
 |
 |
 |
 |
 |
Artigo
| Synthesis, thermal studies and conversion degree of dimethacrylate polymers using new non-toxic coinitiators |
|
Rafael Turra Alarcon1; Bruno Barreto da Cunha Holanda1; Daniel Rinaldo1; Flávio Junior Caires1; Marcos Vinícius de Almeida2; Gilbert Bannach*,1
1. Faculdade de Ciências, Universidade Estadual Paulista, Departamento de Química, 17033-260 Bauru, São Paulo - SP, Brasil Recebido em 01/07/2016 *e-mail: gilbert@fc.unesp.br The aim of this paper is to replace toxic coinitiators (tertiary amines) by non-toxic compounds such as glycerol and
inositol (polyalcohol) in dimethacrylate resins. For this purpose, mid infrared spectroscopy (MIR) was used to calculate
the monomers' degree of conversion (%DC); as well as simultaneous Thermogravimetric Analysis - Differential Thermal
Analysis (TGA-DTA) and Differential Scanning Calorimetry (DSC) were conducted to evaluate thermal stability, degradation
steps, and thermal events. The use of different initiator systems did not modify the thermal events or the thermal
stability of each of the dimethacrylate resins. Results show a substitution of system 2 (toxicity) by system 3 (low
toxicity), which had a good conversion velocity and total conversion in some monomers, is plausible. INTRODUCTION The dimethacrylates resins photopolymers are widely used in dentistry, with applications ranging from restoration material, sealing, and cementation to dental adhesives.1-4 Nowadays, photopolymerization is of high interest, due to its ease of synthesis, and its economic and ecological benefits.4 These dimethacrylates monomers are easily polymerized by radicalar systems and have broad applicability. In particular, the following monomers (shown in Figure 1) are the most commonly used in dentistry: Diurethane dimethacrylate (UDMA), Triethyleneglycol dimethacrylate (TEGDMA), Bisphenol A glycerolate (Bis-GMA), and Bisphenol A ethoxylate (Bis-EMA).
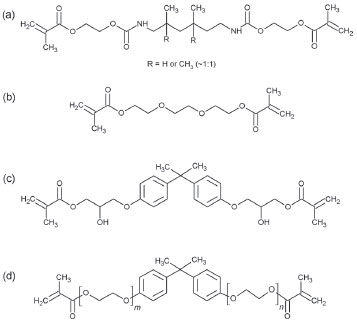 Figure 1. Structure of the Monomers: UDMA (a), TEGDMA (b), Bis‑GMA (c) and Bis-EMA (d)
The polymerization is triggered by initiators, which generate reactive forms that attack the alkene in the monomer structure. The two-component (Type II) method is the most widely used, applying camphorquinone as initiator (CQ, Figure 2-a) and coinitiators such as ethyl-p-dimethylaminobenzoate (Tertiary amine, Figure 2-b). However, it is known that secondary and tertiary amines are toxic and carcinogenic to humans, as they may leach into the human body after being used as bone polymers and dentistry polymers.5-12 To ensure patient safety and regarding green chemistry, these amines should be changed to polyalcohol.
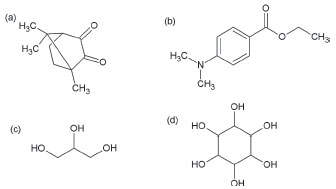 Figure 2. Structure of the Initiator and coinitiators on this study: camphorquinone (a), ethyl-p-dimethylaminobenzoate (b), glycerol (c), and inositol (d)
With this in mind, this study employed only non-toxic polyalcohols such as glycerol (Figure 2-c) and inositol (Figure 2-d) as coinitiators. Glycerol (1,2,3-propanetriol), a colorless, odorless, sweetish and very viscous liquid, is a trihidroxylated compound with three carbons, derived from both natural and petrochemical sources.13-15 Since 1959 it is acknowledged as a non-toxic compound, being considered a "GRAS" (Generally Recognized as Safe) by the American Food and Drug Administration (FDA). Hence, its use is allowed in canned foods.14 Furthermore, in Brazil it is used in food products, assured by resolution 45th, of November 3, 2010.15 Inositol (1,2,3,4,5,6-Hexahydroxycyclohexane) is a cyclic compound hexahydroxylated, present in the growth metabolism of plants and animals. It is also used to treat women with polycystic ovary syndrome and children with respiratory distress syndrome, attesting non-toxic activity.16-21 In the study of these dimethacrylate resins it is important to understand their thermal behavior, thus Thermoanalytical techniques such as Simultaneous Thermogravimetric Analysis - Differential Thermal Analysis (TGA-DTA) and Differential Scanning Calorimetry (DSC) are used in order to determine their thermal stability, degradation steps, glass transition, crystalline transition, crystallization, melt, and polymer cure.22-28 Another important point is to study the dimethacrylate monomers' conversion, using different initiators systems. The Mid Infrared Spectroscopy (MIR) is used to get this information, so that it is possible to measure which system is more advantageous.
EXPERIMENTAL Solution's preparation The solutions of photoinitiators were made by dissolving 0.01 mol of Camphorquinone (CQ) 97% (Aldrich) in 10 mL of acetone P.A (Aldrich) in four different amber little bottles. In the first one just Camphorquinone was added (System 1), in the second one CQ and 0.01 mol of Ethyl-p-dimethylaminobenzoate 99% (Aldrich) were added (System 2), in the third one CQ and 0.01 mol of glycerol P.A (Merck) were added (System 3), and in last one CQ and 0.01 mol of Inositol P.A (Aldrich) were added (System 4). Mixture of each monomer with different initiator system. The monomeric mixtures were made adding monomers (0.01 mol) in 16 different flasks. Thus there were 4 flasks with UDMA, 4 flasks with TEGDMA, 4 flasks with Bis-GMA and 4 flasks with Bis-EMA. In each flask 0.3 mL of each different photoinitiators solutions was added (system 1-4). Synthesis of polymers Blue light was used to induce polymerization. This was obtained using the D-2000 equipment (DMC Ltda, Sao Carlos) that uses a LED to emit light with wavelengths between 430nm and 490nm. The polymerization occurred at room temperature. Each monomeric mixture was placed in a PTFE mold with 1.5 mm of depth and 3 mm of diameter obtaining polymers with average mass of 10mg. Afterwards had thirty two different samples, two samples of each monomer with different photoinitiator solution, to simplify, polymers were named as PMonomer- system number, for example the UDMA polymer with the first solution as called PUDMA-1. Thereby all the samples names are: PUDMA-1, PUDMA-2, PUDMA-3, PUDMA-4, PTEG-1, PTEG-2, PTEG-3, PTEG-4, PGMA-1, PGMA-2, PGMA-3, PGMA-4, PEMA-1, PEMA-2, PEMA-3, and PEMA-4. Characterization Thermal Analysis (TGA-DTA and DSC) The TGA-DTA curves were obtained using a Netzsch equipment, model STA 449 F3, using 70 µL α-alumina open crucibles with samples of about 15 mg and a heating rate of 10.0 ºC min-1 in a dry air atmosphere at flow rate of 50.0 mL min-1 and a temperature range of 30.0-800.0 ºC. The DSC analyses were obtained on a Mettler-Toledo equipment, model DSC 1 Stare System, using 40 µL closed aluminum crucibles with perforated lids, samples of about 9 mg without previous thermal treatment, and at a heating rate of 10.0 ºC min-1 in dry air atmosphere and a 50.0 mL min-1 flow rate. The heating/cooling procedures were performed from -35.0 ºC to the thermal stability of each polymer, with isotherms at -35.0 ºC for 10 minutes. Mid infrared Spectroscopy (MIR) A Bruker equipment, model Vertex 70 spectrophotometer, operating in the range of 1680-1500 cm-1 with a diamond crystal, was used to collect the spectroscopic data of the polymers; one drop of monomers mixture was added to the crystal, and then blue light was irradiated for 350 s, with scans being made in 10-second intervals. Thereby, it was possible to calculate the degree of conversion using the equation 1. 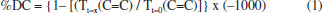 Unconverted double bonds were calculated by comparing the ratio of C=C transmittance near 1640 cm-1. The value of C=C transmittance at some point in time (t=x) is divided by the initial value of C=C monomer transmittance (t=0). With all these data it is possible to plot a graph of Degree Conversion vs. Time.29-41
RESULTS AND DISCUSSION Reaction mechanism The reaction was initiated with free radical photoinitiators. The CQ absorbed visible light in the spectrum of 400-550 nm, reaching an excited state and withdrawing a hydrogen atom from the tertiary amine, producing a primary radical. A resonance (structure) may have occurred in the CQ between the second carbonyl of the molecule and the radical produced in the initiation step, thus forming a new radical in oxygen. This new radical can extract another hydrogen atom from either the tertiary amine or a polyalcohol (initiation step, Figure 3). It is known that the radical in CQ does not start the reaction, due to the resonance (structure), which stabilizes the molecule.42
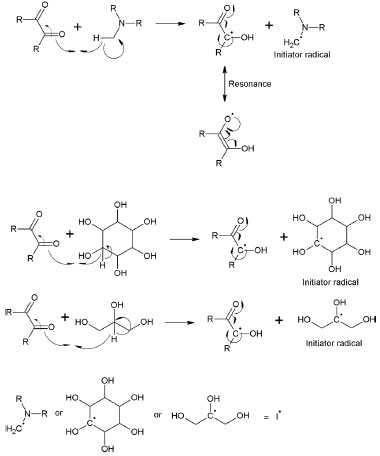 Figure 3. Reaction mechanism of photoinitiators
The primary radical triggers the homolytic cleavage in ϖ bonds (C=C) on dimethacrylate monomers, which produces new radicals (propagation step).43-45 Two different radicals might be formed, a tertiary one [more stable (A), Figure 4-a] or a primary one [less stable (B), Figure 4-a], both of which can initiate and propagate the polymerization (Figure 4-b).
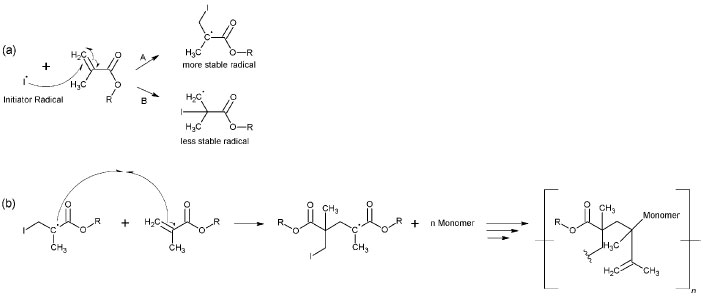 Figure 4. Reaction mechanism: production of new radicals (a); and propagation and termination in UDMA (b)
The polymerization's termination step may have occurred according to two reaction processes in the polymer. In the first process, a bond between the radical UDMA and the radical carbon present in the glycerol molecule may have formed, similarly to the termination step. The second process may have involved the formation of a bond between two radicals present in neighboring UDMA molecules (Figure 4-b). Simultaneous Thermogravimetric Analysis - Differential Thermal Analysis (TGA-DTA) and Differential Scanning Calorimetry (DSC) The TGA/DTG-DTA curves of the polymers with CQ/Glycerol initiator system (system 3) are shown in Figure 5. The TGA/DTG-DTA curves for the other polymers with different coinitiators can be viewed in supplementary material (Figures 1S, 2S and 3S). Due to the similarity of the samples, just the polymers with glycerol (system 3) are shown now. Each polymer, regardless of the monomer, has four steps of mass loss, the first one indicates the volatilization of the acetone and residual monomer and the other three steps indicate its thermal degradation. The coinitiator does not affect these steps and the thermal stability of each polymer. The average stability of PUDMA, PTEG, PGMA and PEMA occurred at 173.75 ºC; 126.70 ºC; 227.37 ºC and 172.85 ºC, respectively. Furthermore, some samples had a carbonaceous residual mass (average residual mass = 0,99%), even though the coinitiator also had no effect on this aspect point. Table 1 shows the details of the volatilization and decomposing steps of all the polymers.
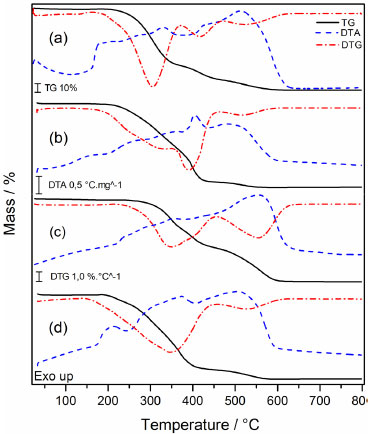 Figure 5. TGA/DTG-DTA curves of polymers with initiator system 3: PUDMA- 3 (a), PTEG-3 (b), PGMA-3(c), and PEMA-3(d)
Due to the similarity of DSC curves of each monomer using different initiator systems (systems 1-4); only curves for system 3 are shown in Figure 6. The DSC curves for other systems can be viewed in supplementary material (Figures 4S, 5S and 6S). The cyclic DSC curves depict the characteristic thermal events of each polymer, which is consistent with the literature.46 Furthermore; it is observed that the coinitiator does not affect the thermal events.
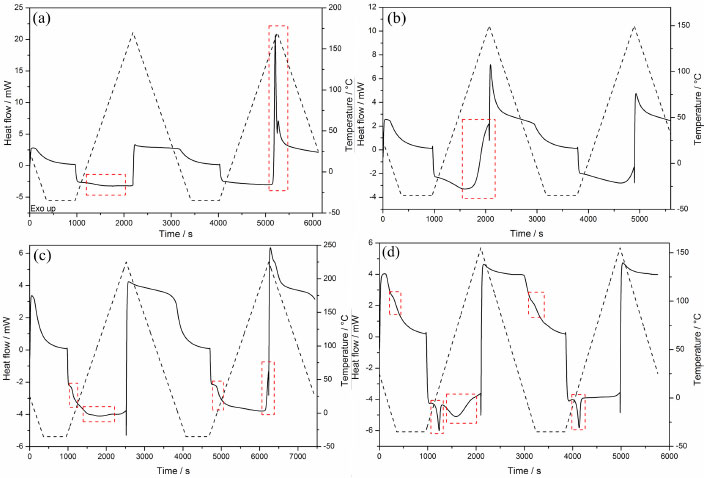 Figure 6. DSC curves of polymers with initiator system 3: PUDMA-3 (a), PTEG-3 (b), PGMA-3(c), and PEMA-3(d)
The DSC curves of UDMA polymers (Figures. 4S-a, 5S-a and 6S-a) did not show any thermal events at any cooling stage. However, on the first heating stage the endothermic event in the range of 20.0-160.0 ºC was observed, indicating the acetone volatilization (PUDMA-3 first heating stage expanded in Figure 7S). On the second heating stage an exothermic event that indicates the degradation of these polymers was observed, due to thermal stress. Hence they became yellowish and brittle. The DSC curves of TEGDMA polymers (Figures. 4S-b, 5S-b and 6S-b) show an exothermic event (130 °C) on the first heating stage, indicating the cure of the residual monomers. As this occurred at a temperature below the thermal stability temperature, and as no thermal events happened on the second heating stage of PTEG-3, this event is in agreement with literature. 46 However, on the second heating stage a glass transition with midpoint at 74.55 ºC was observed only in PTEG-1, PTEG-2 and PTEG-4 (Figures. 4S-b, 5S-b and 6S-b); the polymers had no change in color and did not become brittle. On the cooling stages no thermal events were observed. In the DSC curves of Bis-GMA polymers (Figures. 4S-c, 5S-c and 6S-c) a glass transition (Tg) was observed on the first heating stage, and the midpoint temperatures were calculated following the ASTM47: TgPGMA-1 = -4.06 ºC; TgPGMA-2 = -4.98 ºC; TgPGMA-3 = -7.68 ºC; and TgPGMA-4 = -7.54 ºC.46 Also on the first stage, an endothermic event was observed, due to the volatilization of acetone in the range of 10.0-165.0 ºC, (PGMA-3 first heating stage expanded in Figure 8S). On the second heating stage the same Tg, but slightly displaced to a higher midpoint temperature, was observed: TgPGMA-1 = 4.63 ºC; TgPGMA-2 = 5.27 ºC; TgPGMA-3 = 6.16 ºC; and TgPGMA-4 = 9.15 ºC. Furthermore, an exothermic event (200 ºC) was observed, indicating thermal degradation, as was verified by sample yellowing (PGMA-3 second heating stage expanded in Figure 9S). The Bis-EMA polymers (Figures 4S-d, 5S-d and 6S-d) presented a greater quantity of thermal events (melting, cure, glass transition and volatilization) than other samples in this study. On the first cooling stage, a Tg with an average midpoint at -14.50 ºC (PEMA-3 first cooling stage expanded in Figure 10S) was observed. In addition, on the first heating stage, all the polymers presented an endothermic average peak at 11.08 ºC, indicating the melting of residual monomers and an endothermic event in the range of 20.0-140.0 ºC that indicated the volatilization of the solvent and the partial cure of some of the monomer.47 The melting enthalpies (ΔfusH) on the first heating stage were: ΔfusHPEMA-1 = 3.13 J g-1, ΔfusHPEMA-2 = 5.99 J g-1, Δ fusHPEMA-3 = 8.30 J g-1, and ΔfusHPEMA-4 = 20.39 J g-1. On the second cooling stage, a Tg with an average midpoint at 22.00 ºC was observed (PEMA-3 second cooling stage expanded is shown in Figure 11S). Finally, on the second heating stage, the same melting endothermic average peak at 13.84 ºC (ΔfusHPEMA-1 = 4.74 J g-1, ΔfusHPEMA-2 = 7.75 J g-1, Δ fusHPEMA-3 = 9.78 J g-1, and ΔfusHPEMA-4= 19.23 J g-1), and a few exothermic peaks in the range of 66.0-88.0 ºC, which indicated the polymerization of residual monomers (Figures 4S-d, 5S-d and 6S-d) were observed. However, the latter event is not observed on PEMA-3. Conversion degree by MIR analysis Figure 7 shows the MIR spectra in the range of 1500-1680 cm-1 for each monomer (UDMA, TEGDMA, Bis-GMA and Bis-EMA) mixture with CQ/glycerol system and their corresponding polymers are shown in Figure 7. The MIR measurements depicted a band near 1640 cm-1, evincing the C=C double bond present in the dimethacrylates molecules. As expected, the intensity of this band decreased considerably for the polymers, since this process involves the cleavage of the π bond. Hence, it was possible to calculate the conversion degree of each monomer.
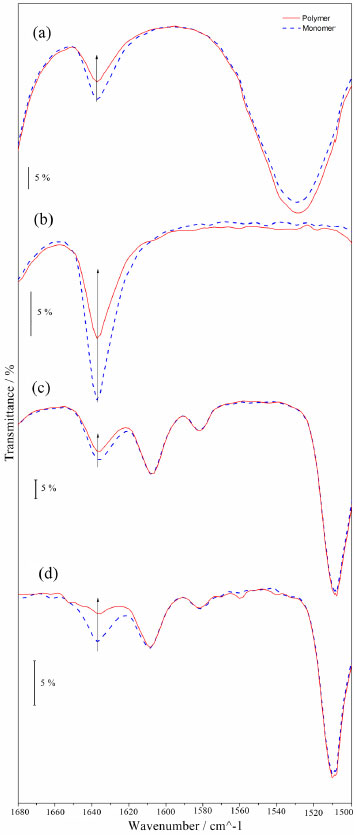 Figure 7. Infrared spectra of polymers used system 3: PUDMA-3 (a), PTEG-3 (b), PGMA-3 (c), and PEMA-3 (d)
To improve the analysis of the results, polymers containing only CQ system (Blank sample) were synthetized, in order to understand if CQ is less or more effective than combined with a coinitiator. Also the standard CQ/Tertiary amine system was used, thus allowing the comparison between the CQ/glycerol system and the CQ/Inositol system. The conversion degree curves of each monomer with a different initiator system are shown in Figure 8 and Table 2.
 Figure 8. Conversion degree curves of all the polymers with different initiator system: UDMA polymers (a), TEGDMA polymers (b), Bis-GMA polymers (c), and Bis-EMA polymers (d)
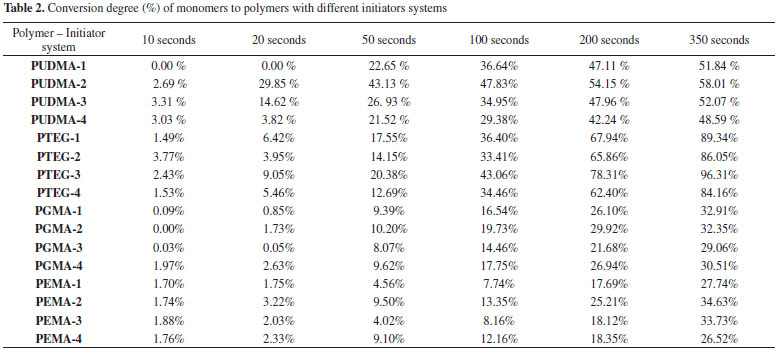
The conversion degree of UDMA polymers (Figure 8-a) shows that the CQ system has a slow conversion degree (0.00 % in the first 20 seconds) with a total conversion degree of 51.84%. As expected, the standard CQ/Amine system had the fastest conversion degree (29.85% in the first 20 seconds) and the highest conversion of all (58.01% in 350 s). The CQ/glycerol system presented itself as a good substitute to the standard system, with a conversion degree of 14.62 % in the first 20 seconds and a total conversion degree of 52.07%. The CQ/inositol system was not as effective as the other systems, as it had a slow conversion and the lowest total conversion, due to the slight solubility of the monomers. The TEGDMA conversions (Figure 8-b) showed no significant difference in the conversion velocity considering each initiator system, despite system 3 being slightly faster. However, the TEGDMA conversions evidence a difference in the total conversion, such as a higher conversion in system 3 (96.31%) as compared to that of the standard system (86.05%), and the poor performance of system 4 with regards same velocity to system 1 and system 2, but with a less conversion. In the Bis-GMA conversions (Figure 8-c) it became evident that there was no difference in the total conversion for each initiator system. However, systems 1 and system 3 had a slow conversion velocity. System 4 had the highest velocity of conversion of all systems in the first 10s (1.97 %), while system 1, 2 and 3 showed no conversion in first 10 s, but after 50s all systems had close relative velocity. However, for comparison purposes it is possible to arrange the systems in terms of a velocity sequence: system 3 < system 1 < system 2 < system 4. Notwithstanding all the systems had similar conversions 32.91% (system 1), 32.35% (system 2), 29.06% (system 3), and 30.51% (system 4). The Bis-EMA conversion (Figure 8-d) showed a similar total conversion in all of the samples. However, the highest total conversion was observed for the CQ/tertiary amine system followed by the CQ/glycerol system. Systems 2 and 4 showed a similar conversion velocity until 100s, after that the system 2 had high conversion velocity. Thus, inositol may be a good choice as a coinitiator for this monomer considering the velocity, however if consider the total conversion the system 3 is the best choice. It was expected that system 1 had a lower velocity and small total conversion, but is did not occur, the system 1 had a similar behavior, which found in other systems. This behavior could be explain considering the chemical structure of monomers, which had some organic functional groups such as urethane, alcohol and ether (see Figure 1). It is known that these organic functional groups may participate as a coinitiator [42]. Thus the radical is generated in the monomer structure and the polymerization is started.
CONCLUSION The TGA/DTG-DTA curves showed that PUDMA, PTEG, PGMA and PEMA (with different systems) had an average stability at 173.75 ºC; 126.70 ºC; 227.37 ºC and 172.85 ºC, respectively. However, the different initiator systems did not modify the thermal stability of each monomer nor their thermal degradation steps. The DSC analysis showed that the many thermal events of each polymer, such as dehydration, glass transition, melting, degradation and cure, are not affected by the choice of initiator. Thus it might be concluded that the initiator system does not modify or influence those thermal events. In summary, the initiator system does not affect the thermal behavior of the polymers. With the MIR analysis, it was possible to calculate the conversion degree of each monomer under study. The results presented the CQ/glycerol system (low toxicity), which had a good conversion velocity and good total conversion in some monomers, such as TEGDMA and UDMA, as a good substitute to the CQ/tertiary amine system (toxic). Considering these results the CQ/glycerol system is a viable choice for formulation of dental resins and materials. In the case of Bis-EMA monomer a good choice is the CQ/Inositol system, as it showed a good conversion velocity in first 10s and similar conversion when compared to other systems, but the best choice is the system 3 (CQ/glycerol) that had a similar total conversion found in the system 2 (CQ/tertary amine).
SUPPLEMENTARY MATERIAL The supplementary material has shown TGA-DTA and DSC of the others polymers that used the coinitiator system 1, 2 and 3; as well as the detailed DSC curve of polymers previous mentioned in results and discussion.
ACKNOWLEDGEMENTS The authors wish to thank CAPES (proc. 024/2012 Pro-equipment), POSMAT/UNESP, PIBIC (proc. 29451) and FAPESP (processes: 2012/21450-1 and 2013/09022-7) for financial support.
REFERENCES 1. Jakubiak, J.; Nie, J.; Linden, L. A.; Rabek, J. F.; J. Polym. Sci., Part A: Polym. Chem. 2000, 38, 876. 2. Lu, H.; Lovell, L. G.; Bowman, C. N.; Macromolecules 2001, 34, 8021. 3. Ye, Q.; Spencer, P.; Wang, Y.; Misra, A.; J. Biomater. Mater. Res. 2007, 80, 342. 4. Rodrigues, M. R.; Neumann, M. G.; Polim.: Cienc. Tecnol. 2003,13, 276. 5. Moszner, N.; Salz, U.; Prog. Polym. Sci. 2001, 26, 535. 6. Vazquez, B.; Levenfeld, B.; Roman, J. S.; Polym. Int. 1998, 46, 241. 7. Albrecht, W. N.; Stephenson, R. L.; Scand. J. Work, Environ. Health 1988, 14, 209. 8. Lijinsky, W.; Cancer Res. 1974, 34, 255. 9. Akintonwa, D. A. A.; Ecotoxicol. Environ. Saf. 1985, 9, 64. 10. Kadmi, F. Y.; Favier, L.; Wolbert, D.; Water Sci. Tecnol: Water Supply 2015, 15, 11. 11. Wu, Q.; Shi, W.; Ma, Y.; Adams, C.; Eichholz, T.; Timmons, T.; Jiang, H.; Talanta 2015, 131, 736. 12. Krasner, S. W.; Mitch, W. A.; Mccurry, D. L.; Hanigan, D.; Westerhoff, P.; Water Res. 2013, 47, 4433. 13. Morrison, L. R. In Encyclopedia of Chemical Technology, Wiley: New York, 1997. 14. http://portal.anvisa.gov.br/documents/33916/391619/Resolu%25C3%25A7%25C3%25A3o%2Bda%2BDiretoria%2BColegiada%2B%2BRDC%2Bn%2B%2B45%2Bde%2B03%2Bde%2Bnovembro%2Bde%2B2010.pdf/23734a57-21c1-4f71-9916-c6fa09edb8c5, accessed February 2017. 15. Jackobson, G.; Kathagen, F. W.; Klatt, M. In Ullmann's encyclopedia of industrial chemistry, VCH: Weinheim, 1989. 16. Govindarajan, C.; Pitchaipillai, R.; Shanmugasundaram, B.; Thangam, S.; Arokiasamy, J.; Pillai, M. S.; World J. Pharm. Pharm. Sci. 2015, 4, 137. 17. Turan, G. A.; Eskicioglu, F.; Sivrikroz, O.N.; Hakan, C.; Adakan, S.; Gur, E. B.; Tatar, S.; Sahin, N.; Yilmaz, O.; Arch. Gynecol. Obstet. 2015, 292, 1163. 18. Cianci, A.; Panella, M.; Fichera, M.; Falduzzi, C.; Bartolo, M.; Caruso, S.; Gynecol. Endocrinol. 2015, 31, 483. 19. Ward, R.M.; Sweeley, J.; Lugo, R. A.; Med. Chem. 2015, 5, 77. 20. Howlett, A.; Ohlsson, A.; Plakkal, N.; The Cochrane Collaboration, John Wiley & Sons, Ltd.: Hoboken, 2015. 21. Wang, M.; Wang, Z. P.; Gao, L. J.; Yang, H.; Zhao, Z. T.; Nutrients 2015, 7, 3067. 22. Hatekayama, T.; Quinn, F. X.; Thermal Analysis: Fundamentals and Applications to Polymer Science, John Wiley: New York, 1995. 23. Wellen, R. M. R.; Rabello, M. S.; Polim.: Cienc. Tecnol. 2007, 17, 113. 24. Scuracchio, C. H.; Waki, D. A.; Bretas, R. E. S.; Polim.: Cien. Tecnol. 2006, 16, 46. 25. Blom, H.; Yeh, R.; Wojnarowski, R.; Ling, M.; Thermochim. Acta. 2006, 442, 64. 26. Guo, R.; Sanders, D. F.; Smith, Z. P.; Freeman, B. D.; Paul, D. R.; Mcgrath, J. E.; J. Mater. Chem. A 2013, 1, 6063. 27. Santos, W. N.; Sousa, J. A.; Gregorio, J. R. R. Polim.: Cien. Tecnol. 2013, 32, 987. 28. Mollova, A.; Androsch, R.; Mileva, D.; Gahleitner, M.; Funari, S. S.; Eur. Polym. J. 2013, 49, 1057. 29. Pfeifer, C. S.; Silva, L. R.; Kawano, Y.; Braga, R. R.; Dent. Mater. 2009, 25, 1136. 30. Xiao, P.; Dumur, F.; Graff, B.; Zhang, J.; Morlet-Savary, F.; Gigmes, D.; Fouassier, J. P.; Lalevée, J.; Polym. Chem. 2015, 53, 567. 31. Albuquerque, P. P. A. C.; Moreira, A. D. L.; Moraes, R. R.; Cavalcante, L. M.; Schneider, L. F. J.; J. Dent. 2013, 41S, e67. 32. Shin, D. H.; Rawls, H. R.; Dent. Mater. 2009, 25, 1030. 33. Albuquerque, P. P. A. C.; Bertolo, M. V. L.; Cavalcante, L. M.; Pfeifer, C.; Schneider, L. F. J.; Journal of Esthetic and Restorative Dentistry 2015, 27, S49. 34. Xiao, P.; Dumur, F.; Zhang, J.; Graff, B.; Gigmes, D.; Fouassier, J. P.; Lalevée, J.; Polym. Chem. 2015, 53, 665. 35. Stansbury, J. W.; Dickens, S. H.; Dent. Mater. 2001, 17, 71. 36. Albuquerque, P. P. A. C.; Bertolo, M. V. L.; Cavalcante, L. M.; Pfeifer, C.; Schneider, L. F. J.; Dent. Mater. 2015, 31, e109. 37. Stansbury, J. W.; Dickens, S. H.; Polym. 2001, 42, 6363. 38. Floyd, C. J. E.; Dickens, S. H.; Polym. 2006, 22, 1143. 39. Ye, Q.; Spencer, P.; Wang, Y.; Misra, A.; J. Biomed. Mater. Res., Part A 2006, 80A, 342. 40. Podgórski, M.; Dent. Mater. 2010, 26, 188. 41. Zhang, J. Z. N.; Dumur, F.; Xiao, P.; Graff, B.; Gigmes, D.; Fouassier, J. P.; Lalevée, J. A.; J. Polym. Sci, Part A: Polym. Chem. 2015, 53, 445. 42. Foussier, J. P.; Lalevée, J.; Photoinitiators for Polymer Synthesis: Scope, Reactivity and Efficiency. Wiley-VCH Verlag Gmbh & Co. KGaA: Weinheim, 2012. 43. Fischer, J. P.; Dean, D.; Engel, P. S.; Mikos, A. G.; Ann. Rev. Mater. Res. 2001, 31 171. 44. Harikrishna, R.; Ponrathnam, S.; Rajan, C. R.; Tambe, S. S.; J. Therm. Anal. Calorim. 2013, 112, 805. 45. Moranch, J. M.; Cadenato, A.; Fernández-Francos, X.; Salla, J. M.; Ramis, X.; J. Therm. Anal. Calorim. 2008, 95, 513. 46. Bannach, G.; Cavalheiro, C. C. S.; Calixto, L.; Cavalheiro, E. T. G.; Brazilian Journal of Thermal Analysis 2015, 4, 28. 47. ASTM International; ASTM E1356, 2014. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access