
 |
 |
 |
 |
 |
Artigo
| Amperometric determination of 8-chlorotheophylline, caffeine, and diphenhydramine in a single run |
|
Jhonys Machado Freitas; Thiago da Costa Oliveira; Rodrigo Alejandro Abarza Munoz; Eduardo Mathias Richter*
Instituto de Química - Universidade Federal de Uberlândia, Av. João Naves de Ávila, 2121 - 38400-902 Uberlândia - MG, Brasil Recebido em 06/12/2016 *e-mail: emrichter@iqufu.ufu.br A simple method for simultaneous determination of 8-chlorotheophylline (CTP), caffeine (CAF), and diphenhydramine (DIP) using batch-injection analysis with multiple pulse amperometric detection (BIA-MPA) is reported. A sequence of three potential pulses (+1.10 V, +1.40 V, and +1.70 V) was applied to the boron-doped diamond working electrode with the acquisition of three distinct amperograms. CTP was detected selectively at +1.10 V, both CTP + CAF at +1.40 V and CTP + CAF + DIP at +1.70 V. The subtraction between the currents detected at the three potential pulses (with the use of correction factors) was the strategy used to achieve selectivity for the determination of CAF and DIP. The proposed method is simple, inexpensive, fast (120 injections h-1), presents good precision (RSD < 0.9%; n = 20), and adequate limits of detection (0.31, 0.49, and 0.76 µmol L-1 for CTP, CAF and DIP, respectively). The results obtained by BIA-MPA were similar to those found by high-performance liquid chromatography (95% confidence level). INTRODUCTION Dimenhydrinate (DIM) is an over-the-counter drug commonly used to prevent motion sickness related to vomiting and nausea.1 DIM is a salt composed by the combination of two active pharmaceutical ingredients in equimolar ratio (1:1): 8-chlorotheophylline (CTP) and diphenhydramine (DIP).2 The ability to reduce nausea mainly occurs due to the presence of DIP, however, the concomitant presence of CTP (stimulant of the central nervous system) diminishes somewhat drowsiness induced by DIP.3 In addition, the existence of synergistic effects (sum of the effects of their individual constituents) between DIP and CTP has also been reported.4According to this study, DIM presents pleasing properties which are not present if DIP or CPT is used individually. Despite the presence of CTP, side effects, such as somnolence, decrease in the capacity of concentration and coordination, remain common in patients which make use of medications containing DIM (DIP + CTP). Therefore, in some formulations containing DIM,5,6 caffeine (CAF) is added in order to reduce the sedative effects of DIP.7 To our knowledge, there is only one previous work which reported an analytical method for the simultaneous determination of DIP, CTP and CAF. The method is based on HPLC with UV-Vis detection.7 Apart from this method, procedures which demonstrated the possibility of determination of two from the three target analytes were reported previously, such as DIM (DIP + CTP) by spectrophotometry,8 by batch injection analysis (BIA),9 and by HPLC-UV.10 The possibility of determination of both CAF and DIP was demonstrated by spectrophotometry.5 Medicines containing the combination of three target compounds (DIP, CTP, and CAF) are commonly used in different countries. However, only a single method that allows the simultaneous determination of the three compounds was found in the literature.7 Therefore, the development of an alternative analytical method is of great importance. The system known as batch-injection analysis with multiple-pulse amperometric detection (BIA-MPA) has been used successfully for simultaneous analyses. In this system, a small volume (typically 10-150 µL) of sample or standard solution is directly injected using an electronic micropipette on a working electrode surface, which is submerged in a large-volume of supporting electrolyte (blank solution).11-15 This approach allows the development of analytical methods with several desirable characteristics, such as the need of small sample volumes (minimal waste production), high sensitivity, high speed, low cost, possibility of multi-component analysis (good selectively) and easily applicable in laboratories with minimal infrastructure or on-site analysis.11,12,16,17 In this work, we propose a new method using batch-injection analysis with multiple pulse amperometric detection (BIA-MPA) for the simultaneous determination of three compounds (CTP, DIP, and CAF). The purpose was achieved by the continuous application of three potential pulses (acquisition of three separate amperograms) and a fast (60 s) and single injection step.
MATERIAL AND METHODS Reagents and samples Deionized water with resistivity not less than 18 MΩ cm (Millipore Direct-Q3 system) was used for preparation of all aqueous solutions. Acetic acid, phosphoric acid, sulfuric acid, and caffeine (CAF) were obtained from Synth (Diadema - Brazil), sodium hydroxide from Dinamica (Diadema, Brazil), ethanol from Química Moderna (Barueri, Brazil), diphenhydramine (DIP) from Sigma-Aldrich (St. Louis, United States) and 8-chlorotheophylline (CTP) from Alfa Aesar (Ward Hill, USA). All reagents were used without further purification. The stock solutions of CTP and DIP were prepared just before the experiments by dissolving the exact amount of the substances in 20% (v/v) ethanol-buffer solution. The stock solution of CAF was prepared in water. Standard solutions were prepared by dilution of the stock solutions in supporting electrolyte (0.05 mol L-1 acetic acid/acetate buffer; pH 4.7) just before analysis. Pharmaceutical samples were purchased from a local compounding pharmacy. Regular samples containing a combination of the target compounds (some different brands) are only marketed in some European countries. Six tablets of each sample were weighed and converted into uniform powder. Next, an adequate amount of the powder, to obtain a solution with suitable concentration for the linear range studied, was dissolved (under sonication for 10 min) in 20% (v/v) ethanol-buffer solution. Sample solutions were diluted in supporting electrolyte for subsequent injection in the BIA system. Instrumentation and apparatus Voltammetric and amperometric measurements were carried out using a µ-Autolab Type III potentiostat (Metrohm Autolab B.V., The Netherlands) interfaced to a microcomputer and controlled by GPES 4.9.007 software. A mini Ag/AgCl (saturated KCl)18 and platinum wire were used as reference and auxiliary electrodes, respectively. A boron-doped diamond (BDD) thin film deposited on silicon substrate with 1.0 mm of thickness (Neocoat SA, La Chaux-de-Fonts, Switzerland) was used as working electrode. The thickness of the BDD film was ~1.2 µm with doping level of ~8000 ppm. The initial activation of the BDD electrode (new electrode) was performed using two steps: (1) application of +0.01 A for 1000 s in 0.04 mol L−1 Britton-Robinson buffer solution (anodic pretreatment); (2) application of −0.01 A for 1000 s in a 0.1 mol L−1 H2SO4 solution (cathodic pretreatment).19,20 The current application time may be shorter if potentiostats with the possibility of applying larger currents (e.g. 1 A) are used. In this work, the maximum applicable current was 0.01 A (µ-Autolab Type III potentiostat). After the first pretreatment (activation), the BDD electrode was pretreated only cathodically at the beginning of each working day. If the electrode is not used for a longer period (few days) or studies were performed using higher concentrations (e.g. studies by cyclic voltammetry), both pretreatments (anodic and cathodic) are commonly required. In our research group, the BDD electrode is considered suitable for use when the background current reaches values below 1 µA in cyclic voltammetric (0.0 to +1.4 V; 50 mV s-1) experiments using 0.1 mol L−1 H2SO4 solution as supporting electrolyte. The same BDD electrode was used in all results presented in the proposed work. The batch injection analysis (BIA) system consisted of a motorized electronic micropipette (Eppendorf Multipetter stream) and a home-made BIA cell in the three-electrode configuration. The cell was constructed with polypropylene material, similar to that used in previous work.14 A piece of BDD (0.7 x 0.7 cm) was pressed on an organic solvent resistant O-ring (internal diameter of 5 mm; electrode area = 0.2 cm2) positioned over a hole located at the center of the bottom of the cell. The electric contact between the BDD electrode and the potentiostat was made with a metal plate (stainless steel) positioned under the BDD piece. The metal plate was fixed on the bottom of the cell with the use of screws. The solution inside the BIA cell was stirred using a micro DC-motor (3 - 24 V).17 The stirring rate was kept constant at 280 ± 10 rpm in all studies. The distance between the Multipetter combitipr and the working electrode was kept constant (≈ 2 mm) 11 in all injection procedures. All electrochemical measurements were carried out at room temperature without removing dissolved oxygen. Results for the simultaneous determination of CTP, CAF, and DIP by BIA-MPA were compared to those found by HPLC.7 The HPLC analysis was performed using a Shimadzu LC-10 VP equipped with an UV-Vis detector (SPD-10AV), a LC column (Phenomenex MAX-RP-C12, 250 mm x 4.6 mm, 4 µm), a manual injector (20 µL) and a pump (LC-10AD-VP). The mobile phase consisted of a mixture of acetonitrile and 0.01 mol L-1 H3PO4 + triethylamine up to pH 2.8 (22:78, v/v). The detector and flow rate were fixed at 229 nm and 1.0 mL min-1, respectively. The retention times were 4.6, 5.3, and 16.7 min for CAF, CTP, and DIP, respectively. All results obtained by multiple pulse amperometry (MPA) were presented without baseline correction. The limit of detection (LOD) was considered according to IUPAC definition (LOD = 3sB/S, in which sB is the standard deviation of baseline noise and S is the analytical sensitivity of the calibration curve). The limit of quantification (LOQ) was calculated as 3.3 LOD.
RESULTS AND DISCUSSION First, the electrochemical performance of CAF, DIP, and CTP was examined by cyclic voltammetry in four electrolyte solutions (0.1 mol L-1 H2SO4 - pH = 1.0; 0.05 mol L-1 acetic acid/acetate buffer - pH 4.7 and 0.1 mol L-1 phosphate buffers - pH 2.1 and 7.2). Taking into account some key features, such as separation between oxidation peaks, sensitivity, and stability, the solution composed by 0.05 mol L-1 acetic acid/acetate buffer showed the best conditions for simultaneous determination of the three compounds. In Figure 1a, characteristic cyclic voltammograms obtained at BDD electrode in 0.05 mol L-1 acetic acid/acetate buffer solution (pH 4.7), before, and after addition of CAF, DIP, and CTP are shown. In order to evaluate the electrochemical behavior of the three compounds in hydrodynamic conditions, experiments using the BIA-MPA system were also carried out (Figure 1b). Only the faradaic current is shown in the Figure 1B (the capacitive current was subtracted).
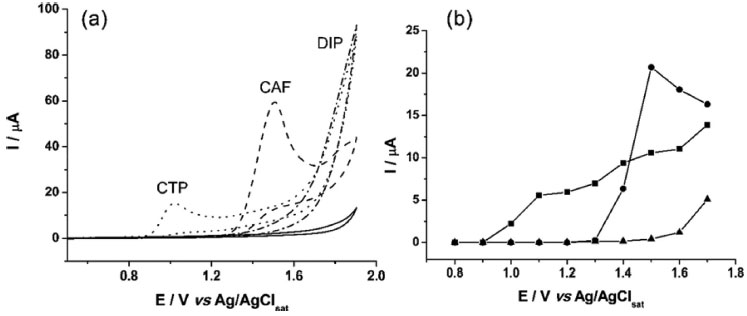 Figure 1. (a). CVs of BDD electrode in 0.05 mol L-1 acetic acid/acetate buffer (pH 4.7) before (─) and after addition of 1.0 mmol L-1 CTP (···), CAF (---) or DIP (-·-). Scan rate: 50 mV s−1; step potential: 5 mV. (b) Hydrodynamic voltammograms for CTP (■; 100 µmol L-1), CAF (●; 100 µmol L-1) and DIP (▲; 100 µmol L-1) obtained with the BIA system and BDD electrode as detector. Injected volume: 150 µL; dispensing rate: 75 µL s-1
In both studies (Figure 1a and 1b), the behavior of the three compounds was similar. Irreversible oxidation processes were observed by cyclic voltammetry at around +1.10 V, +1.40 V, and above +1.60 V for CTP, CAF, and DIP, respectively. As reported in previous works, an equal number of electrons and protons is transferred in the electrochemical oxidation of CTP (4e- and 4H+)21,22 and CAF (4e- and 4H+).23-25 In addition, scan rate studies carried out by cyclic voltammetry show that the electrode processes for both CTP and CAF are controlled by diffusion.9,23 On the other hand, investigations on the electrochemical mechanism of DIP were difficult to be obtained due to its poorly defined oxidation peak (near to background discharge potentials). As previously suggested, the oxidation mechanism of a tertiary aliphatic amine (DIP) tends to generate a N-radical with fast cleavage to aldehydes and aliphatic amines.26,27 Based on the results presented in Figure 1, a potential-pulse sequence (Figure 2a) was selected in order to detect CTP, CAF and DIP independently by BIA-MPA. Figure 2b shows amperometric responses recorded at three potentials pulses (+1.10 V/50 ms, +1.40 V/50 ms and +1.70 V/50 ms) for sequential injection of four different solutions: (1) 50 µmol L-1 DIP; (2) 120 µmol L-1 CAF; (3) 50 µmol L-1 CTP; and (4) 50 + 120 + 50 µmol L-1 of DIP, CAF, and CTP, respectively. A fourth a potential pulse (+0.70 V/150 ms) was also applied throughout the experiment in order to avoid contamination/passivation of the BDD electrode surface. The potential and time of application (+0.7 V/ 150 ms) were selected through experimental tests in order to obtain the best stability in the system response. The current detected at +0.70 V/150 ms was not presented.
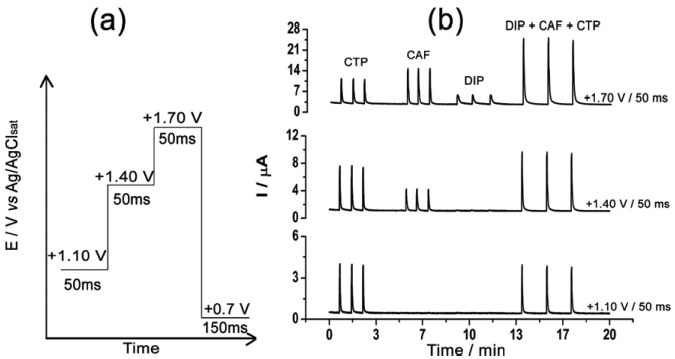 Figure 2. (a). Potential-pulse scheme for application of the four potential pulses; (b) BIA-MPA responses (n = 3) for injection of four different solutions: (1) 50 µmol L-1 CTP; (2) 120 µmol L-1 CAF; (3) 50 µmol L-1 DIP; (4) 50 + 120 + 50 µmol L-1 of CTP + CAF + DIP, respectively. Cleaning/activation potential pulse: 0.7 V/150 ms (amperogram not shown); supporting electrolyte: 0.05 mol L-1 acetic acid/acetate buffer (pH 4.7); dispensing rate: 75 µL s-1; injection volume: 150 µL
In Figure 2b it can be observed that only CTP is oxidized at +1.10 V (50 ms) once the current response kept constant even in the presence of the three species (DIP + CAF + CTP). At +1.40 V (50 ms), both CTP and CAF are oxidized. CAF can only be quantified using the current detected at this potential pulse if the current from oxidation of CTP is previously subtracted. At +1.70 V (50 ms), the three analytes of interest were oxidized. DIP can only be quantified using the current detected at +1.70 V if the currents from oxidation of both CTP and CAF are previously subtracted. However, as can be noted in Figure 2b, the current responses for CTP at +1.10 V, +1.40 V, and +1.70 V and for CAF at +1.40 V and +1.70 V are not equal. These results are in agreement with the current variation observed in Figures 1A and 1B. Therefore, simple subtraction between the currents detected in the respective potentials pulses is not an option if the accurate determination of the analytes is required. As already shown in previous works, this situation can be circumvented using simple correction28-31factors (CF). For CAF determination at +1.40 V without interference of CTP, the CF can be obtained by a simple injection of a standard solution containing only CTP in the proposed BIA system and the using the equation 1: 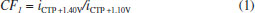 Then, if a solution containing both CTP and CAF is analyzed with the BIA-MPA system, the current from CAF oxidation at +1.40 V can be obtained using the CF1 value and the equation 2: 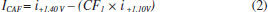 In order to obtain selectivity in the amperometric detection of DIP at +1.70 V in the presence of both CAF and CTP, the use of two CFs is needed. The CF2 is related to the difference between the current detected for CTP oxidation at +1.10 V and +1.70 V, and the CF3 is related to the difference between the current detected for CAF oxidation at +1.40 V and +1.70 V. The CF2 value can be achieved by injection of a solution containing only CTP and using the equation 3: 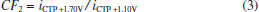 The CF3 value can be achieved by injection of a standard solution containing only CAF and using the equation 4: 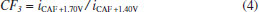 When a sample or standard solution containing all three compounds is injected in the BIA-MPA system, the current from the oxidation of DIP at +1.70 V can be calculated using the equation 5: 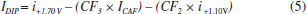 In the development of new analytical methods, studies in order to identify the linear working range (concentration range in which the sensitivity of the detector is relatively constant) should be carried out. In the proposed work, apart from the linearity, the CFs values should also have relatively constant values in the selected concentration intervals. In the concentration intervals between 10 to 30 µmol L-1 for CTP and 20 to 60 µmol L-1 for CAF, the following CF values were found (n = 6): CF1 = 1.63 ± 0.01, CF2 = 1.97 ± 0.02, and CF3 = 3.45 ± 0.03. In all cases, adequate RSD values (≤ 2%) were found. As well as the calibration curves, it is recommended to check the CFs values in each work-day. In order to improve the precision and the signal to noise ratio (analytical signal) for simultaneous determination of CTP, CAF and DIP by BIA-MPA, parameters as injected volume and dispensing rate were optimized. Better results were achieved for an injection volume of 150 µL and dispensing rate of 75 µL s-1. These parameters were used in the further studies. The signal stability of the BDD electrode positioned in the BIA-MPA system was evaluated by repeated injections of standard solutions containing 20 + 10 + 10 µmol L-1 (a) or 60 + 30 + 30 µmol L-1 (b) of CAF, DIP, and CTP, respectively (Figure 3).
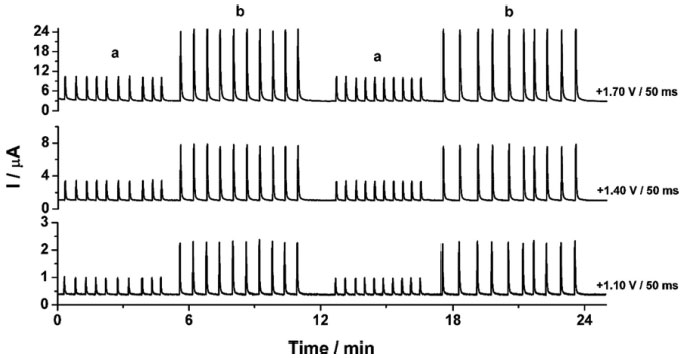 Figure 3. Amperograms obtained at +1.10V, +1.40 V, and +1.70 V from repeated injections (n = 20) of standard solutions containing 20 + 10 + 10 µmol L-1 (a) or 60 + 30 + 30 µmol L-1 (b) of CAF, DIP, and CTP, respectively. Other conditions see Figure 2
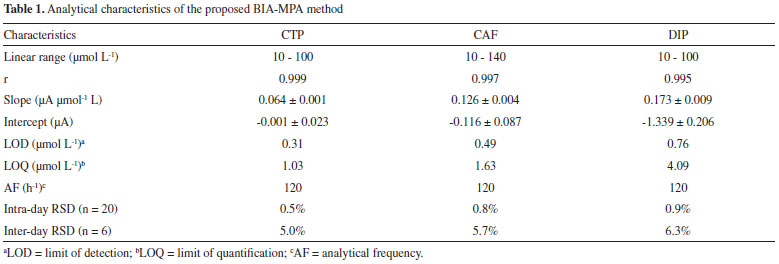
From the data shown in Figure 3, the following RSD values (n = 20) were obtained: 0.6% (a) and 0.4% (b) for CTP (at +1.10 V), 0.7% (a) and 0.8% (b) for CAF (at +1.40 V using the CF1) and 0.9% (a) and 0.9% (b) for DIP (at +1.70 V using the CF2 and CF3). These results revealed that the proposed BIA-MPA system presents excellent repeatability and high-throughput analysis (120 injections h-1). In addition, the system shows no memory effect (without contamination) even employed standard solutions with different concentrations. These satisfactory results were achieved with the solution into the BIA cell under constant stirring (280 rpm) and by application of an additional potential pulse (+0.7 V/ 150 ms) throughout the experiment (cleaning potential pulse). When the additional potential pulse and stirring were not used, RSD values in stability studies were higher than 4.0%. Furthermore, with the solution in the BIA cell under stirring, a considerably increase in the analytical frequency was observed (from 30 to 120 injections h-1). After optimization of the BIA-MPA parameters, the linear working ranges for the three analytes were investigated. A linear response (r > 0.99) was observed between 10 to 140 µmol L-1 for CAF and between 10 to 100 µmol L-1 for CTP and DIP. The calibration curves were recorded in smaller concentration ranges, due to the higher stability of the CFs values in these conditions. Figure 4 shows the amperometric recordings at + 1.10 V, + 1.40 V and +1.70 V for injection of a solution containing CTP (used to obtain the CF1 and CF2), CAF (used to obtain the CF3), and five solutions with increasing concentrations of the three analytes: CTP (c-g: 10 to 30 µmol L-1), CAF (c-g: 20 to 60 µmol L-1), and DIP (c-g: 10 to 30 µmol L-1). Two samples (1 and 2) diluted in supporting electrolyte were also injected.
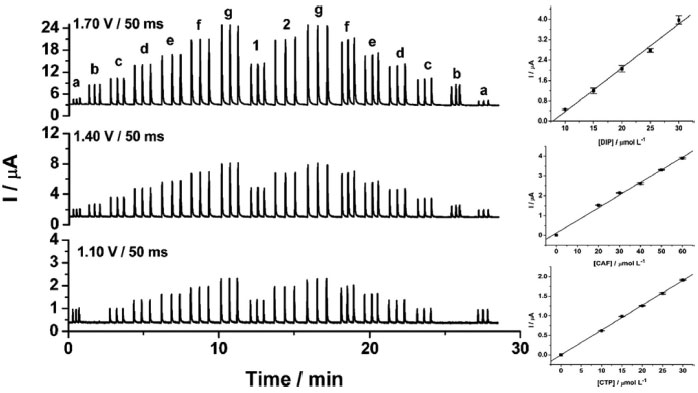 Figure 4. Amperometric recordings for injections of solutions containing CTP (a: 15 µmol L-1), CAF (b: 30 µmol L-1), five standard solutions (c-g) containing simultaneously increasing concentration of CTP (10-30 µmol L-1) + CAF (20-60 µmol L-1) + DIP (10-30 µmol L-1), two diluted sample solutions (1 and 2), and the analytical curves for each analyte with the respective SD bars (n = 6) for each measurement. Other conditions see Figure 2
The calibration curves showed good linearity in the concentration range used in the present study (r ≥ 0.995). In addition, similar slope values (difference < 2.9%) were obtained for the standard solutions injected in ascending (c - g) and descending (g - c) concentration order. The small variation in the slope values again confirms that the electrode contamination and memory effect phenomena were avoided. Table 1 shows the analytical characteristics of the BIA-MPA method here proposed.
Two compounded drugs samples acquired in the local market were also analyzed in order to evaluate the performance BIA-MPA for simultaneous determination of CTP, CAF, and DIP. For comparison purposes, the same samples were also analyzed by HPLC (Table 2).
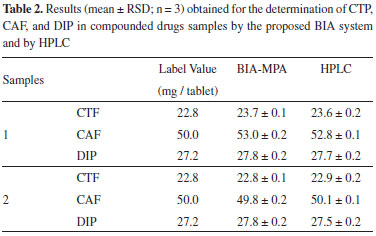
The results achieved with the proposed BIA-MPA method were in agreement with the results found by HPLC. At the 95% confidence level, the calculated t values (paired Students t-Test) were lower than the critical value (2.78, n = 3). This indicates that there are no significant differences between the results obtained with the BIA-MPA method and the HPLC method. In addition, these results also demonstrated that proposed method has adequate accuracy for simultaneous determination of CTP, CAF, and DIP.
CONCLUSION In this work, we have demonstrated that the BIA-MPA system can be used successfully for fast and simultaneous determination CTP, CAF, and DIP in pharmaceutical samples. The proposed method presents advantages when compared to others techniques (e.g. chromatography), concerning simplicity, cost, speed of analysis, waste generation (environmentally friendly), and samples pretreatment (only dilution in electrolyte solution prior to analysis). There are good prospects for this method to be applied in routine analysis in substitution of expensive chromatographic separation systems.
ACKNOWLEDGEMENTS The authors are grateful to FAPEMIG (CEX - APQ-02118-15) and CAPES (PRO FORENSES-Process number: 23038.007073/2014-12) for financial support. EMR (Process number: 307333/2014-0) and RAAM (Process number: 308174/2013-5) thank CNPq for the fellowship.
REFERENCES 1. Özkan, C. K.; Taşdemir, U.; Taş, Ç.; Savaşer, A.; Erol, H.; Özkan, Y.; Chromatographia 2013, 76, 1521. 2. Halpert, A. G.; Olmstead, M. C.; Beninger, R. J.; Pharmacol. Biochem. Behav. 2003, 75, 173. 3. Halpert, A. G.; Olmstead, M. C.; Beninger, R. J.; Neurosci. Biobehav. Rev. 2002, 26, 61. 4. Nguyen, T.-L.; You, I.-J.; Oh, S.; Yang, C. H.; Lee, S.-Y.; Jang, C.-G.; Neurosci. Lett. 2010 471, 38. 5. Cañada, M. J. A.; Reguera, M. I. P.; Díaz, A. M.; Capitán-Vallvey, L.; Talanta 1999, 49, 691. 6. Spataru, N.; Sarada, B. V.; Tryk, D. A.; Fujishima, A.; Electroanalysis 2002, 14, 721. 7. Barbas, C.; García, A.; Saavedra, L.; Castro, M.; J. Chromatogr. A 2000, 870, 97. 8. Kar, A.; Aniuha, G. I.; J. Pharm. Sci. 1981, 70, 690. 9. Freitas, J. M.; Oliveira, T. C.; Silva, P. L.; Gimenes, D. T.; Munoz, R. A. A.; Richter, E. M.; Electroanalysis 2014, 26, 1905. 10. Döge, U.; Eger, K.; Pharmazie 2007, 62, 174. 11. Quintino, M. S. M.; Angnes, L.; Electroanalysis 2004, 16, 513. 12. Wang, J.; Taha, Z.; Anal. Chem. 1991, 63, 1053. 13. da Silva, R. A. B.; Gimenes, D. T.; Tormin, T. F.; Munoz, R. A. A.; Richter, E. M.; Anal. Methods 2011, 3, 2804. 14. Tormin, T. F.; Cunha, R. R.; Richter, E. M.; Munoz, R. A. A.; Talanta 2012, 99, 527. 15. Gimenes, D. T.; Cunha, R. R.; Ribeiro, M. M. A. C.; Pereira, P. F.; Muñoz, R. A. A.; Richter, E. M.; Talanta 2013, 116, 1026. 16. Brett, C. M. A.; Brett, A. M. O.; Mitoseriu, L. C.; Electroanalysis 1995, 7, 225. 17. Pereira, P. F.; Marra, M. C.; Munoz, R. A. A.; Richter, E. M.; Talanta 2012, 90, 99. 18. Pedrotti, J. J.; Angnes, L.; Gutz, I. G. R.; Electroanalysis 1996, 8, 673. 19. Terashima, C.; Rao, T. N.; Sarada, B. V.; Kubota, Y.; Fujishima, A.; Anal. Chem. 2003, 75, 1564. 20. Salazar-Banda, G. R.; de Carvalho, A. E.; Andrade, L. S.; Rocha-Filho, R. C.; Avaca, L. A.; J. Appl. Electrochem. 2010, 40, 1817. 21. Spataru, N.; Sarada, B. V.; Tryk, D. A.; Fujishima, A.; Electroanalysis 2002, 14, 721. 22. Dryhurst, G. J.; Electrochem. Soc. 1972, 119, 1659. 23. Švorc, L.; Tomčík, P.; Svítková, J.; Rievaj, M.; Bustin, D.; Food Chem. 2012, 135, 1198. 24. Švorc, Ľ.; Int. J. Electrochem. Sci. 2013, 8, 5755. 25. Eisele, A. P. P.; Clausen, D. N.; Tarley, C. R. T.; Dall'Antonia, L. H.; Sartori, E. R.; Electroanalysis 2013, 25, 1734. 26. Surmann, P.; Peter, B.; Electroanalysis 1996, 8, 685. 27. Fuchigami, T.; Iwaoka, T.; Nonaka, T.; Sekine, T.; Bull. Chem. Soc. Jpn. 1980, 53, 2040. 28. Freitas, J. M.; Oliveira, T. C.; Gimenes, D. T.; Munoz, R. A. A.; Richter, E. M.; Talanta 2016, 146, 670. 29. Pereira, P. F.; Marra, M. C.; Cunha, R. R.; da Silva, W. P.; Munoz, R. A. A.; Richter, E. M.; J. Electroanal. Chem. 2014, 713, 32. 30. Pereira, P. F.; da Silva, W. P.; Muñoz, R. A. A.; Richter, E. M.; J. Electroanal. Chem. 2016, 766, 87. 31. Gimenes, D. T.; Marra, M. C.; Freitas, J. M.; Muñoz, R. A. A.; Richter, E. M.; Sens. Actuators B 2015, 212, 411. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access