
 |
 |
 |
 |
 |
Artigo
|
|
| Avaliação da liberação de elementos traço em solos tratados com xisto retortado Evaluation of trace elements releasing from soils treated with retorted oil shale |
|
Jeniffer V. dos Santos1; Rafael J. B. De Presbiteris1; Vanessa C. G. dos Santos1; Marco T. Grassi1; Iara Messerschmidt1; Betânia F. Pereira1; Rosane Martinazzo2; Gilberto Abate1,*
1. Departamento de Química, Universidade Federal do Paraná, Centro Politécnico, 81531-990, Curitiba - PR, Brasil Recebido em 10/08/2016 *e-mail: gilberto@quimica.ufpr.br Soils that had received successive applications of retorted oil shale (ROS) in an experiment under field conditions were used in this study, aiming to evaluate the availability of trace elements. In the batch tests, we observed that toxic elements present in the ROS are not desorbed from the matrix in significant amounts to contaminate soil and groundwater. The element Ca represents the greatest nutrient contribution, which has high mobility and ease release into the soil solution. Concerning the results obtained from the soil extracts analysis; we observed that the undesirable elements released into the extracts are not directly related to the presence of ROS in the soil samples. Comparing the behavior of soil samples with and without ROS it is possible to infer that many of the trace elements are derived from soil constituents, from previous fertilizer and pesticides applications. The study suggested that the ROS can be used as a soil conditioner for agricultural purposes without adding harmful elements to the environment. INTRODUÇAO O folhelho pirobetuminoso (xisto) é uma rocha sedimentar rica em querogênio, que é um composto orgânico de composiçao indefinida, gerado pela açao química e de microrganismos sobre a matéria orgânica ao longo de milhoes de anos.1 O Brasil possui a segunda maior reserva de xisto no mundo, sendo os Estados Unidos detentor das maiores reservas mundiais,1,2 além de outros países que merecem destaque como Austrália, Canadá, China, Estônia, Itália, Jordânia, Marrocos, Rússia e Zaire.3 Em Sao Mateus do Sul, no estado do Paraná, é explorada a extraçao de xisto da mais importante reserva de xisto do Brasil, a Formaçao Irati, a qual abrange também os estados de Mato Grosso do Sul, Goiás, Sao Paulo, Santa Catarina e Rio Grande do Sul.3 A Unidade de Negócios da Industrializaçao do Xisto realiza a mineraçao e o processamento do xisto extraído neste local. O processamento do xisto produz óleo e gás em condiçoes de alta temperatura e alta pressao, além de outros subprodutos, sendo um deles o xisto retortado (XR) produzido em grandes quantidades, representando cerca de 80 a 90% da quantidade de matéria prima inicial do processo.1 O XR é uma matriz dependente de sua origem e do processo de extraçao de óleo e gás, e pode conter compostos orgânicos e elementos traços indesejáveis ao meio ambiente. A composiçao química e as características do XR têm sido objeto de estudo de vários países, variando conforme a regiao de origem.1,4-10 Na matriz do XR estao presentes minerais tais como quartzo, feldspatos, argilas (principalmente ilita e clorita), carbonatos (calcita e dolomita), pirita e outros minerais.11-13 Outros elementos químicos também estao presentes no XR como Fe, Al, Mg, Ca, K e P, em quantidades majoritárias, além de elementos em menores proporçoes como Mn, Zn, Ba, Pb, Mo, Cu, Ni, Cr, Co e Cd, entre outros. As quantidades relativas de cada elemento dependem da localidade da mina, das quantidades de elementos no xisto cru, composiçao do querogênio e tecnologia empregada no beneficiamento.6,8,14,15 O XR tem sido reaproveitado na indústria de cerâmica e cimento,3 estudado como sorvente para íons Pb2+,1,16 e utilizado para produçao de energia.17 Aspectos como a perda de nutrientes por conta da lixiviaçao ocasionada pelas chuvas, culturas que absorvem determinados nutrientes em maiores quantidades, falta de rotaçao de culturas ou mesmo atividades antrópicas ocasionam a necessidade de adiçao de nutrientes adequados ao solo.18-20 Mais recentemente, estudos têm sido dedicados para a avaliaçao do emprego do XR como condicionador de solos em decorrência da sua elevada porosidade e capacidade de troca catiônica (CTC), composiçao mineral contendo macro e micronutrientes, bem como os possíveis efeitos no crescimento das plantas. Pereira e Vitti,21 aplicaram 0, 3, 6, 9 e 12 t de XR ha-1 ao solo e avaliaram os efeitos no plantio de tomates. Foi constatado aumento nos teores de S, Ca e Mg nas amostras de solos, ao passo que para o C e micronutrientes nao foram observadas mudanças significativas nas concentraçoes, embora um aumento expressivo tenha sido observado para o Mo. Também nao foram constatados aumentos significativos nas concentraçoes de P, K, Ca e Mg e de micronutrientes em funçao das diferentes doses de XR aplicadas nas amostras de folhas, ao passo que para o Si, um aumento significativo foi observado. Em outro estudo, foi constatado que a adiçao de quantidades crescentes de XR favoreceu a fixaçao de carbono proveniente de folhas e talos de soja em solos.22 Cabe ressaltar também que a presença de XR em solo reduziu a emissao de CO2 e nao influenciou negativamente na atividade microbiana e enzimática do solo, pelo menos até o limite de 3 ton de XR ha-1 de solo.23 Apesar da possibilidade de uso do XR na agricultura, é importante levar em consideraçao a presença de compostos orgânicos indesejáveis que podem estar presentes no XR. Nicolini et al.9 constataram a presença de oito diferentes hidrocarbonetos policíclicos aromáticos (HPA), dentre os dezesseis HPA estabelecidos como poluentes prioritários, nas amostras coletadas no interior das pilhas de XR, ao passo que para amostras de XR coletadas na superfície das pilhas, as concentraçoes observadas foram comparativamente inferiores. Dessa forma, os autores sugerem o uso de XR disposto em camadas rasas, a fim de favorecer a degradaçao dos HPA. Outros compostos tóxicos que podem ser liberados no solo sao os íons provenientes de metais tóxicos, entretanto, poucos estudos têm sido direcionados a esse assunto. Pereira e Vitti21 propuseram o uso de XR em solo e estudaram a liberaçao de íons de Ni, Cd, Cr, Pb, Co, Ti, Ba, V, Cu e Mo em solo e, de modo geral, nao foram observadas modificaçoes nas concentraçoes de diversos íons metálicos indesejáveis em funçao do aumento nas quantidades de XR, em comparaçao com solo testemunha, exceto em um dos experimentos para o titânio. Além disso, houve a diminuiçao do teor de bário em decorrência da presença de sulfato proveniente do próprio XR. De acordo com esses autores, a baixa concentraçao de metais tóxicos no XR propicia o seu uso como condicionador de solos, sem causar riscos. Adamson et al.24 avaliaram a liberaçao de íons dos metais Cd, Cr, Ni, Pb e Zn, pela aplicaçao de cinzas de XR em duas amostras de solos. De acordo com esse estudo, os íons de Ni, Cr e Zn foram aqueles de maior mobilidade; contudo, de acordo com os autores, as concentraçoes dos íons metálicos nos lixiviados foram bem abaixo dos padroes legislados, sendo o processo de lixiviaçao dependente do tipo de solo e das cinzas, além das condiçoes de lixiviaçao. De acordo com o que foi exposto, há a possibilidade de emprego do XR como insumo agrícola, porém, é preciso avaliar o potencial de contaminaçao do solo por elementos tóxicos. Dessa forma, o foco do presente trabalho foi investigar a possível contaminaçao por diversos elementos para o solo tratado com adiçoes crescentes de XR. Foi estudada a liberaçao de diversos íons nutrientes ou tóxicos, em batelada, sendo os resultados obtidos confrontados com os valores orientadores em águas subterrâneas da legislaçao do Conselho Nacional do Meio Ambiente (CONAMA) em sua Resoluçao n° 420.25
PARTE EXPERIMENTAL Area de estudo e amostragem O estudo foi realizado na área experimental da Universidade Federal de Santa Maria - UFSM, no Rio Grande do Sul. O solo em que foram conduzidos os experimentos foi caracterizado como argissolo vermelho distrófico arênico. Suas principais características, segundo o Sistema Brasileiro de Classificaçao de Solos,26 sao textura arenosa desde a superfície até um mínimo de 50 cm e um máximo de 100 cm de profundidade e horizonte B textural (conteúdo de argila maior em relaçao ao horizonte A) dentro de 200 cm do perfil do solo. Foi utilizado XR originário da unidade de beneficiamento de xisto localizada em Sao Mateus do Sul - PR, com tamanho de partícula menor que 0,30 mm.23 O solo estudado recebeu diferentes tratamentos em campo, com aplicaçao superficial de doses crescentes de XR, lançado em superfície, além da adubaçao de base (NPK - fontes de nitrogênio, fósforo e potássio). O experimento foi instalado em 2009, sendo que as coletas de solo foram realizadas em Novembro de 2011, Junho de 2012 e Janeiro de 2013, nas profundidades de 0 - 5 cm e de 5 - 10 cm. O procedimento de coleta foi feito com abertura de covas com uma pá, separando-se os solos de cada profundidade. A coleta para cada tratamento foi feita em quadruplicata e as fraçoes coletadas foram homogeneizadas para compor uma amostra representativa, totalizando cerca de 5,0 kg de solo. Em seguida as amostras foram homogeneizadas e peneiradas com peneira de 2 mm. As amostras foram secas ao ar, reservadas em sacos de polietileno, etiquetadas e transportadas para o Departamento de Química da Universidade Federal do Paraná. As amostras foram quarteadas para a realizaçao dos experimentos em batelada, a partir de 1,0 kg de solo até se obter a massa desejada para o experimento (2 g), separando-se as triplicatas para o experimento em batelada. Deste modo, todas as granulometrias que compoem a amostra foram repartidas igualmente, compondo uma amostra representativa. A Tabela 1 apresenta as doses anuais de XR aplicadas em cada um dos tratamentos e, com base nisso, a quantidade de XR acumulado no momento de cada uma das três coletas.
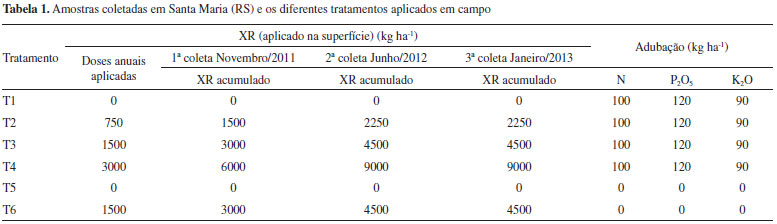
Cabe informar que os valores citados na Tabela 1 foram definidos com base em estudos anteriores do grupo de pesquisa vinculado ao Projeto Xisto Agrícola (Termo de Cooperaçao entre Embrapa Clima Temperado, PETROBRAS/SIX e FAPEG). Há uma recomendaçao de uma dose anual de 500 kg ha-1, sendo que no presente estudo foram aplicadas quantidades bem superiores a esse valor (conforme Tabela 1), para se ter o experimento em condiçoes extremas de aplicaçao de XR. Equipamentos, reagentes e soluçoes As amostras de solo e de XR foram previamente caracterizadas por difraçao de raio X (XRD), sendo utilizado um equipamento da marca Shimadzu, modelo XRD-6000, utilizando radiaçao Cu Kα (λ = 1,5418 Å) com parâmetros de 40 kV e 40 mA. Os ângulos de varredura foram entre 10 e 80° (2θ) com velocidade de 2° min-1. Também foram determinados alguns elementos por fluorescência de raio X (XRF) empregando um equipamento da marca Panalytical, modelo Axios Max. Para as determinaçoes dos elementos foi utilizado um Espectrômetro de Emissao Otica com Plasma Indutivamente Acoplado (ICP OES) da marca Thermo Scientific, modelo iCAP 6500. As medidas de pH foram efetuadas utilizando um pHmetro marca MS Tecnopon, modelo MPA 210 previamente calibrado com soluçoes tampao de pH 7,00 e 4,00, ao passo que as determinaçoes de condutividade foram feitas empregando um condutivímetro da marca MS Tecnopon, modelo MCA 150, calibrado com uma soluçao de KCl 0,01 mol L-1. A água empregada em todos os experimentos, preparo de soluçoes e enxágue de vidrarias foi obtida por sistema de osmose reversa (Marca Quimis, Q842-210) seguida da purificaçao em um equipamento da marca Millipore modelo Simplicity, dotado de lâmpada UV. Esse sistema proporcionou água com resistividade superior a 18 MΩ cm, sendo essa água designada no presente trabalho como água tipo 1. As soluçoes padrao dos elementos estudados foram preparadas a partir de sais solúveis na forma de nitratos, ou empregando soluçoes padrao multi-elementares para ICP OES. Os sais ou as soluçoes foram adquiridos de fornecedores como Merck, Sigma-Aldrich, Carlo Erba ou de qualidade similar, bem como o ácido nítrico, ácido acético e peróxido de hidrogênio. Toda a vidraria, tubos de centrífuga e frascos plásticos empregados no presente trabalho foram lavados com água e detergente Extran neutro, e após o enxágue foram mantidos por pelo menos 24 h em soluçao de ácido nítrico 5%, e novamente enxaguados com água tipo 1. Os experimentos de dessorçao foram conduzidos a 25 ºC, sendo utilizada uma incubadora refrigerada com agitaçao orbital, da marca Tecnal, modelo TE-421. Outros equipamentos e materiais auxiliares como centrífuga, refrigerador (4 ºC), estufa e micropipetas com volumes variáveis também foram utilizados. Procedimento de lixiviaçao e de digestao do XR Foram realizados experimentos de dessorçao por batelada em água tipo 1, com as amostras T1 a T6 (Tabela 1), com base na norma DIN 38914 (S4)27 de acordo com Delay et al.28 Em torno de 2,0 g (± 0,1 mg) das amostras (base seca) foram acondicionadas em tubos plásticos de centrífuga, adicionando-se 40,0 mL de água tipo 1, sendo os tubos mantidos sob agitaçao por 24 h a 25 ºC. Também foram conduzidos experimentos similares, porém, empregando ácido acético (pH 5,00 ± 0,05), segundo a norma brasileira da ABNT NBR 10005 (procedimento para obtençao de extrato lixiviado de resíduos sólidos),29 exceto que, nesse caso, foi utilizado tempo de agitaçao de 18 h. Após o período de agitaçao as amostras foram centrifugadas por 20 minutos, a 3000 rpm. Nas fases sobrenadantes obtidas pelos extratos em água tipo 1 foram determinados os valores de pH e condutividade. Todas as fases aquosas foram acondicionadas em frascos plásticos de polietileno em meio de ácido nítrico 1,0% (v/v) e mantidas a 4 °C até o momento de efetuar as determinaçoes. Todos os experimentos foram conduzidos em triplicata. Foi realizada a digestao da amostra de XR, sendo utilizada uma massa de 1,0 g em HNO3 concentrado e H2O2 30% com base na norma da Environmental Protection Agency - EPA 3050B.30 Os extratos obtidos foram diluídos até 100 mL com água tipo 1 e armazenados como os demais extratos para a determinaçao dos elementos de interesse. Esse procedimento de digestao foi conduzido apenas para a amostra de XR e nao para as amostras de solo, com o intuito de determinar o teor dos elementos no XR. Experimentos de dessorçao em meio aquoso também foram conduzidos para o XR, de modo análogo para as amostras de solo. Determinaçao dos elementos por ICP OES As curvas analíticas por ICP OES foram estabelecidas com sete diferentes concentraçoes para cada elemento de interesse em faixas de concentraçao de acordo com a necessidade, respeitando o LQ de cada um dos elementos. A faixa de concentraçao para as curvas analíticas da maioria dos elementos foi entre 1,0 µg L-1 e 1000 µg L-1. Um estudo prévio de recuperaçao foi efetuado, sendo adicionado em amostras de extrato de solo 100 µg L-1 de cada um dos elementos estudados. As concentraçoes dos elementos nos extratos obtidos em meio de água tipo 1, ácido acético e proveniente da digestao do XR foram determinadas com base nas referidas curvas analíticas por ICP OES. Para a determinaçao de Hg por ICP OES foi utilizado o método de geraçao de vapor a frio proposto por dos Santos et al.31 e para quantificar o As foi utilizado o método de geraçao de hidretos.32
RESULTADOS E DISCUSSAO Caracterizaçao das amostras de solos e de XR As amostras de solos da primeira coleta e na profundidade de 0 - 5 cm, e as amostras de XR foram inicialmente caracterizadas por XRD, sendo constatada uma constituiçao majoritária de quartzo para todas as amostras de solo. Também foram observados alguns picos referentes à hematita (Fe2O3), berlinita (AlO4P), brucita (Mg(OH)2) e rutilo (TiO2), cujas intensidades dos picos sao reduzidas pela presença do quartzo (SiO2).33,34 O difratograma do XR apresentou picos referentes à albita (NaAlSi3O8), ortoclásio (KAlSi3O8) e carbonatos de cálcio e magnésio, além de picos característicos de quartzo. Alguns elementos foram determinados por XRF, sendo observados valores similares entre as amostras de solo (T1 - T6) e assim foram calculados os valores médios. Nesse caso foram utilizadas somente as amostras de solo da 1ª coleta (Tabela 1). Esses resultados bem como os resultados obtidos para o XR, todos expressos na forma de óxidos, sao apresentados na Tabela 2. Além disso, sao mostrados dois resultados da literatura para o XR, também da regiao de Irati, para fins de comparaçao.
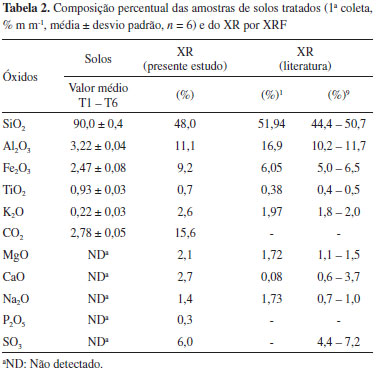
Com base nas determinaçoes por XRF, foi observado um elevado teor de SiO2 para as amostras de solo de 90,0% e de 48% para o XR, estando esse valor para o XR em boa concordância com a literatura para amostras procedentes da mesma regiao e de diferentes períodos.1,9 De modo geral, pode-se constatar uma boa similaridade entre os resultados deste trabalho em comparaçao com aqueles da literatura para a maioria dos elementos na forma de óxidos. Os resultados obtidos por XRF mostraram teores mais elevados de alguns elementos em relaçao ao solo estudado, tais como Al, Fe, K, C, Mg, Ca, Na, P e S, sugerindo a possibilidade de liberaçao de tais elementos no solo. Foram determinados diversos parâmetros físico-químicos para as amostras de solo de acordo com os resultados mostrados na Tabela 3, sendo nesse caso avaliadas as amostras da primeira coleta de solos.
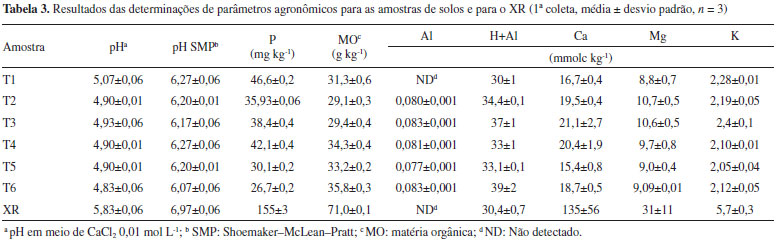
De modo geral, nao foram constatadas diferenças significativas entre os tratamentos feitos em campo (T1 - T6). Isso é indicativo que o XR nao causou qualquer modificaçao nas amostras de solo. Apesar disso, cabe salientar que o XR apresentou maior valor de pH em relaçao às amostras de solo, o que também foi observado por Pereira e Vitti,21 além de maiores conteúdos de nutrientes e matéria orgânica (Tabela 3). É possível que o processo de dessorçao de elementos provenientes do XR nao seja significativo a ponto de ser mensurável, nao sendo suficiente a quantidade de XR incorporada ao solo para a liberaçao dos elementos estudados. Todavia, esses resultados superiores em relaçao às amostras de solo, aliado aos maiores teores observados para alguns elementos por XRF, sugerem que o XR possui potencial para o fornecimento de nutrientes ao solo. Para o Ca foram observados teores significativamente maiores para as amostras que receberam XR (T2,T3,T4,T6). Determinaçao de pH e condutividade elétrica nos extratos Antes da determinaçao da concentraçao de íons metálicos foram efetuadas medidas de pH e condutividade elétrica (CE) dos extratos obtidos em água tipo 1. Para a determinaçao desses dois parâmetros as três coletas de solos foram avaliadas. Nos extratos obtidos a partir dos experimentos em água tipo 1 foram verificados valores muito similares de pH para as três coletas efetuadas, sendo o valor médio para de pH = 6,2 ± 0,3 (0 - 5 cm) e de 5,9 ± 0,1 (5 - 10 cm) ao passo que para o extrato de XR o pH verificado foi de 6,9, embora de acordo com a literatura, o XR apresenta características alcalinas.21,35 Apesar do valor de pH mais elevado para o extrato de XR, nao foram constatadas tendências de elevaçao nos valores de pH em funçao das adiçoes de XR nos solos, para as três coletas, o que está concordante com a literatura.21 A medida de CE de extratos de solo é importante, pois é um indicativo do aumento ou diminuiçao do teor de sais dissolvidos nos solos.26,36 De modo geral, os valores de CE se situaram entre 15 e 40 µS cm-1, para as duas profundidades nas coletas 1 e 3. Na coleta 2, um comportamento bem similar foi observado, embora dois valores mais elevados de 75 µS cm-1 e 58 µS cm-1 foram constatados para T3 e T6, respectivamente, para a profundidade de 5 - 10 cm. Apesar disso, é provável que isso nao esteja relacionado com a presença de XR, visto que para T4, que teve a maior adiçao de XR, nao foram verificados valores mais elevados em nenhuma das três coletas para as duas profundidades. Dessa forma, mesmo com teores mais elevados de alguns elementos no XR, pode-se inferir que esses elementos nao foram liberados, pelo menos em 24 h, ou as quantidades liberadas nao foram suficientes para causar uma alteraçao perceptível nos valores de CE. Determinaçao de Al, Ca, Fe, Mg, As, Ba, Cd, Co, Cr, Cu, Hg, Mn, Mo, Ni, Pb e Zn Inicialmente foi avaliada a linearidade do método empregado, que é determinada pelo coeficiente de determinaçao (r2), o qual deve ser superior a 0,90, segundo a literatura.37 Foi observada boa linearidade entre as curvas avaliadas, com r2 variando de 0,9957 até 0,9999. Testes de hipóteses de t e F foram realizados para avaliar a significância da regressao e do desvio da linearidade. Valores bastante elevados de F confirmaram uma regressao significativa na faixa de concentraçao estudada.38 Os perfis dos gráficos de resíduos de regressao demonstraram que nao houve tendências de heteroscedasticidade ou de desvio de linearidade. Os valores de limite de detecçao (LD) e limite de quantificaçao (LQ) foram calculados pelas expressoes: LD = 3s/S e LQ = 10s/S, sendo s a estimativa do desvio padrao de dez provas em branco e S é a inclinaçao da curva analítica. Os valores de LD se situaram entre 0,003 µg L-1 e 4,3 µg L-1 para Mn e Al, respectivamente, ao passo que para o LQ, valores entre 0,01 µg L-1 e 14,2 µg L-1 foram verificados para esses mesmos elementos. No caso do Pb, o limite máximo é de 10 µg L-1 para águas subterrâneas,25 sendo esse o único elemento dentre os que foram estudados no presente trabalho que o LQ verificado (12,3 µg L-1) foi superior ao valor legislado. Foram também realizados testes de recuperaçao com adiçao de padrao dos elementos na concentraçao de 100 µg L-1 às amostras de extratos de solo. Esses experimentos foram conduzidos visando verificar se poderia haver interferência na etapa de quantificaçao, em decorrência dos extratos obtidos pelas matrizes de solo. Foram constatados valores de recuperaçao entre 82,7% (Pb) e 118,1% (Cu) e, além disso, a maioria dos valores de recuperaçao se situaram muito próximo de 100%, sugerindo nao haver interferência significativa, o que permitiu a continuidade dos estudos posteriores. Teores dos elementos no XR e avaliaçao dos extratos de XR A Tabela 4 apresenta os teores dos elementos contidos no XR, e as concentraçoes nos extratos obtidos a partir do XR em meio aquoso, além da comparaçao com os valores máximos permitidos pelo CONAMA,25 considerando águas subterrâneas e o fator de mobilidade (FM). Cabe salientar que essa avaliaçao foi conduzida apenas para o XR, na ausência das amostras de solo, visando verificar se a presença dos elementos de interesse nos extratos de solo poderia estar relacionada com o XR.
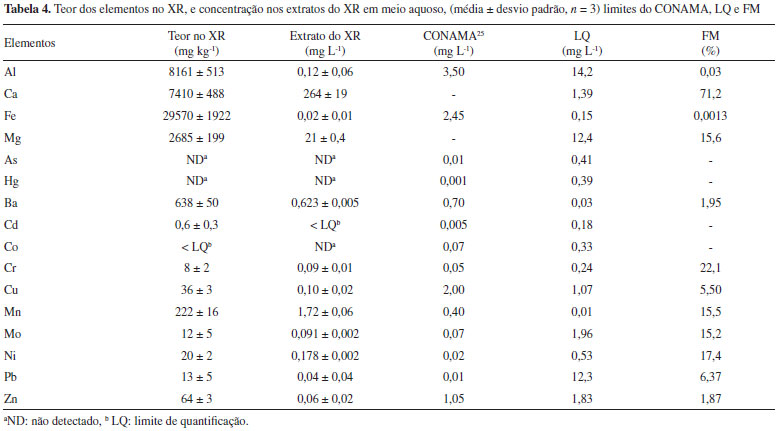
Como pode ser observado na Tabela 4, tanto no XR quanto no extrato da batelada do XR nao foram detectados os elementos Hg e As, que sao altamente tóxicos, conforme observado por Maranhao et al.39 Apesar de ter sido determinado no XR, a concentraçao de Cd no extrato da batelada ficou abaixo do LQ, indicando a baixa mobilidade desse elemento a partir da matriz do XR. Nao foi constatada a presença de Co no XR, e em consequência nao foi detectado no extrato em meio aquoso. O XR apresentou altos teores de Al, Fe, Ca e Mg, o que está em concordância com os resultados obtidos por XRF (Tabela 2) e menores teores dos demais elementos, mas ainda com altas concentraçoes de Ba e Mn, dados que estao em concordância com o que foi observado por Pereira e Vitti.21 No extrato aquoso do XR foram observados maiores teores de Ca e Mg em relaçao aos demais elementos determinados, sugerindo a possibilidade de fornecimento desses elementos como nutrientes ao solo. A aplicaçao de interesse do XR é voltada para o fornecimento de nutrientes ao solo, o que, contudo, poderia ocasionar a contaminaçao de águas subterrâneas e, por tal motivo, os resultados obtidos dos extratos de XR foram comparados com a Resoluçao do CONAMA.25 De acordo com os resultados (Tabela 4), o elemento Ba apresentou uma concentraçao de 0,623 mg L-1, que é muito próxima ao limite máximo permitido em águas subterrâneas de 0,70 mg L-1. Os elementos Cr, Mo, Ni e Pb apresentaram resultados um pouco superiores aos respectivos limites, e o Mn apresentou uma elevada concentraçao no extrato, em comparaçao com a referida Resoluçao.25 Para todos os demais elementos, os resultados foram inferiores aos limites da legislaçao. Foi calculada a razao entre a concentraçao dos elementos que foi lixiviada, com a concentraçao assumida como total na constituiçao do XR, em decorrência da digestao ácida em meio de H2O2 (método EPA 3050B),30 sendo adotado expressar como FM em %. Dessa forma, com base nos resultados obtidos, foi calculado o FM de acordo com o volume utilizado para a dessorçao de 40 mL e a massa de 2,0 g de XR, conforme a equaçao:  Esse cálculo foi efetuado, visando verificar a potencialidade de liberaçao dos elementos estudados no solo e, consequentemente, de atingir águas subterrâneas. Os valores obtidos em forma de fator de mobilidade calculados indicam maior disponibilidade de Ca dentre todos os elementos. Al e Fe praticamente nao sao mobilizados, provavelmente porque fazem parte da matriz estrutural e nao sao solúveis em pH próximo da neutralidade.40 Assim, também, Ba e Zn estao pouco disponíveis na soluçao. Os elementos Mg, Mn, Mo e Ni demonstraram mobilidades semelhante (15 a 17%), próxima do Cr (22,1%), assim como o Cu em relaçao ao Pb. O Ca demonstrou uma mobilidade de 71,2% e, sendo um macronutriente importante para o solo, tem potencial para estar biodisponível aos vegetais através da adiçao de XR ao solo. Com base nos valores de FM é possível inferir que os elementos mais preocupantes quanto à toxicidade para o meio foram Cr, Mn, Ni e Pb, e assim todos os elementos foram avaliados quanto à dessorçao em extratos de solo. Extratos dos solos Os resultados para os elementos determinados nos extratos de solos obtidos em água tipo 1 e em ácido acético sao apresentados nas Figuras 1 a 3 para os elementos Fe, Al, Ca, Mg, Mn e Ba. Cabe informar que alguns gráficos foram omitidos, pois os valores de concentraçao obtidos foram inferiores ao LQ (Tabela 4), ou as concentraçoes foram muito inferiores aos limites permitidos pelo CONAMA,25 como no caso para os elementos Zn e Cu. Os experimentos utilizando ácido acético foram conduzidos a fim de verificar a possível extraçao de nutrientes disponíveis para as plantas, deslocando íons metálicos ligados à matéria orgânica e às superfícies de minerais de silicatos e de óxidos de Mn, Fe e Al que requerem prótons para efetivar processos de troca iônica.41 Visando avaliar se havia diferenças significativas entre os valores médios das determinaçoes, para os diferentes tratamentos com XR (Figuras 1 - 3), foi empregado o critério da diferença menos significativa (DMS) para o número de réplicas N = 3 e um nível de confiança de 95%, de acordo com a literatura.41 Cabe ainda informar que para todos os extratos de solo obtidos em água pura ou em ácido acético, para ambas as profundidades investigadas, nao foram observadas correlaçoes de aumento, ou mesmo de diminuiçao da concentraçao dos elementos em funçao dos diferentes tratamentos efetuados (T1-T6) ou em termos do tempo decorrido, ou seja, para as três coletas (C1-C3).
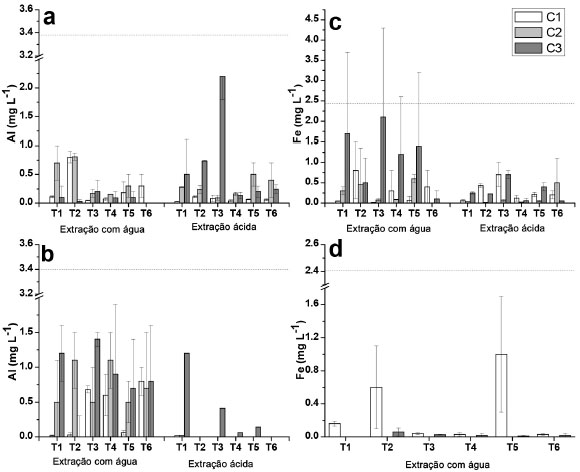 Figura 1. Concentraçoes de Al e Fe determinadas nos extratos aquosos e ácidos provenientes de amostras dos solos das profundidades de 0 - 5 cm e de 5 - 10 cm, para as coletas C1, C2 e C3. (a) Al (0 - 5 cm); (b) Al (5 -10 cm); (c) Fe (0 - 5 cm); (d) Fe (5 - 10 cm). A linha pontilhada indica o limite estabelecido pelo CONAMA25 de acordo com a Tabela 4
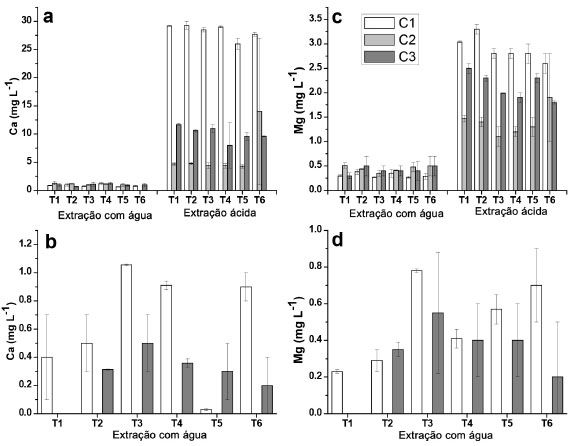 Figura 2. Concentraçoes de Ca e Mg determinadas nos extratos aquosos e ácidos provenientes de amostras dos solos das profundidades de 0 - 5 cm e de 5 - 10 cm, para as coletas C1, C2 e C3. (a) Ca (0 - 5 cm); (b) Ca (5 -10 cm); (c) Mg (0 - 5 cm); (d) Mg (5 - 10 cm)
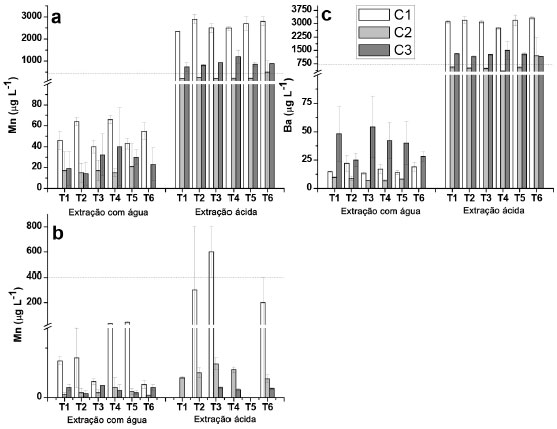 Figura 3. Concentraçoes de Mn e Ba determinadas nos extratos aquosos e ácidos provenientes de amostras dos solos das profundidades de 0 - 5 cm e de 5 - 10 cm, para as coletas C1, C2 e C3. (a) Mn (0 - 5 cm); (b) Mn (5 -10 cm); (c) Ba (0 - 5 cm). A linha pontilhada indica o limite estabelecido pelo CONAMA25 de acordo com a Tabela 4
Alumínio e ferro De acordo com a Figura 1, nao foi observada uma tendência de diminuiçao ou aumento da concentraçao de Al e Fe liberados para a maior profundidade estudada na extraçao com água, embora para o Fe, na coleta 2 e profundidade de 5 - 10 cm, as concentraçoes verificadas foram abaixo do LQ. Concentraçoes relativamente elevadas foram observadas para esses elementos nos extratos (0,9 mg L-1 para Al e 2,1 mg L-1 para Fe), o que seria esperado visto que ambos sao integrantes da matriz do solo, principalmente na forma de óxidos e hidróxidos,20,40 embora elevadas estimativas de desvio padrao foram constatadas, o que dificultou avaliaçao. Cabe informar que houve um longo período de seca que antecedeu a coleta 2, o que poderia explicar os resultados relativamente mais elevados em alguns casos em comparaçao às coletas 1 e 3. Os resultados observados para o Al, e especialmente para o Fe nos extratos obtidos em ácido acético foram relativamente inferiores em comparaçao com aqueles extratos obtidos em água tipo 1, sendo que na grande maioria das determinaçoes na profundidade de 5 - 10 cm os resultados foram inferiores ao LQ, e por isso nao foram apresentados. Apesar de ser esperada uma maior lixiviaçao dos íons na presença de ácido acético, esse aspecto poderia estar relacionado com a interaçao entre o acetato com os íons, e a consequente interaçao com a matéria orgânica do solo. No processo de centrifugaçao, esses íons ficariam retidos na fase sólida, com a diminuiçao das concentraçoes na fase lixiviada. Para o Al e Fe, considerando os extratos obtidos em água tipo 1 e ácido acético, as concentraçoes foram inferiores àquelas estipuladas pelo CONAMA de 3,50 mg L-1 e de 2,45 mg L-1 para Al e Fe, respectivamente.25 Na maioria dos extratos nao foi possível verificar uma tendência de aumento nas concentraçoes de Al e Fe com o aumento das doses de XR aplicado (T2 - T4) para as diferentes coletas, o que em um primeiro momento seria esperado pelas elevadas concentraçoes desses elementos em comparaçao com os solos (Tabela 2). Todavia, o fator de mobilidade para Al e Fe foi de 0,03 e 0,0013%, respectivamente, o que pode ser considerado desprezível. Por outro lado, foi observado um teor significativamente maior para amostra T5 (testemunha absoluta, sem adiçao de adubaçao de base e XR) da segunda coleta comparando com a amostra T4 que recebeu maior adiçao de XR. Somente para a profundidade 5 - 10 cm para primeira coleta foi verificado um teor de Al significativamente maior para as amostras que receberam XR em relaçao à T5. Cálcio e magnésio Foi constatada uma tendência geral de maiores concentraçoes de Ca e Mg (Figura 2) na camada superficial em todas as coletas, para as extraçoes em água, já que sao íons trocáveis na matriz do solo,20 apresentando comportamento equivalente entre tratamentos e entre coletas, sem grandes variaçoes das concentraçoes. Em meio ácido Ca e Mg foram detectados apenas na camada de 0 - 5 cm nas três coletas. Para a profundidade 0 - 5 cm, os resultados obtidos nos extratos em ácido acético foram muito superiores em comparaçao com água tipo 1, em especial para o Ca. O íon Al3+ poderia substituir o Ca2+ e Mg2+ nas intercamadas de minerais, liberando esses íons para o meio,42 e talvez por esse motivo o Al tenha sido determinado em menores concentraçoes em meio de ácido acético. Apesar disso, nao foi constatada correlaçao entre as concentraçoes de Ca e Mg em funçao das concentraçoes de Al. Por outro lado, para a camada de 5 - 10 cm, esses elementos apresentaram concentraçoes inferiores aos respectivos valores de LQ, o que sugere que a presença desses elementos em elevadas concentraçoes na camada superior poderia ser decorrente da presença do XR. A dessorçao de Mg em relaçao ao Ca foi inferior em todas as coletas, provavelmente porque o Ca seja mais facilmente trocável do que o Mg.43 O XR apresentou concentraçoes relativamente elevadas determinadas por XRF (Tabela 2) e nos extratos obtidos por digestao em meio ácido (Tabela 4), bem como fatores de mobilidade de 71,2% (Ca) e 15,6% (Mg), sugerindo um bom potencial para liberaçao desses elementos como nutrientes ao solo. Entretanto, com base nos resultados obtidos, nao seria possível afirmar que a adiçao de XR ao solo poderia propiciar um ganho nos teores de Ca e Mg nos extratos, visto que os resultados nao apresentaram uma tendência de aumento que possa ser considerada significativa, em decorrência dos tratamentos efetuados. Para o Ca foi observada diferença significativa entre os tratamentos somente na primeira coleta na profundidade 0 - 5 cm, em que o resultado para T4 foi significativamente maior que T1 e T5 (amostras testemunhas), porém, esta diferença nao foi significativa para as demais coletas. Na profundidade 5 - 10 cm, T3 e T4 apresentaram um teor de Ca significativamente maior que T1 e T5. Para o Mg os resultados para T3 e T6 foram significativamente maiores que para as outras amostras só na primeira coleta, porém, o resultado para T4 nao foi significativamente maior que para T1 e T5. No caso do Ca com elevado FM de 71,2%, é possível que o mesmo permaneça fortemente retido nas camadas de argilominerais, possivelmente precipitado como carbonato de cálcio.40 Apesar disso, cabe salientar que para o Ca e Mg os valores observados em meio ácido para as coletas 1 e 3 que receberam a adiçao de XR mostraram diferenças significativas em relaçao à coleta 2, sugerindo de fato a liberaçao desses elementos ao solo. Isso foi constatado em meio ácido para a camada superficial, e poderia ser decorrente da solubilizaçao de CaCO3 e MgCO3 em decorrência dos menores valores de pH em ácido acético em torno de 5,0, em comparaçao com os extratos obtidos em água tipo 1, cujos valores de pH se situaram entre 6,0 e 6,7. Manganês e bário O Mn é essencial a todos os organismos vegetais e animais, além de influenciar na solubilidade e disponibilidade de outros elementos traços no solo por estar comumente presente na forma de óxido na matriz mineral.44 Para os extratos em água tipo 1 (Figura 3), os resultados obtidos foram superiores na camada superficial, chegando a 66 µg L-1 (T4, coleta 1, significativamente maior que T1 e T5), embora esse valor esteja abaixo do limite máximo permitido em águas subterrâneas de 400 µg L-1.25 Por outro lado, na camada de 10-5 cm, T5 apresentou um teor de Mn significativamente maior que todas as outras amostra para a primeira coleta. Foram observadas concentraçoes elevadas do Mn nos extratos ácidos, sendo a própria estrutura do solo a principal fonte de Mn,40 sugerindo uma parcial solubilizaçao do Mn devido ao ácido acético. Este elemento foi determinado em maiores concentraçoes nos extratos ácidos da camada superficial do solo (0 - 5 cm). Em contrapartida, para a camada de 5 - 10 cm os valores foram relativamente baixos, ou menores que o LQ, sugerindo que as concentraçoes mais elevadas na camada superficial poderiam ser advindas do XR, sendo que nessa camada as concentraçoes de Mn excederam o limite para as coletas 1 e 3, e para T6 na coleta 2, sendo significativamente maior que os outros tratamentos, e T4 na coleta 3 foi significativamente maior que as testemunhas (T1 e T5). A solubilidade do Mn tem alta dependência com o pH. Em meio mais ácido está mais disponível, por isso foram observadas concentraçoes maiores deste elemento nos extratos em soluçao ácida, de todas as amostras da camada superficial, na qual se apresenta sorvido à superfície de minerais e em complexos com a matéria orgânica.44 Apenas uma pequena parte do Mn está biodisponível no solo, sorvido a minerais ou formando quelatos com a matéria orgânica.44 Esse é o provável motivo de sua maior concentraçao nos extratos das camadas superficiais dos solos estudados. Para a primeira coleta, T2 e T4 apresentaram um teor de Mn significativamente maior que as testemunhas em extratos de água na camada 0 - 5 cm. Para a profundidade 5 - 10 cm, T5 demonstrou um teor de Mn significativamente maior em comparaçao com as outras amostras para a primeira coleta em relaçao aos extratos em água. O Ba é um dos elementos mais abundantes no solo superficial,45 e foi determinado nos extratos das amostras superficiais (0-5 cm) em todas as coletas, com máxima concentraçao de 54 µg L-1 (coleta 3, T3), em extratos de água tipo 1 (Figura 3). Nos extratos obtidos em ácido acético também foram observadas concentraçoes de Ba na camada superficial, porém, em valores muito superiores. Essas elevadas concentraçoes dos extratos em ácido acético podem ser devido à dissoluçao do BaCO3,40,44 assim como comentado anteriormente para o Ca e Mg, devido aos valores de pH inferiores para os extratos obtidos em ácido acético (em torno de 5,0) frente à faixa de pH entre 6,0 e 6,7 observada para os extratos obtidos em água tipo 1. Isso ocasionou que em todos os extratos os limites permitidos pela legislaçao (700 µg L-1)25 tenham sido excedidos. Os resultados obtidos para os extratos em água tipo 1 e em ácido acético demonstraram valores mais elevados para as coletas 1 e 3, sugerindo, assim como para o Mn, que o XR contribuiu para aumentar a concentraçao de Ba na camada superficial, especialmente pela elevada concentraçao de Ba verificada na digestao do XR, (638 mg kg-1) apesar de um valor de FM de 1,95% (Tabela 4). Por outro lado, para todos os extratos as concentraçoes de Ba foram inferiores aos valores de LQ (5 - 10 cm), indicando mais uma vez que o Ba é proveniente do XR, visto que as concentraçoes de Ba para essa camada deveriam ser também elevadas caso o Ba fosse originário do solo, embora nao tenha sido constatado um aumento significativo da concentraçao de Ba em funçao das quantidades crescentes de XR aplicado, ou superiores em relaçao à amostra testemunha (T5). Esse elemento tem baixa mobilidade no solo, podendo se fixar a sítios de troca em argilas e facilmente precipitado na forma de carbonato ou sulfato. Os sítios de troca catiônica apresentam seletividade para a incorporaçao de Ba2+ frente aos íons Ca2+ e Mg2+,45,46 provavelmente por isso nao foi detectado nos extratos de amostras na maior profundidade. Apesar de o XR em meio de ácido acético causar um aumento de Ba na camada superficial, o fato de nao ter sido detectado Ba na camada inferior sugere nao haver migraçao para as camadas inferiores, e em consequência esse elemento nao causaria a contaminaçao de águas subterrâneas. Zinco e cobre A presença de Zn no solo está relacionada à presença de Cd e à razao Cd/Zn, já que suas propriedades sao semelhantes no solo e podem precipitar juntos. O Zn tende a inibir a biodisponibilidade de Cd, evitando que este seja absorvido pelos vegetais, sendo os fertilizantes fosfatados considerados uma das maiores fontes de Cd e Zn no solo.44 As maiores concentraçoes de Zn foram obtidas nos extratos das amostras superficiais (0 - 5 cm), em especial para os extratos obtidos em meio de ácido acético. Para todos os extratos obtidos em água tipo 1 (5 - 10 cm), as concentraçoes de Zn foram inferiores ao LQ (1,83 µg L-1) assim como para a maioria dos extratos obtidos em meio de ácido acético. Nas coletas 1 e 2 a presença de Zn predominou nos extratos ácidos, por ser mais solúvel em condiçoes de maior acidez.44 Considerando que houve período de estiagem, antes da coleta 2, possivelmente os íons formaram precipitados ou permaneceram sorvidos aos minerais presentes, como óxidos de Al e Fe.20,41 Apesar da presença de Zn no XR (Tabela 4), este nao parece ter o papel principal na liberaçao do Zn para a soluçao do solo, em especial pelo baixo FM (1,87%). A amostra testemunha absoluta (T5) apresentou teores similares aos observados nos solos com NPK e XR, indicando que sua presença está relacionada à formaçao do solo local,20 com exceçao do extrato ácido para T6 coleta 2, em que o teor de Zn foi significativamente maior, mas o desvio padrao relativo foi relativamente alto, e para a amostra T4 da terceira coleta, para a qual foi observada a maior concentraçao (20 ± 3 µg L-1) em comparaçao com todos os tratamentos para as três coletas. Ainda assim, a concentraçao de Zn foi muito inferior ao limite máximo legislado de 1050 µg L-1,25 indicando nao haver riscos de contaminaçao pelo Zn devido à aplicaçao do XR no solo. As maiores concentraçoes de Cu determinadas nos extratos em água tipo 1 foram de 3 µg L-1 (coleta 2, T5 e coleta 3, T3, 0-5 cm) e de 4,8 µg L-1, (coleta 1, T3, 5-10 cm) sendo que para algumas amostras as concentraçoes de Cu foram inferiores ao LQ (1,07 µg L-1) para a maior profundidade. Em meio de ácido acético as concentraçoes de Cu ficaram abaixo do LQ nos extratos de todas as amostras, para a profundidade de 5 - 10 cm e para algumas profundidades da coleta 1 (0 - 5 cm). O Cu é um elemento de baixa mobilidade no solo em pH neutro, e o íon Cu2+ tem alta afinidade e interage fortemente com a matéria orgânica, argilas e óxidos metálicos. A maior parte do Cu disponível no solo está na forma complexada com a matéria orgânica nas camadas mais superficiais.40,46,47 O Cu pode ainda formar compostos como CuS, o que diminui sua toxicidade.44 Os resultados observados para o cobre (meio aquoso) para a profundidade de 5 - 10 cm (T3 e T4) foram significativamente maiores em comparaçao com T1 e T5 (amostras testemunhas), ao passo que para o extrato obtido em ácido acético, o resultado observado para T4 (terceira coleta) foi significativamente maior em relaçao às amostras testemunhas. A maior concentraçao de Cu observada entre todos os extratos foi de 5,5 ± 0,3 µg L-1 (coleta 2, T5), sendo, portanto, muito inferior ao valor máximo permitido de 2,00 mg L-1,25 sugerindo nao haver tendência da presença do Cu em decorrência da adiçao do XR. Demais elementos Para o Pb, nos extratos obtidos em água tipo 1 foram verificadas concentraçoes de 24 ± 2 µg L-1 para T3 e 21 ± 6 µg L-1 para T6 (profundidade 5-10 cm, coleta 1) e para aqueles obtidos em meio de ácido acético 19,8 ± 0,9 µg L-1 (T2, 0-5 cm, coleta 1). Esses resultados, assim como os demais que foram inferiores ao LQ de 12,3 µg L-1 requerem certa atençao, pois o limite estabelecido pela legislaçao é de 10 µg L-1,25 havendo, portanto, a possibilidade de liberaçao de Pb para o meio aquoso. Adicionalmente, o Pb está presente no XR (Tabela 4) e apresentou fator de mobilidade de 6,37%, o que reforça chance de dessorçao de Pb. Apesar disso, nao foi possível constatar se a presença do Pb nesses extratos foi decorrente do XR ou do próprio solo. O Mo foi determinado apenas em três amostras dos extratos obtidos em ácido acético, na camada superficial, e somente na coleta 2, com os valores de 12 ± 5 µg L-1 (T3) 6 ± 2 µg L-1 (T5) e 8 ± 1 µg L-1 (T6). Para todos os demais extratos os valores foram abaixo do LQ (1,96 µg L-1) e, portanto, abaixo também do valor máximo permitido de 70 µg L-1, pelo CONAMA.25 Os elementos As, Cd, Co, Cr, Hg e Ni nao foram detectados ou apresentaram resultados abaixo dos respectivos valores de LQ, embora Cr e Ni tenham sido determinados na matriz de XR, bem como o Cd, porém nesse caso o XR apresentou 0,6 mg kg-1 (Tabela 4). Isso sugere que o XR nao contribuiria com esses elementos ao solo, ou as concentraçoes liberadas seriam muito baixas, e assim a aplicaçao de XR ao solo nao seria uma fonte de contaminaçao significativa.
CONCLUSAO De acordo com o estudo realizado com extratos de solos utilizando como extratores a água tipo 1 e soluçao de ácido acético foi possível observar que a adiçao de XR ao solo visando o fornecimento de nutrientes nao alterou suas propriedades, pelo menos com base nas avaliaçoes efetuadas. Os elementos traços detectados nos extratos sao provenientes do próprio solo, visto que o solo testemunha apresentou características semelhantes às observadas nos solos tratados com diferentes quantidades de XR, sendo que para a maioria dos casos, os resultados observados nao diferiram significativamente em relaçao às amostras testemunha. Os resultados permitem inferir que o XR pode contribuir no fornecimento de Ca e Mg ao solo, especialmente o Ca, e que de modo geral nao contribuiu na lixiviaçao e biodisponibilidade de elementos traço indesejáveis nas amostras de solos avaliados neste experimento, como As, Cd, Cr, Hg, Mo, Ni e Pb, pelo menos com base nas doses de XR aplicadas. Além disso, para a maior parte dos elementos, as concentraçoes observadas foram inferiores àquelas estipuladas pela legislaçao vigente. Apesar disso, deve-se levar em consideraçao a possibilidade de heterogeneidade do XR em termos de diferenças de composiçao dos elementos estudados, o que sugere a necessidade da realizaçao de estudos adicionais.
AGRADECIMENTOS Os autores agradecem o suporte logístico, financeiro, e a bolsa de estudos concedida à Jeniffer V. dos Santos, ao Projeto Xisto Agrícola (Termo de Cooperaçao entre Embrapa Clima Temperado, PETROBRAS/SIX e FAPEG) e ao LAMIR pelas análises de fluorescência de raio X.
REFERENCIAS 1. Pimentel, P. M.; Silva, C. N.; Melo, D. M. A.; Melo, M. A. F.; Maldonado, G.; Henrique, D. M.; Cerâmica 2006, 52, 194. 2. Altun, N. E.; Hiçylmaz, C.; Hwang, J. Y.; Suat Bagci, A.; Kök, M. V.; Oil Shale 2006, 23, 211. 3. dos Santos, M. M.; Matai, P. H. L. S.; R. Esc. Minas 2010, 63, 673. 4. Shirav, M.; Zimmels, Y.; Halicz, L.; Eldad, H.; Fuel 1987, 66, 281. 5. Krol, A. A., Bell, P. R. F., Greenfield, P. F.; Water Res. 1993, 2, 277. 6. Jones, D. R., Chapman, B. M., Jung, R. F.; Water Res. 1990, 24, 131. 7. Ballice, L.; Fuel Proces. Technol. 2005, 86, 673. 8. Orupold, K.; Habicht, J.; Tenno, T.; Oil Shale 2008, 25, 267. 9. Nicolini, J.; Pereira, B. F.; Pillon, C. N.; Machado, V. G.; Lopes, W. A.; Andrade, J. B.; Mangrich, A. S.; J. Anal. Appl. Pyrolysis 2011, 90, 112. 10. Gütlein, A.; Kersten, M.; Feinstein, S.; Illner, P.; Procedia Earth Planet. Sci. 2013, 7, 413. 11. Yen, T. F.; Chilingarian, G. V.; Oil shale. Elsevier: Amsterdan,1976. 12. Snape, C.; Composition, Geochemistry and Conversion of Oil Shales. Springer: UK, 1993. 13. Speight, J. G.; Shale oil production processes. Elsevier: USA, 2012. 14. Bell, P. R. F.; Krol, A. A.; Greenfield, P. F.; Water Res. 1986, 20, 741. 15. Jaber, J. O.; Probert, S. D.; Appl. Energy 1999, 62, 169. 16. Ibrahim, K. M.; Jaber, J. O.; Environ. Geol. 2007, 52, 979. 17. Zhang, L.; Zhang, X.; Li, S.; Wang, Q.; Energy Procedia 2012, 17, 39. 18. Bohn, H. L.; Mcneal, D. L.; O'connor, G. A.; Soil Chemistry, 3rd ed., John Wiley & Sons: New York, 2001. 19. Sparks, D. L.; Environmental Soil Chemistry. Academic Press, 2nd ed., Elsevier Science, 2003. 20. Sposito, G.; The chemistry of soils, 2nd ed., Oxford University Press: New York, 2008. 21. Pereira, H. S., Vitti, G. C.; Hortic. Bras. 2004, 2, 317. 22. Leao, R. E.; Giacomini, S. J.; Redin, M.; Souza, E. L.; Silveira, C. A. P.; Pesq. Agropec. Bras. 2014, 49, 818. 23. Doumer, M. E.; Giacomini, S. J.; Silveira, C. A. P.; Weiler, D. A.; Bastos, L. M.; Freitas, L. L.; Pesq. Agropec. Bras. 2011, 46, 1538. 24. Adamson, J.; Irha, N.; Adamson, K.; Steiness, E.; Kirso, U.; Oil Shale 2010, 27, 250. 25. Conama; Resoluçao nº 420, 28 de dezembro de 2009. 26. Embrapa; Sistema Brasileiro de Classificaçao de Solos. Embrapa SPI: Rio de Janeiro, 2009. 27. DIN 38414-4: German Standard Methods for the Examination of Water, Waste Water and Sludge; Sludge and Sediments (Group S). Determination of Leachability by Water (S4), 1984. 28. Delay, M., Lager, T., Schulz, H. D., Frimmel, F. H.; Waste Manage. 2007, 27, 248. 29. ABNT NBR 10005, Procedimento para obtençao de extrato lixiviado de resíduos sólidos, 2004. 30. EPA. Method 3050b, Acid digestion of sediments, sludges, and soils, 1996. 31. dos Santos, V. C. G.; Grassi, M. T.; Campos, M. S.; Peralta-Zamora, P. G.; Abate, G.; Analyst 2012, 137, 4458. 32. Shi, J.; Tang, Z.; Jin, Z.; Chi, Q.; He, B.; Jiang, G.; Anal. Chim. Acta 2003, 477, 139. 33. Bhargava, S.; Awaja, F.; Subasinghe, N. D.; Fuel 2005, 84, 707. 34. Nayak, P. S.; Singh, B. K.; Bull. Mater. Sci. 2007, 30, 235. 35. Chaves, L. H. G.; Vasconcelos, A. C. F.; Revista Brasileira de Engenharia Agrícola e Ambiental 2006, 10, 84. 36. Van Raij, B.; Andrade, J. C.; Cantarella, H.; Quaggio, J. A.; Análise Química para Avaliaçao da Fertilidade de Solos Tropicais, Campinas, Instituto Agronômico, 2001. 37. INMETRO. DOQ-CGCRE-008. Orientaçao sobre validaçao de métodos analíticos, 4ª revisao 2011. 38. Skoog, D. A.; West, D. M.; Holler, F. J.; Crouch, S. R.; Fundamentos de Química Analítica, trad. 9ª ed. norte-americana, Sao Paulo, 2014. 39. Maranhao, T. A.; Silva, J. S. A.; Andrade, R. M.; Bascuñan, V. L. A. F.; Oliveira, F. J. S.; Curtius, A. J.; Microchem. J. 2013, 106, 139. 40. McBride, M. B.; Environmental Chemistry of Soils, Oxford University Press: New York, 1994. 41. Amaral Sobrinho, N. M. B.; Barra, C. M.; La, O. R.; Em Química e Mineralogia do Solo - Parte II - Aplicaçoes; Alleoni, L. R. F., Melo, V. de F., eds.; SBCS: Viçosa, 2009, cap XVI. 42. White, R. E.; Princípios e práticas da ciência do solo, 4ª ed., Andrei, Sao Paulo, 2009. 43. Osman, K. T.; Soils: principles, properties and management, Springer: New York, 2013. 44. Hooda, P. S.; Trace elements in soil, Wiley: United Kingdom, 2010. 45. Alloway, B. J.; Heavy metals in soils: Trace Metals and Metalloids in Soils and their Bioavailability, 3rd ed., Springer: UK, 2013. 46. Andrade, M. G.; Tese de Doutorado, Universidade Estadual Paulista, Brasil, 2011. 47. Foth, H. D.; Fundamentals of Soil science, 8nd ed., John Wiley & Sons: New York, 1990. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access