rotaxano 28, mostrado no Esquema 8, a plataforma tetracatiônica 26 move-se entre as estações de reconhecimento por estímulos externos de temperatura. Essas estações são unidades 1,4-benzenodioxi dispostas simetricamente dentro do eixo de poliéter 27, apresentando grupos triisopropilsilila volumosos nas extremidades (Esquema 8).17
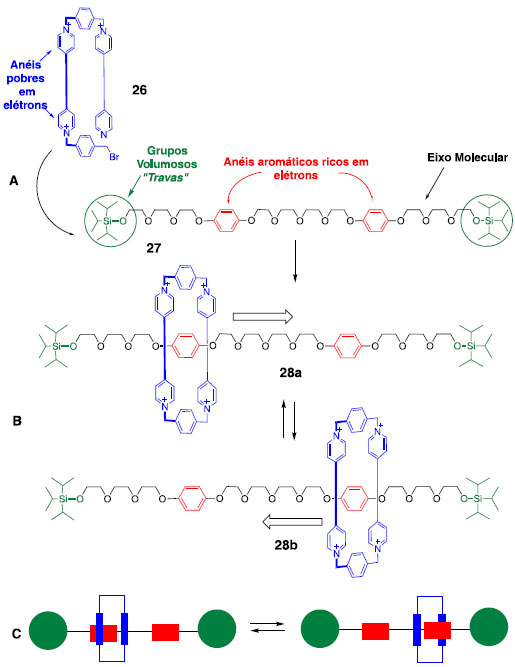 Esquema 8. A) Síntese do rotaxano desenvolvido por Stoddart; B) Movimento da plataforma tetracatiônica ao longo do eixo molecular; C) Desenho ilustrando o movimento do rotaxano16
Inspirados pelos promissores resultados dos primeiros rotaxanos, em 1995, Stoddart surpreendeu com o desenvolvimento de uma máquina molecular que se comporta como um elevador em escala nanométrica (Esquema 9). As dimensões do elevador são 2,5 nm de altura e diâmetro de 3,5 nm. Esta máquina molecular consiste da plataforma 31, contendo três anéis (macrocíclicos) presos por uma base, e o eixo 32, contendo três pernas de cadeias aromáticas (Esquema 9-C). Em cada uma das três pernas do eixo 32 existem dois níveis, que são interligados pela plataforma 31, a qual pode parar em qualquer um dos diferentes níveis. As pernas do eixo trifurcado 32 são protegidas por grupos volumosos 33 que funcionam como travas para não perder a plataforma.
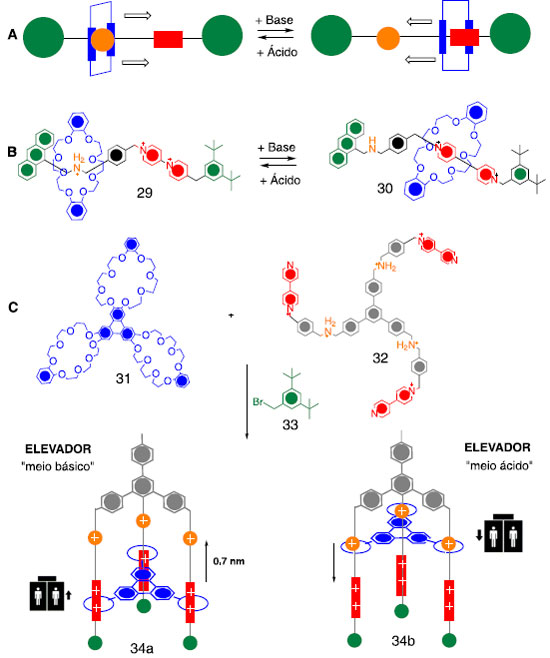 Esquema 9. A) Esquema representativo do rotaxano, contendo duas diferentes estações de reconhecimento e seu movimento ao longo do eixo molecular; B) Rotaxano contendo as unidades de reconhecimento dialquilamônio e anel bipirídinio; C) Síntese do Elevador e seu movimento de subida e descida decorrente das reações ácido-base17
A energia necessária para levantar e abaixar a plataforma entre os dois níveis dos eixos (pernas do equipamento) provém de uma reação ácido-base, sendo 0,7 nm a distância percorrida entre os níveis do elevador, envolvendo uma força de 200 pN. O elevador molecular foi inspirado no movimento de transporte molecular de um rotaxano contendo duas diferentes estações. Nesse sistema, a estrutura do anel envolve preferencialmente uma das duas estações de reconhecimento no eixo molecular (Esquema 9-B). O funcionamento ocorre por estímulos externos, movendo o anel de forma controlada ao longo do eixo pelas duas estações. O princípio de funcionamento do elevador é dado por reações ácido-base. Em meio ácido, as fortes ligações de hidrogênio [N+-H---O] permitem à plataforma permanecer na região superior do elevador (estação do dialquilamônio - NH2+ -, 34b). Já quando está em meio básico, ocorre a desprotonação da estação (- NH2+ -), levando à ruptura da forte ligação de hidrogênio, fazendo com que a plataforma se mova para a estação do anel bipiridínio (34a) onde é estabilizada por ligações de hidrogênio [C-H---O] e interações eletrônicas π-stacking. Após a adição de ácido, a plataforma move-se novamente para a estação superior do elevador (34b).18 Expandindo os estudos sobre os rotaxanos, foi criado o "músculo molecular" para mimetizar os movimentos de contração e extensão do músculo esquelético (Esquema 10).
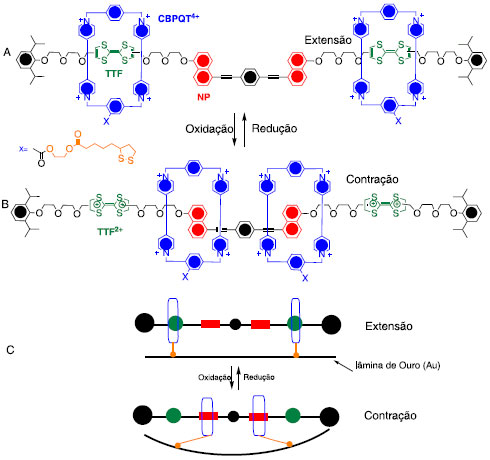 Esquema 10. A) Movimento de extensão do músculo molecular; B) Movimento de contração do músculo molecular; C) Esquema representativo do mecanismo de operação do músculo molecular ancorado na superfície do ouro18
O design foi pensado na bem estabelecida plataforma tetracatiônica (CBPQT4+) em volta de duas estações tetratiofulvaleno (TTF) ao invés das duas estações de naftaleno (NP). O funcionamento ocorre pela oxidação química das estações (TTF) para sua forma dicatiônica (TTF2+) dirigindo assim o anel (CBPQT4+) para a estação de naftaleno, a "forma contraída do músculo". Essa "força motriz" surge principalmente das repulsões eletrostáticas das estações (TTF2+). Após redução das duas estações (TTF2+), a plataforma volta para sua forma inicial, a "forma estendida do músculo". Uma aplicação desse sistema foi a incorporação de uma cadeia dissulfeto em cada plataforma (CBPQT4+), que fornece um ponto de ancoragem em superfície metálica de ouro, na forma de uma monocamada. Desta forma, o ciclo de contração e extensão mimetiza o movimento que ocorre dentro das fibras musculares naturais.19 Contribuições do Feringa: Motores moleculares unidirecionais Assim como foi dito anteriormente, o desenvolvimento de sistemas contendo ligações duplas isomerizáveis, capazes de girar em uma única direção, foi um dos avanços científicos que possibilitou o desenvolvimento das máquinas moleculares. Tais sistemas poderiam ser classificados como motores moleculares graças a sua característica de giro unidirecional, a exemplo do funcionamento dos motores elétricos com os quais estamos familiarizados. Até então, alguns cientistas já haviam estudado a possibilidade de controlar a rotação de ligações simples que apresentassem uma alta barreira energética,20 porém nenhum destes estudos levou ao desenvolvimento de sistemas que se comportassem como um verdadeiro motor molecular. Em 1999, o grupo do professor Bernard (Ben) L. Feringa demonstrou que era possível obter um movimento giratório unidirecional, característico de um motor. O motor unidirecional 35 é um composto denominado de "overcrowded alkene", cuja tradução livre para a língua portuguesa seria "alceno super congestionado". Tal motor é constituído de estruturas cíclicas unidas por uma ligação dupla e apresenta quiralidade. Vale a pena ressaltar que o fato de o motor ser quiral é de suma importância para o seu movimento unidirecional (Esquema 11).21
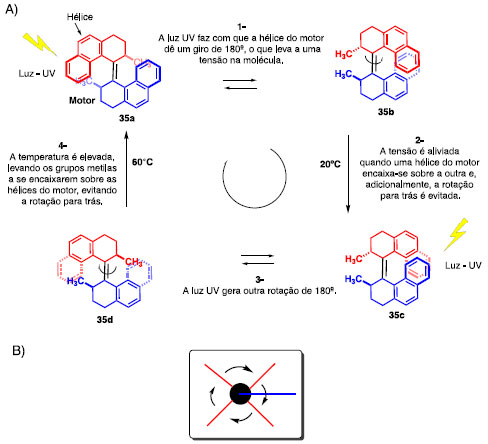 Esquema 11. A) Motor Unidirecional Molecular e seu princípio de funcionamento; B) Representação esquemática do movimento rotatório unidirecional21,22
O seu funcionamento decorre de quatro etapas ativadas por irradiação de luz e alteração na temperatura do sistema. O primeiro passo é uma isomerização da ligação dupla por luz de comprimento de onda 280 nm. Isso faz com que a conversão de 35b para 35a não possa ocorrer nas condições utilizadas, pois o composto 35b só pode ser excitado com luz UV de menor energia (λ = 380 nm). Em seguida, ocorre a inversão da hélice por ação da temperatura, levando ao isômero 35c. Já o terceiro passo é uma segunda isomerização da ligação dupla em 35c, levando ao 35d e, por último, outra inversão da hélice retornando a situação inicial (35a), configurando um movimento completo em uma única direção (Esquema 11).21 Este desenvolvimento representou um avanço, pois não existia, até então, uma conversão controlada de energia em movimento rotativo unidirecional, ou seja, máquinas que giravam em uma direção particular.21 O surgimento do motor serviu como inspiração para o grupo do Feringa e outros cientistas e, após sua criação, foram desenvolvidas outras classes de motores muito mais eficientes, além do ancoramento desses motores em superfícies metálicas para realizar tarefas específicas.23 Uma aplicação muito interessante desses sistemas ocorreu quando Feringa e seu grupo desenvolveram um motor que é incorporado a um filme de cristal líquido, capaz de girar objetos colocados sobre o filme que excedem o tamanho da molécula do motor por um fator de 10.000 vezes.24 O grupo do professor Feringa também desenvolveu um carro molecular, denominado "Nanocar", uma estrutura contendo quatro motores moleculares funcionando como as rodas do carro em um chassi molecular formado por ligações triplas.25 Esse sistema foi sublimado e depositado em uma superfície de cobre para que se pudesse excitar e visualizar seu movimento, com o auxílio de um microscópio de tunelamento eletrônico. Quando a ponta da agulha do microscópio se aproxima da estrutura do carro, a diferença de potencial entre a agulha e a superfície de cobre transfere energia às unidades motoras que são excitadas e passam pelo mesmo processo mostrado no esquema anterior, levando a um giro unidirecional dos anéis fluorenos (rodas), resultando no movimento translacional do carro (Esquema 12).25
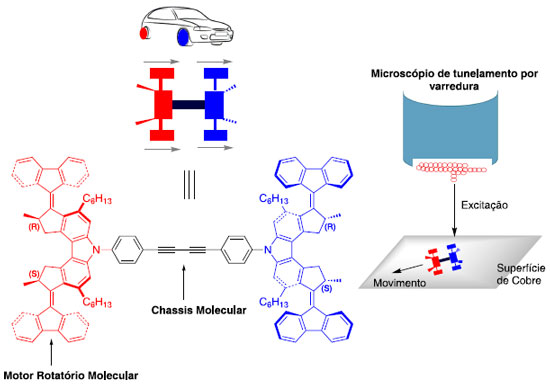 Esquema 12. Carro molecular desenvolvido pelo grupo do Feringa25
PERSPECTIVAS De acordo com Stoddart, agora que as máquinas moleculares já foram (e continuam sendo) desenvolvidas, é hora de torná-las úteis. Segundo ele mesmo, existem dois caminhos a serem seguidos: podemos continuar nano, projetando e construindo novas máquinas moleculares para que elas realizem tarefas que nenhuma outra máquina do mundo macro seja capaz, ou passarmos para a escala macro, onde trilhões destas máquinas podem trabalhar de maneira conjunta, remoldando materiais já existentes ou até mesmo movendo cargas, como se fossem um exército de formigas. Ben Feringa, em umas das suas declarações, disse acreditar que dentro de cinco anos já teremos os primeiros materiais inteligentes, baseados em máquinas moleculares.26 Atualmente já é possível ver uma aplicação comercial das máquinas moleculares no mundo macro. Em 2001, Ito e colaboradores sintetizaram um hidrogel contendo rotaxanos nos pontos de ligação entre as cadeias poliméricas.27 Este polímero tem sido utilizando desde 2011 pela Nissan e outras montadoras automotivas como forma de recobrimento da pintura dos veículos, protegendo-os contra eventuais arranhões. A presença dos rotaxanos nesses pontos da estrutura polimérica faz com que as cadeias possam deslizar facilmente umas sobre as outras, conferindo alta elasticidade ao polímero. Dessa forma, deformações provocadas por agentes externos podem ser facilmente absorvidas, fazendo com que o mesmo se recupere facilmente. Isso só é possível devido às ligações mecânicas presentes nos rotaxanos, as quais permitem que o macrociclo deslize sobre o eixo molecular central. Por fim, de acordo com Balzani,16 existem duas aplicações emergentes: 1) transporte molecular, como, por exemplo, os braços mecânicos e similares, que podem solucionar problemas de transporte em escala nano e, 2) no campo da lógica, os processadores moleculares de informações, que no futuro poderão levar a computadores moleculares. Embora seja difícil neste momento prever as aplicações dessas máquinas moleculares que irão, de fato, materializar-se, não devemos nos esquecer que muitas das ideias transformadoras tiveram sua gênese na curiosidade científica. Nesse sentido, é inquestionável que os avanços proporcionados pelos grupos de Sauvage, Stoddart e Feringa tornaram disponíveis os conhecimentos básicos necessários às aplicações futuras.
BIBLIOGRAFIAS Jean-Pierre Sauvage28 Jean-Pierre Sauvage é um químico francês nascido em Paris, em 21 de outubro de 1944. Ele obteve seu título de PhD em 1971 na Universidade Louis-Pasteur, sob a orientação do professor Jean-Marie Lehn, o qual foi um dos laureados do Premio Nobel de 1987. Durante seu doutorado, Sauvage contribuiu para a primeira síntese dos ligantes do tipo criptandos, tema pelo qual Lehn foi indicado ao Premio Nobel. Assim que terminou seu doutorado, Sauvage se juntou ao National Center for Scientific Research (CNRS) na Universidade de Strasbourg, como um pesquisador colaborador (research fellow). De 1973 a 1974, Sauvage realizou um estágio de pós-doutorado com o professor Malcolm L. H. Green, na universidade de Oxford, Inglaterra. Após o término de seu pós-doutorado, Sauvage retornou à Strasbourg onde hoje é professor emérito.
 Figura 4. Fotografia de Jean-Pierre-Sauvage. Créditos: Caroline Schneider / ISIS
Os principais interesses de pesquisa do professor Sauvage giram em torno da química de coordenação e da química supramolecular. Sua indicação ao Prêmio Nobel foi justificada com base na sua contribuição para o design e síntese de sistemas contendo ligações mecânicas, algo que foi considerado como ponto de partida e fonte de inspiração para o desenvolvimento de máquinas moleculares. James Fraser Stoddart29 Fraser Stoddart é um químico escocês, nascido em Edimburgo em 1942. Ele recebeu seu Bacharelado em 1964 e seu PhD em 1966 pela Universidade de Edinburgo. Em 1967 mudou-se para o Canadá, onde realizou seus estudos de pós-doutorado pela "National Research Council Postdoctoral Fellow" na "Queen's University". Em 1970, recebeu uma bolsa de pesquisador da "Imperial Chemical Insdustries" (ICI), na Universidade de Sheffield, antes de ingressar no corpo docente como professor de Química.
 Figura 5. Fotografia de James F. Stoddart. Créditos: Northwestern University
Após algumas passagens pela Universidade da Califórnia (UCLA) em 1978, e um período de sabático (1978-81) no "ICI Corporate Laboratory" em Runcorn, Inglaterra, ele retornou para Sheffield, onde foi promovido para "Readership", em 1982. Ele foi premiado com um grau DSc (Doutorado) pela Universidade de Edimburgo em 1980 por sua pesquisa em estereoquímica molecular. Em 1990, ele assumiu a Cadeira de Química Orgânica na Universidade de Birmingham. Em 1997 ele se mudou para a UCLA, onde assumiu a cátedra "Saul Winstein". Em janeiro de 2008, transferiu-se para a Universidade Northwestern, EUA. Bernard L. Feringa30 Bernard L. Feringa obteve seu PhD na Universidade de Groningen na Holanda, sob a supervisão do professor Hans Wynberg. Após um período trabalhando como pesquisador científico na empresa Shell, na Holanda, e também no "Biosciences Centre" no Reino Unido, ele se tornou professor na Universidade de Groningen em 1988. Em 2004, ele foi nomeado "Jacobus H. van't Hoff Distinguished Professor" de ciências moleculares. Ele também foi eleito membro estrangeiro honorário da Academia Americana de Artes e Ciências e é membro e vice-presidente da Real Academia Holandesa de Ciências. Em 2008, ele foi nomeado cavaleiro pela rainha da Holanda.
 Figura 6. Fotografia de Bernard L. Feringa. Créditos: Feringa
A pesquisa de Feringa foi reconhecida com vários prêmios como: o "Koerber European Science Award" (2003), o "Spinoza Award" (2004), o "Prelog gold medal" (2005), o "Norrish Award" da ACS (2007), o "Paracelsus medal" (2008), a medalha de Quiralidade (2009) e o RSC "Organic Stereochemistry Award" (2011), entre outros. Os interesses de pesquisa de Feringa incluem: síntese orgânica, catálise assimétrica e nanossistemas em geral, como interruptores e motores moleculares.
REFERÊNCIAS 1 . Cheng, C.; Stoddart, J. F.; Chem. Phys. Chem. 2016, 17, 1780. 2. Drexler, K. E.; Engines of Creation, 6th ed., Anchor Press/Doubleday: Garden City, 1986. 3. Wasserman, E.; J. Am. Chem. Soc. 1960, 82, 4433. 4. Dietrich-Buchecker, C. O.; Sauvage, J. P.; Tetrahedron Lett. 1983, 24, 5095. 5. Laing, J.; Laing, L.; Art of the Celts: From 700 BC to the Celtic revival, Thames and Hudson: New York, 1992. 6. Meehan, B.; The Book of Kells: An Illustrated Introduction to the Manuscript in Trinity College Dublin, Thames and Hudson: New York, 1994. 7. https://www.nobelprize.org/nobel_prizes/chemistry/laureates/2016/advanced-chemistryprize2016.pdf, acessado em janeiro de 2017. 8. Forgan, R. S.; Sauvage, J. P.; Stoddart, J. F.; Chem. Rev. 2011, 111, 5434. 9. Dietrich-Buchecker, C. O.; Sauvage, J. P.; Angew. Chem., Int. Ed. Engl. 1989, 28, 189. 10. Mohr, B.; Weck, M.; Sauvage, J.-P.; Grubbs, R. H.; Angew. Chem., Int. Ed. Engl. 1997, 36, 1308. 11. Dietrich-Buchecker, C. O.; Sauvage, J.-P.; Decian, A.; Fischer, J.; J. Chem. Soc. Chem. Commun. 1994, 2231. 12. Ferreira, V. F.; da Silva, F. de C.; Quim. Nova Esc. 2005, 22, 3. 13. Amabilino, D. B.; Dietrich-Buchecker, C. O.; Livoreil, A.; Pérez-García, L.; Sauvage, J. P.; Stoddart, J. F.; J. Am. Chem. Soc. 1996, 118, 3905. 14. Van Houten, J.; Watts, R. J. Inorg. Chem. 1978, 17, 3381; Rillema, D. P.; Taghidiri, D. S.; Jones, D. S.; Keller, C. D.; Worl, L. A.; Meyer, T. J.; Levy, H. A.; Inorg. Chem. 1987, 26, 578; Wang, R.; Vos, J. G.; Schmehl, R. H.; Hage, R.; J. Am. Chem. Soc. 1992, 114, 1964. 15. Mobian, P.; Kern, J. M.; Sauvage, J. P.; Angew. Chem., Int. Ed. 2004, 43, 2392. 16. Raymo, F. M.; Stoddart, J. F. Chem. Rev. 1999, 99, 1643; Balzani, V.; Credi, A.; The Chemical Record 2001, 1, 422. 17. Anelli, P. L.; Stoddart, J. F.; J. Am. Chem. Soc. 1991, 113, 5131. 18. Badjic, J. D.; Balzani, V.; Credi, A.; Silvi, S.; Stoddart, J. F.; Science 2004, 303,1845. 19. Huang, T. J.; Brough, B.; Ho, C-M.; Liu, Y.; Flood, A. H.; Bonvallet, P. A.; Tseng, H-R.; Stoddart, J. F.; Baller, M.; Magonov, S.; Appl. Phys. Lett. 2004, 85, 5391. 20. Blount, J. F.; Finocchiaro, P.; Gust, D.; Mislow, K.; J. Am. Chem. Soc. 1973, 95, 7019; Iwamura, H.; J. Chem. Soc., Chem. Commun. 1973, 232; Mislow, K. Acc. Chem. Res. 1976, 9, 26; Ōki, M.; Angew. Chem., Int. Ed. 1976, 15, 87. 21. Koumura, N.; Zijlstra, R. W. J.; van Delden, R. A.; Harada, N.; Feringa, B. L.; Nature 1999, 401, 152. 22. Feringa, B. L.; Koumara, N.; van Delden, R. A.; ter Wiel, M. K. J.; Appl. Phys. A 2002, 75, 301. 23. Vicario, J.; Meestma, A.; Feringa, B. L. Chem Commun., 2005, 5910; Feringa, B. L.; Acc. Chem. Res. 2001, 34, 504; van Delden, R. A.; ter Wiel, M. K. J.; Pollard, M. M.; Vicario, J.; Koumura, N.; Feringa, B, L.; Nature, 2005, 437, 1337; Vachon, J.; Carrol, G. T.; Pollard, M. M.; Mes, E. M.; Brouwer, A. M.; Feringa, B. L.; Photochem, Photochem. Photobiol. Sci. 2014, 13, 241. 24. Eelkema, R.; Pollard, M. M.; Vicario, J.; Katsonis, N.; Ramon, B. S.; Bastiaansen, C. W. M.; Broer, D. J.; Feringa, B. L.; Nature, 2006, 440, 163. 25. Kudernac, T.; Ruangsupapichat, N.; Parschau, M.; Macia, B.; Katsonis, N.; Harutyunyan, S. R.; Ernst, K. H.; Feringa, B. L., Nature, 2011, 479, 208. 26. Peplow, M. Nature, 2015, 525, 18. 27. Okumura, Y.; Ito, K.; Adv. Mater. 2001, 13, 485. 28. https://news.cnrs.fr/articles/jean-pierre-sauvage-a-nobel-for-molecular-machines; http://starsunfolded.com/jean-pierre-sauvage/; https://en.wikipedia.org/wiki/Jean-Pierre_Sauvage; https://global.britannica.com/biography/Jean-Pierre-Sauvage, acessadas em janeiro de 2017. 29. http://stoddart.northwestern.edu/Index.php?View=Fraser_Stoddart/General.php, acessada em janeiro de 2017. 30. http://www.benferinga.com/biography.php, acessada em janeiro de 2017.
#This paper is part of the PubliSBQ Special Issue in honor of the late Prof Angelo da Cunha Pinto. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access