
 |
 |
 |
 |
 |
Revisão
|
|
| Avanços recentes na hidroaminação de compostos insaturados Recent advances on hydroamination of unsaturated compounds |
|
Luiz F. T. Novaes; Julio C. Pastre*
Departamento de Química Orgânica, Instituto de Química, Universidade Estadual de Campinas, CP 6154, 13084-971 Campinas - SP, Brasil Recebido em 23/12/2016 *e-mail: juliopastre@iqm.unicamp.br This review presents thermodynamics and kinetics aspects of the hydroamination reaction, recent advancements on this field for non-activated alkenes, alkynes and allenes, employing transition metal catalysis or organocatalysis, including activation by hydrogen bonding or Bronsted acids. Selected syntheses that contain a strategic hydroamination step will be discussed, and advancements on the asymmetric version will also be highlighted. The full coverage of scientific activity about hydroamination is beyond this review, therefore the examples presented are from the last ten years, except when there are historical reasons for the discussion of previous works. INTRODUÇAO Os compostos orgânicos nitrogenados sao onipresentes em nossa sociedade. Aparecem na forma de biomoléculas, como proteínas, ácidos nucléicos e alcaloides, além de uma diversidade de materiais de uso diário, como polímeros, pigmentos, flavorizantes e fármacos (Figura 1).
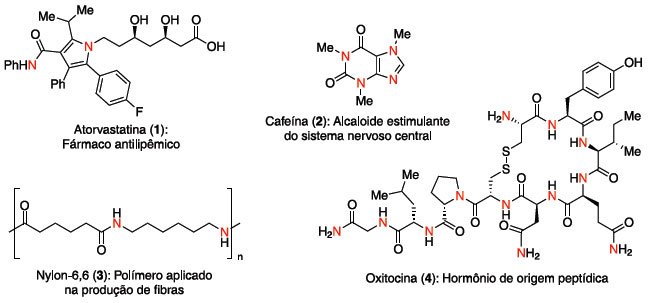 Figura 1. Exemplos de compostos nitrogenados
Para a síntese dessa classe de compostos, diversas metodologias para formaçao de ligaçao C-N foram desenvolvidas, dentre essas destacam-se N-alquilaçao,1,2 aminaçao redutiva com compostos carbonílicos,3,4 N-acilaçao5,6 e acoplamento cruzado C-N.7,8 Entre os métodos modernos para construçao de compostos nitrogenados complexos, a hidroaminaçao de compostos insaturados vem ganhando destaque e desperta interesse da comunidade científica.9,10 Essa abordagem impacta na economia de átomos do processo, o que é benéfico do ponto de vista da química verde.11 O presente texto engloba uma breve discussao sobre aspectos termodinâmicos e cinéticos da reaçao de hidroaminaçao, avanços recentes na área para reaçao com alcenos nao ativados, alcinos e alenos, sob catálise de metais de transiçao ou com uso de organocatalisadores, envolvendo ativaçao por ligaçao de hidrogênio ou por ácidos de Bronsted. Sínteses selecionadas que empregam de forma estratégica o uso de reaçoes de hidroaminaçao serao abordadas, e avanços no desenvolvimento de abordagens assimétricas também serao contemplados. A cobertura completa da atividade científica sobre hidroaminaçao está além do escopo deste texto, assim, foi dada preferência para exemplos dos últimos dez anos, exceto quando houver razoes históricas para discussao de trabalhos anteriores.
ASPECTOS TERMODINAMICOS E CINÉTICOS A reaçao de hidroaminaçao de compostos insaturados envolve a quebra de uma ligaçao σ N-H e uma ligaçao π C-C, com a formaçao das ligaçoes σ N-C e σ C-H. A termodinâmica da hidroaminaçao do estireno (5) com N-metilanilina (6) sob catálise de paládio foi estudada por Hartwig e colaboradores12 (Esquema 1).
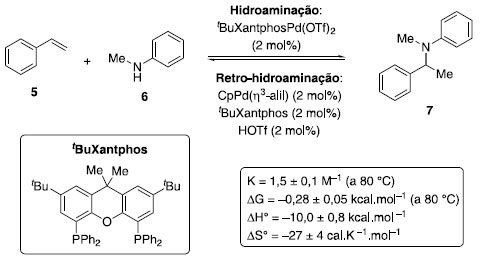 Esquema 1. Aspectos termodinâmicos da hidroaminaçao de estireno (5) com amina 6
As constantes de equilíbrio encontradas para essa reaçao foram K = 1,5 ± 0,1 L mol−1 a 80 °C e K = 0,52 ± 0,05 L mol−1 a 110 °C. Após avaliaçao em temperaturas intermediárias e com uso da equaçao de van't Hoff (Equaçao 1), foram calculados ΔH° e ΔS°, mostrados no Esquema 1. 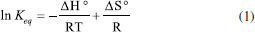 A partir desses dados e de análise similar para outras reaçoes de hidroaminaçao, foi generalizado que essa transformaçao usualmente é favorecida entalpicamente (exotérmica), porém esse efeito é contrabalanceado pelo termo T.ΔS em intensidade similar, fazendo com que o processo seja aproximadamente ergoneutro (ΔG = 0). Apesar do fator termodinâmico, a adiçao de aminas em ligaçoes múltiplas C-C é cineticamente difícil, uma vez que há repulsao entre as duas espécies eletronicamente ricas. Adicionalmente, os orbitais σ N-H e π C-C possuem diferentes simetrias e energias. O uso de altas temperaturas, para cruzar a elevada energia de ativaçao dessa transformaçao, causa um deslocamento do equilíbrio em direçao aos materiais de partida, em vista da entropia negativa. A barreira energética pode ser modulada com uso de bases fortes, para gerar amidetos de metais que podem ser adicionados em ligaçoes múltiplas C-C de modo facilitado, ou pelo uso de ácidos fortes que podem protonar ligaçoes múltiplas C-C, tornando mais fácil o ataque por um nucleófilo nitrogenado. Por fim, o grande desenvolvimento da hidroaminaçao ocorreu com a utilizaçao de catálise por metais de transiçao atuando na ativaçao da ligaçao π C-C ou ativando a espécie nitrogenada.9 Hidroaminaçao com catalisadores à base de metais de transiçao O uso de transformaçoes catalíticas está contemplado nos 12 princípios de química verde,13 por impactarem em uma reduçao na geraçao de resíduos, quando comparados aos métodos que utilizam reagentes estequiométricos. Nesse contexto, a utilizaçao de complexos de metais de transiçao como catalisadores é atrativa, pois usualmente baixa carga catalítica é necessária para a transformaçao. Para as reaçoes de hidroaminaçao, esses catalisadores podem atuar tanto na ativaçao do sistema π dos compostos insaturados, quanto na ativaçao da espécie nitrogenada. Hidroaminaçao com amônia Um dos grandes desafios da reaçao de hidroaminaçao é a utilizaçao de amônia, um reagente barato e produzido industrialmente em larga escala, mas que usualmente reage com espécies metálicas para gerar complexos ácido-base de Lewis inertes. Trabalhos de Hartwig14 e Buchwald15 sobre acoplamento de haletos de arila com amônia, via catálise de paládio, abriram caminho para estudos do seu uso em reaçoes de hidroaminaçao. Em 2008, Bertrand e colaboradores16 reportaram o uso de catalisadores de Au (I) com ligante do tipo (alquil)(amino)carbeno cíclico para ativaçao de amônia e alcinos em reaçoes de hidroaminaçao (Esquema 2). Para o alcino 8, ocorreu uma reaçao de hidroaminaçao, com adiçao Markovnikov de NH3, e posterior tautomerizaçao, gerando a imina 11. Para o diino 9, ocorreu inicialmente a hidroaminaçao de uma ligaçao tripla, seguida de uma segunda hidroaminaçao com a espécie enamina intermediária, para gerar o pirrol substituído 12.
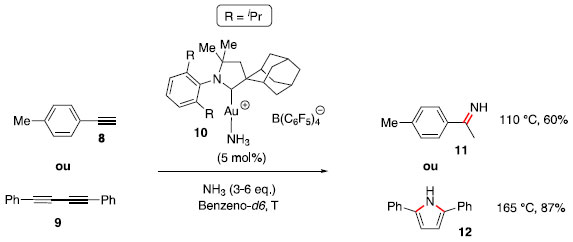 Esquema 2. Reaçoes de hidroaminaçao com uso de amônia
Um mecanismo possível para essa transformaçao se baseia na proposta de Mizushima, Hayashi e Tanaka,17 em que o catalisador 10 se coordena com o alcino 13, na sequência, ocorre adiçao de NH3 sobre a ligaçao tripla ativada de 14. Em seguida, o catalisador 10 é regenerado, liberando a enamina, que pode ser tautomerizada na imina 16 (Esquema 3).
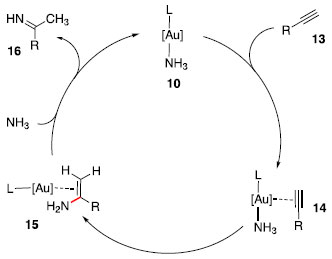 Esquema 3. Possível mecanismo para hidroaminaçao com NH3 e catalisador de ouro 10
Até entao, apenas condiçoes drásticas haviam sido utilizadas para a reaçao de hidroaminaçao com amônia. Um exemplo é a utilizaçao de uma mistura de etileno e amônia em temperaturas superiores a 170 °C e pressoes acima de 800 bar com sódio metálico catalítico para gerar uma mistura de etilamina, dietilamina e trietilamina.18 Uso de aminas primárias: metais do grupo 4 A hidroaminaçao de alcinos e alenos com aminas primárias foi extensamente estudada com catalisadores e pré-catalisadores a base de titânio e zircônio (grupo 4 da tabela periódica).19-21 O mecanismo proposto para essa transformaçao é mostrado a partir do titanoceno 17 e alcino 22 no Esquema 4. A espécie catalítica ativa nessa reaçao é o complexo metal-imido 20, que realiza uma cicloadiçao [2+2] com o alcino 22 gerando o azametalociclobuteno 23. Em seguida, esse ciclo é protonado e forma-se a espécie bis-amido 24, que sofre α-eliminaçao da enamina, regenerando a espécie ativa metal-imido 20. A enamina 25 pode ser tautomerizada para a imina 26.
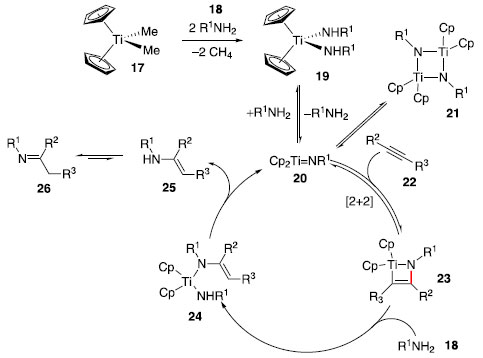 Esquema 4. Mecanismo proposto para hidroaminaçao catalisada por Cp2Ti=NR1
O pré-catalisador Cp2TiMe2 (17) foi empregado na síntese total do alcaloide (−)-xilopinina por Mujahidin e Doye.22,23 A partir do composto 27 foi realizada uma hidroaminaçao intramolecular para gerar a imina 28, após tautomerizaçao. Uma sequência de reduçao assimétrica de Noyori e ciclizaçao de Pictet-Spengler foi utilizada para a conclusao da síntese da (−)-xilopinina (Esquema 5).
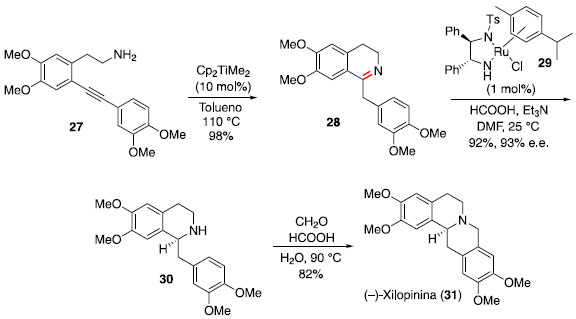 Esquema 5. Síntese total da (−)-xilopinina via hidroaminaçao intramolecular
Como consequência do mecanismo apresentado, a grande aplicaçao de catálise com os metais do grupo 4 para reaçoes de hidroaminaçao se limita ao uso de aminas primárias, necessárias para formaçao da espécie metal-imido. Sob condiçoes similares, a utilizaçao de alcenos nao ativados usualmente se limita a reaçoes intramoleculares para formaçao de pirrolidinas e piperidinas substituídas.24 Síntese de alilaminas: Catálise de paládio e ródio A síntese de alilaminas é particularmente interessante pela versatilidade desse bloco construtor, permitindo o uso de reaçoes de funcionalizaçao tanto da amina quanto da olefina. Alguns métodos desenvolvidos para a síntese desse motivo incluem substituiçao alílica,25 rearranjos de Overman,26 aminaçao C-H alílica27 e vinilaçao de iminas.28 A hidroaminaçao aparece como uma alternativa ou método complementar, que nao exige pré-instalaçao de um grupo de saída e pode ser realizada por catálise de metais de transiçao. Yamamoto e colaboradores29 reportaram a hidroaminaçao de alcinos internos do tipo 1-aril-1-propino com anilinas substituídas. O sistema catalítico composto de Pd(PPh3)4 e ácido benzoico inicialmente gera a espécie hidreto de paládio 32 por meio de uma adiçao oxidativa. O intermediário 32 participa de dois ciclos catalíticos, no primeiro realiza uma hidropaladaçao seguida de β-eliminaçao, para isomerizar o alcino 33 no aleno 35. No segundo ciclo, há formaçao do complexo paládio π-alila 36, que pode reagir com a amina 37, gerando a alilamina 38 e o hidreto de paládio 32 (Esquema 6).
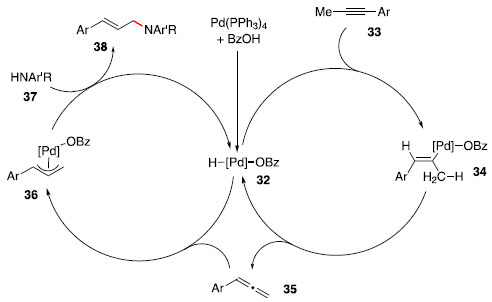 Esquema 6. Proposta mecanística da hidroaminaçao de 1-aril-1-propino
Essa reaçao foi avaliada com diferentes grupos substituintes no nitrogênio da anilina. A ausência de substituinte (39), uso de grupos alquílicos (40 e 41) ou tosila (42) forneceram rendimentos iguais ou superiores a 89% (Esquema 7). No entanto, o uso de grupos terc-butoxicarbonila (Boc, 43) e acetila (44) nao levaram à formaçao das respectivas alilaminas, provavelmente por tornarem o composto nitrogenado menos nucleofílico.
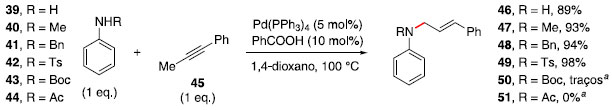 Esquema 7. Avaliaçao de escopo na formaçao de alilaminas. a Recuperaçao completa do material de partida
A hidroaminaçao catalisada por paládio também foi realizada empregando alenos como material de partida, com passagem por uma espécie paládio π-alila.30 Para a síntese assimétrica de alilaminas, Breit e colaboradores31 propuseram uma hidroaminaçao de alenos com uma forma mascarada de amônia, a imina derivada da benzofenona, e um catalisador de Rh (I). Dentre os ligantes bis-fosfinas avaliados, Josiphos apresentou o melhor resultado entre rendimento e excesso enantiomérico, além disso, o uso do aditivo p-toluenossulfonato de piridínio (PPTS) mostrou-se essencial para obtençao de rendimentos elevados. A aplicabilidade desse método foi demonstrada em uma reaçao em escala de grama do aleno 52 (Esquema 8). Na sequência, foi realizada a hidrólise da imina 54, gerando o cloridrato de amina 55 com recuperaçao da benzofenona (56).
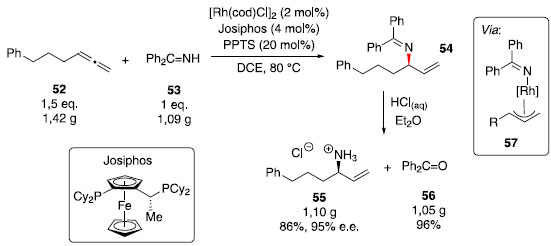 Esquema 8. Síntese assimétrica do cloridrato da alilamina 55
O mecanismo proposto para essa transformaçao passa pela adiçao oxidativa da imina 53 e hidrometalaçao do aleno 52, formando o complexo ródio π-alila 57, a eliminaçao redutiva leva à formaçao regiosseletiva do isômero ramificado 54 e regenera o catalisador de ródio (I). Síntese de enaminas: Catálise de rutênio com seletividade Markovnikov e anti-Markovnikov As enaminas sao blocos de construçao bastante versáteis, a alquilaçao desses compostos foi explorada por Stork e colaboradores,32 e grande atençao foi dada à química de enaminas como um modo de ativaçao em organocátalise.33 Diversos métodos para a síntese de enaminas se baseiam na condensaçao de um compostos carbonílicos com uma amina, empregando uso de ácidos ou bases. Para compostos sensíveis a essas condiçoes, a construçao dessa funçao também é possível via hidroaminaçao de alcinos com aminas secundárias. Uchimaru relatou o primeiro uso de hidroaminaçao intermolecular de alcinos terminais com aminas secundárias catalisada por rutênio.34 Um dos exemplos estudados foi a formaçao da enamina 59, através da adiçao Markovnikov da amina 6 sobre o alcino 58 (Esquema 9).
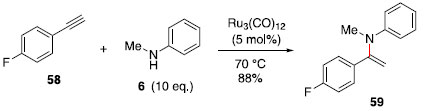 Esquema 9. Hidroaminaçao Markovnikov de alcino catalisada por rutênio
Essa transformaçao envolve a adiçao oxidativa da ligaçao N-H da amina 6 sobre uma espécie de Ru (0), gerando o intermediário hidreto de amido-rutênio 61. Na sequência, o alcino 62 se complexa ao centro metálico e ocorre ataque do nitrogênio sobre a ligaçao tripla ativada, gerando a espécie 64, que após eliminaçao redutiva gera a enamina 65 e a espécie catalítica de Ru (0) (Esquema 10).
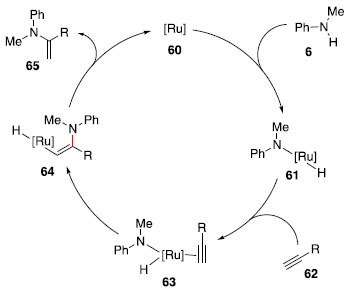 Esquema 10. Mecanismo de hidroaminaçao de alcinos via catálise de Ru (0)
A catálise de rutênio também foi utilizada para realizar hidroaminaçao com seletividade oposta, anti-Markovnikov, utilizando alcinos terminais, aminas secundárias e o pré-catalisador 68 (Esquema 11).35
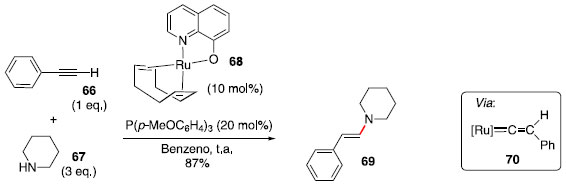 Esquema 11. Hidroaminaçao anti-Markovnikov de alcino catalisada por rutênio
Durante o desenvolvimento dessa metodologia baseada em catálise de Ru (I), observou-se que alcinos internos nao sofreram hidroaminaçao sob as condiçoes mostradas no Esquema 11 e que a adiçao de aminas ocorre exclusivamente no carbono terminal, indicando que o mecanismo passa pela formaçao de uma espécie vinilideno-rutênio (70).36 Na sequência, a piperidina (67) atacaria o carbono ligado diretamente no centro metálico e a protonaçao da ligaçao Ru-C levaria à enamina 69 e à espécie catalítica de rutênio. Síntese total da (−)-quinocarcina via hidroaminaçao de alcino catalisada por ouro (I): Regiosseletividade controlada pelo substrato O núcleo piperazina-tetrahidroisoquinolina aparece em uma variedade de alcaloides de arquitetura complexa, como (−)-quinocarcina, tetrazomina, lemonomicina e ecteinascidinas.37 Dentre esses, a (−)-quinocarcina atraiu interesse de químicos sintéticos por possuir atividade antitumoral.38 Esse produto natural foi isolado em 1983 por Takahashi e Tomita.39 Em 2012, Fujii, Ohno e colaboradores40 reportaram uma estratégia convergente para a síntese total desse alcaloide natural empregando um acoplamento cruzado de Sonogashira e uma hidroaminaçao intramolecular catalisada por ouro (I). Inicialmente foi realizada a síntese de quatro substratos modelos, que foram testados na etapa de hidroaminaçao. A reaçao a partir do alcino 72 forneceu exclusivamente a formaçao do anel de 5 membros 74. Assim, a modificaçao de uma cadeia flexível presente em 72 por um anel de 7 membros fundido ao sistema aromático em 75 e 78 levou a uma mistura dos produtos 6-endo e 5-exo. Finalmente, a modificaçao por um anel fundido de 5 membros (81) levou exclusivamente ao produto 6-endo desejado (Esquema 12). A ausência de formaçao do produto 83 provavelmente se deve à alta tensao anelar desse sistema tricíclico.
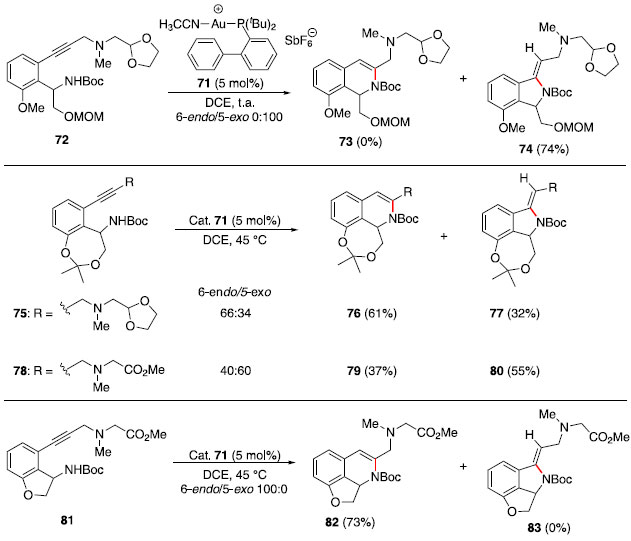 Esquema 12. Reaçoes modelo de hidroaminaçao catalisadas por 71
Em face desses resultados, foi realizada a síntese do composto 84,41 contendo um anel de 5 membros fundido ao sistema aromático, assim a etapa de hidroaminaçao levou à formaçao exclusiva do anel de 6 membros, que foi reduzido na sequência para o composto 86 em 90% de rendimento para 2 etapas. Em seguida, interconversao de grupos funcionais e uso de protocolo relatado previamente por Allan e Stoltz42 para transformaçao de 89 em 90 levou à formaçao do alcaloide (−)-quinocarcina (Esquema 13).
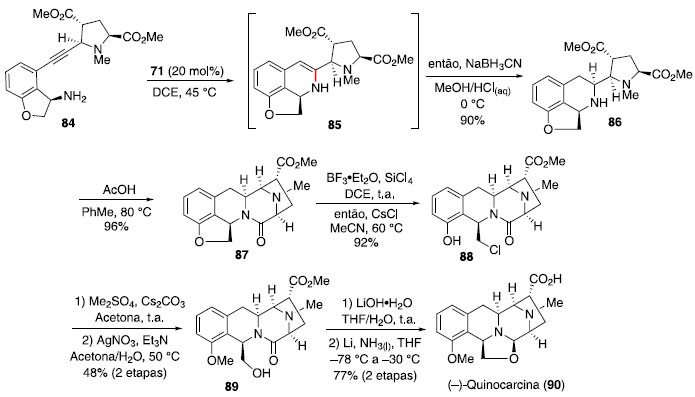 Esquema 13. Síntese da (−)-quinocarcina via hidroaminaçao
Hidroaminaçao fotoredox para construçao de N-heterociclos saturados: catálise de irídio Um dos grandes objetivos no campo da catálise é desenvolver novos modos de ativaçao de pequenas moléculas, e uma abordagem que vem ganhando espaço é o uso de catálise fotoredox com luz visível. Essa estratégia se baseia no uso de complexos metálicos ou pigmentos orgânicos, que participam de um processo de transferência única de elétrons, após fotoexcitaçao com luz visível.43 Complexos de polipiridinas de rutênio ou irídio sao usualmente empregados como catalisadores nessas transformaçoes. Apesar desses compostos serem oxidantes e redutores pouco eficazes pela transferência única de elétron nos estados fundamentais, tornam-se bastante efetivos quando no estado excitado. Dessa forma Knowles e colaboradores44 se basearam no mecanismo da reaçao de Hofmann-Löffler-Freytag e ciclizaçao de olefina com cátion-radical nitrogenado para desenvolver um protocolo fotoredox catalítico, que utiliza luz visível para formaçao de N-heterociclos saturados. Na reaçao de Hofmann-Löffler-Freytag, a amina de partida é convertida em um análogo N-halogenado de forma estequiométrica, que é submetido à fotólise com radiaçao ultravioleta na presença de um ácido de Bronsted forte. Para evitar uso de um oxidante estequiométrico, Knowles propôs o uso de um fotocatalisador para formar a espécie cátion-radical nitrogenado, que poderia entao reagir com um alceno de modo intramolecular formando os N-heterociclos de interesse. O catalisador que forneceu o melhor resultado foi Ir(ppy)2(dtbbpy)PF6. O mecanismo proposto é mostrado no Esquema 14 a partir da amina 91.
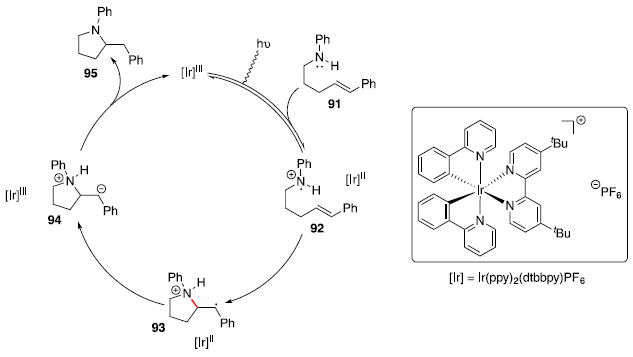 Esquema 14. Proposta mecanística para ciclizaçao fotocatalisada
Essa metodologia foi utilizada com sucesso para a preparaçao de diversos N-heterociclos substituídos: pirrolidinas (96-98), octaidroindol (99), piperidinas (100-101), morfolina (102) e piperazina (103). A regiosseletividade pode ser atribuída à formaçao de um radical benzílico intermediário, representado pela estrutura 93 no Esquema 14, tornando a ciclizaçao exo favorecida em relaçao à ciclizaçao endo. Nos casos em que mais de um centro estereogênico foi criado (99 e 101), baixa diastereosseletividade foi observada. Síntese de aminas quirais com estereocentros nas posiçoes α, β, γ ou δ: Hidroaminaçao formal catalisada por hidretos de cobre A hidrofuncionalizaçao de alcenos internos é um desafio sintético por apresentar dificuldades quanto ao controle da regio e estereosseletividade. A presença de grupos diretores nos substratos foi utilizada com sucesso para controlar a seletividade de hidrogenaçoes,45,46 e esse conceito tem sido expandido para hidrosililaçao,47 hidroboraçao48 e hidroacilaçao,49 em que um substituinte polar orienta a qual carbono será ligado o grupo funcional. Apesar dos avanços na reaçao de hidroaminaçao, o uso de grupos diretores para essa transformaçao ainda é raro. Recentemente, Hartwig e colaboradores reportaram o uso de Cu(PPh3)H catalítico, ligante (S)-DTBM-SEGPHOS (DTBM = 3,5-di-terc-butil-4-metoxi),50 HSiMe(EtO)2 e uma fonte eletrofílica de grupo amino (Bn2N-OBz) para realizar hidroaminaçao de olefinas 1,2-dissubstituídas com a posiçao homoalílica funcionalizada.51 Esses resultados fornecem uma rota simples para acessar fragmentos 1,3-aminoálcoois. Foi observada correlaçao entre efeito indutivo do grupo OR e a seletividade entre os produtos proximal:distal, quanto mais retirador de elétrons o grupo OR, maior a seletividade observada em favor do produto proximal (Esquema 16).
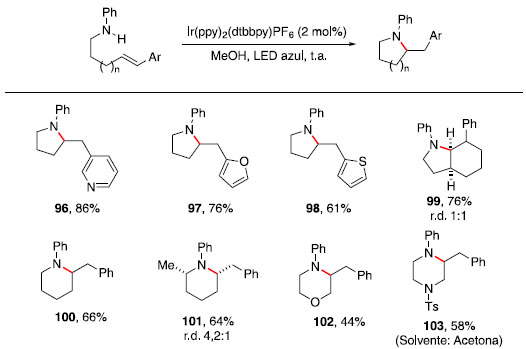 Esquema 15. Escopo para ciclizaçao catalítica fotoredox
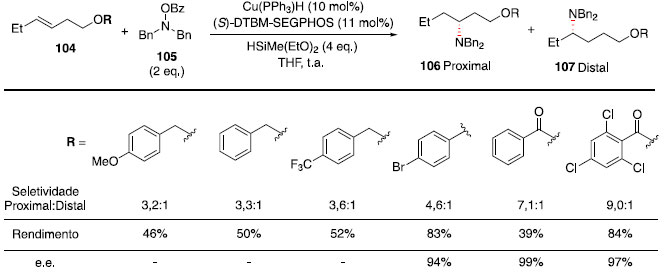 Esquema 16. Avaliaçao de grupos oxigenados para hidroaminaçao regiosseletiva
Para o composto com benzoato, foi obtido um rendimento de 39%, por conta da reduçao do grupo éster tanto do material de partida quanto do produto de hidroaminaçao. Assim, a adiçao de grupos na posiçao orto contribuiu para evitar a reduçao da carbonila do éster, como foi observado com o grupo 2,4,6-triclorobenzoato, que forneceu um rendimento de 84%. Para distinguir se essa regiosseletividade ocorre por efeito indutivo do grupo retirador de elétrons OR ou por coordenaçao direta com o catalisador metálico, foi realizada a hidroaminaçao do composto cíclico 108. Assim, se ocorresse coordenaçao com o centro metálico, o produto majoritário deveria ter substituiçao cis, e se o efeito indutivo fosse o principal, a reaçao com hidreto de cobre ocorreria pela face menos impedida estericamente, fornecendo o produto trans. Ao realizar a reaçao, o produto foi obtido na forma majoritariamente trans (r.d. >95:5, Esquema 17),52 confirmando que o efeito indutivo é o principal na regiosseletividade observada anteriormente (Esquema 16).
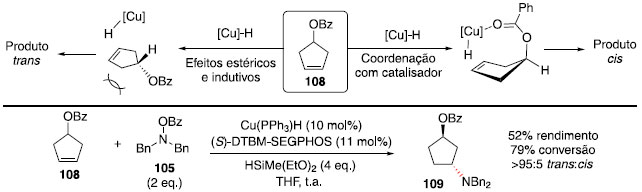 Esquema 17. Origem da seletividade da hidroaminaçao catalisada por hidreto de cobre
Um sistema similar ao mostrado foi aplicado por Buchwald e colaboradores para a síntese de aminas com estereocentro na posiçao β, a partir da hidroaminaçao de olefinas 1,1-dissubstituídas.53 Posteriormente, para formaçao de aminas com estereocentros remotos (posiçoes γ e δ) foi utilizada uma estratégia de hidroaminaçao redutiva de transferência (do inglês, reductive relay hydroamination).54 Exemplos dessas abordagens estao mostrados no Esquema 18.
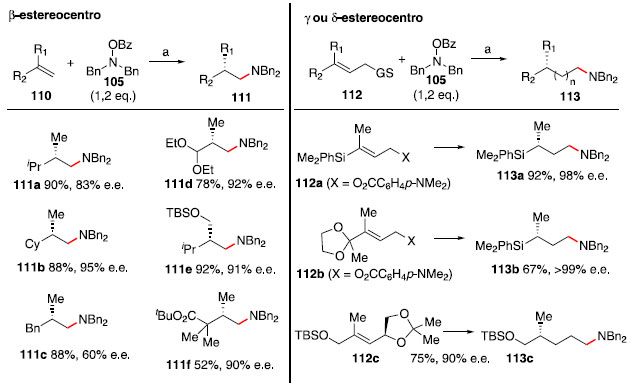 Esquema 18. Formaçao de estereocentros nas posiçoes β, γ e δ a partir de hidroaminaçao de alcenos com catálise de cobre. Condiçao a: Cu(OAc)2 (2 mol%), (R)-DTBM-SEGPHOS (2,2 mol%), HSiMe(EtO)2 (2,0 - 5,0 eq.), THF. GS = grupo de saída
A regiosseletividade anti-Markovnikov observada na formaçao de 111a-111f foi atribuída a fatores estéricos.55 O cobre se ligaria à posiçao menos substituída, e após etapas de adiçao oxidativa com o composto 105 e eliminaçao redutiva, a amina se instalaria no carbono anteriormente ligado ao cobre. A proposta de mecanismo para a formaçao dos compostos 113a-113c passa por uma hidrocupraçao assimétrica de 112 seguida de eliminaçao do alcóxido ou carboxilato em β, formando o alceno terminal 116 (para 112c, ocorreria uma segunda sequência de hidrocupraçao e β-eliminaçao). Na próxima etapa, a olefina 116 sofreria uma hidrocupraçao com seletividade anti-Markovnikov,55 e após uma sequência de adiçao oxidativa/eliminaçao redutiva o fragmento dibenzilamina seria instalado. O hidrosilano é responsável pela reduçao dos intermediários 117 e 120, permitindo o uso catalítico do hidreto de cobre 114 (Esquema 19).
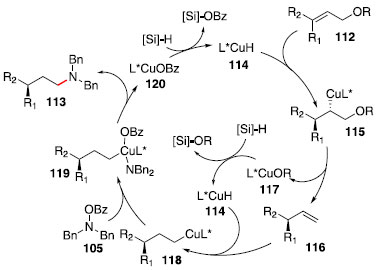 Esquema 19. Proposta mecanística para hidroaminaçao redutiva de trans ferência
Hidroaminaçao formal de olefinas com nitroarenos: catálise por hidretos de ferro Em 2014, Baran e colaboradores relataram um protocolo para formaçao de ligaçao C-C via acoplamento redutivo de olefinas.56,57 Esse método notável utilizou etanol como solvente, catalisadores de ferro (III) como Fe(acac)3 e Fe(dibm)3 (acac = acetilacetonato, dibm = diisobutirilmetano) e fenilsilano como redutor, o que impacta em uma reaçao de baixo custo e que nao necessita de exclusao de umidade ou oxigênio. Em 2015, durante a aplicaçao desse protocolo em um composto nitroaromático, foi observada a formaçao de um subproduto com a formaçao de uma ligaçao C-N, uma anilina alquilada. Ao investigar essa reaçao inesperada, foi observado que o grupo nitro estaria sendo reduzido para uma espécie nitrosoareno 124 e, após acoplamento com o radical derivado da olefina 122, formaria a ligaçao C-N. Uma etapa adicional de reduçao com zinco metálico e soluçao de HCl(aq) foi realizada no mesmo frasco reacional para otimizar o rendimento (Esquema 20).58
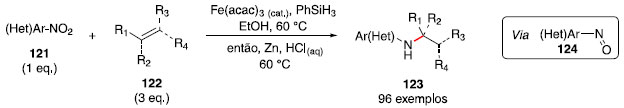 Esquema 20. Acoplamento entre nitroaromáticos e olefinas
Essa metodologia se mostrou compatível com diversos grupos funcionais, incluindo triflatos e iodetos de arila, tioéteres, álcoois, aminas e cetonas, porém apresentou incompatibilidade com substratos contendo fenóis e tiofenóis. Um dos substratos estudados (composto 125) se destaca pelo fato de ser um intermediário na síntese de um inibidor de transcriptase reversa HIV-1, e a metodologia estudada permite a transformaçao de 125 em 126 em apenas uma etapa, em rendimento comparável ao método convencional, que utiliza três etapas e reagentes mais caros, incluindo o cloreto de propargila 127, que também é sensível à umidade (Esquema 21). Outro exemplo é a preparaçao do fragmento 130, em menos etapas e tempo, em rendimento similar à rota convencional (Esquema 21).
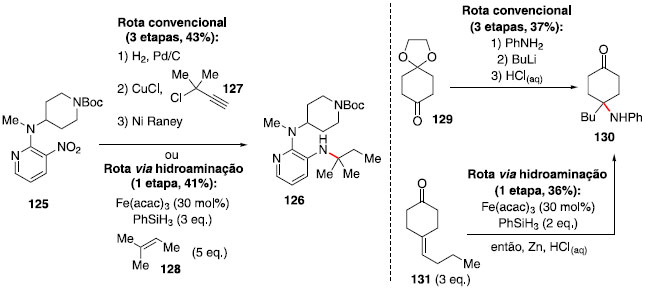 Esquema 21. Síntese dos compostos 126 e 130: rota convencional e hidroaminaçao
O mecanismo proposto para essa hidroaminaçao ainda nao está totalmente esclarecido, Baran e colaboradores propuseram a combinaçao de uma espécie nitrosoareno com um radical, derivados do composto nitroaromático e da olefina, respectivamente. Dois subprodutos foram identificados, uma hidroxilamina N-alquilada e uma hidroxilamina N,O-alquilada, assim a etapa de reduçao desses intermediários forneceria a anilina substituída desejada. Em 2016, Zhu, Shaver e Thomas59,60 relataram o uso do catalisador amino-bis(fenolato) de ferro (III) (132) para realizar a hidroaminaçao formal investigada por Baran. Esse catalisador (Figura 2) foi empregado com sucesso em reaçoes de hidrosililaçao61 e polimerizaçao radicalar.62 A carga catalítica pôde ser reduzida de 30 mol% de Fe(acac)3 para apenas 2 mol% de 132, e a temperatura reacional reduzida de 60 °C para temperatura ambiente, mantendo rendimentos similares, mostrando a vantagem com a troca da espécie de ferro (III).
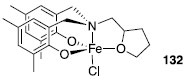 Figura 2. Catalisador amino-bis(fenolato) de ferro (III) (132)
Hidroaminaçao sem metais de transiçao Processos sintéticos que evitam o uso de metais de transiçao sao desejados na indústria farmacêutica, em que mesmo concentraçoes baixas de metais devem ser evitadas no produto final. Assim, a ausência de metais de transiçao na metodologia escolhida pode impactar em um menor custo de purificaçao do material. No caso de organocatalisadores,63 como ácidos fosfóricos, tiouréias, esquaramidas ou derivados de prolina, usualmente nao há problemas com umidade ou oxigênio durante sua manipulaçao, característica que usualmente nao é encontrada em diversos métodos baseados em metais de transiçao. Catálise por ligaçao de hidrogênio Hidroaminaçoes tipo Cope foram aceleradas com uso de solventes próticos,64 e estudos computacionais identificaram ligaçoes de hidrogênio que estabilizariam o estado de transiçao polar dessa reaçao.65 Dessa forma, Jacobsen e colaboradores avaliaram o uso de uma tiouréia simples como doadora de ligaçao de hidrogênio aplicando a teoria do funcional da densidade (DFT - density functional theory). Os autores observaram com uso da tiouréia, uma energia de ativaçao para a etapa de ciclizaçao concertada 3,1 kcal mol-1 menor do que para a reaçao sem a tiouréia, indicando que tal espécie poderia ser utilizada como um catalisador desta reaçao (Figura 3).66
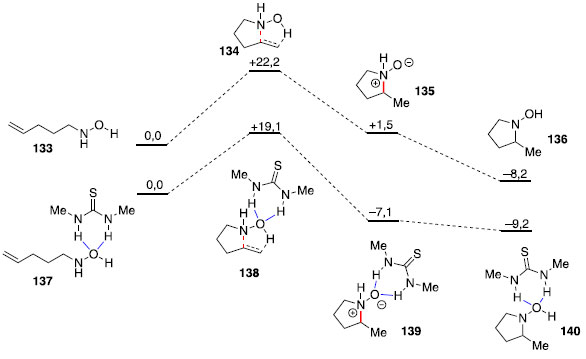 Figura 3. Energias calculadas para estruturas otimizadas da reaçao de hidroaminaçao de Cope, método B3LYP/6-311+G(d,p), energias em kcal.mol-1. Li gaçoes de hidrogênio estao destacadas em azul
Em um trabalho prévio de Uyeda e Jacobsen, foi identificado um efeito combinado de ligaçao de hidrogênio e estabilizaçao secundária do estado de transiçao dipolar em rearranjos enantiosseletivos de Claisen.67 Assim, diversas tiouréias contendo anéis aromáticos ricos eletronicamente (que poderiam estabilizar a carga parcial positiva desenvolvida no nitrogênio durante a ciclizaçao) foram avaliadas para a hidroaminaçao de Cope. Dentre os catalisadores avaliados, o melhor resultado foi obtido com a tioureia 142, que contém um substituinte diarilpirrol. Esse catalisador foi avaliado para hidroaminaçao intramolecular do composto 141, fornecendo a pirrolidina 143 em 83% de rendimento e elevado excesso enantiomérico (Esquema 22).
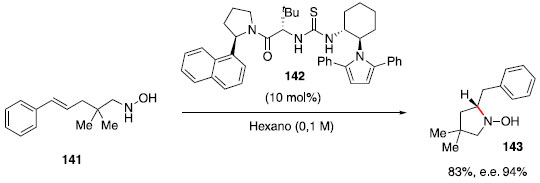 Esquema 22. Hidroaminaçao tipo Cope catalisada pela tiouréia 142
O uso de substituintes p-Me, p-Cl, m-Cl, o-Cl e p-MeO no anel aromático de 141 nao influenciaram significativamente na alteraçao do rendimento e excesso enantiomérico dos produtos ciclizados. Catálise por ácido de Bronsted Outra frente explorada para reaçoes de hidroaminaçao sem uso de metais de transiçao faz uso de ácidos de Bronsted. Em 2002, Schlummer e Hartwig relataram o uso de ácidos para a ciclizaçao de aminoalcenos contendo o nitrogênio ligado a um grupo tosila,68 e identificaram que os ácidos sulfúrico e tríflico seriam suficientemente fortes para catalisar a reaçao. Um possível mecanismo passaria pela protonaçao do alceno 144, na sequência, o carbocátion 146 seria atacado pelo nitrogênio de forma intramolecular e após perda do próton, seria formada a pirrolidina 148 (Esquema 23). O uso de aminas protegidas na forma de carbamatos ou aminas livres com os ácidos tríflico ou sulfúrico nao levaram ao produto de hidroaminaçao.
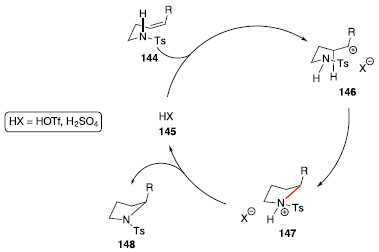 Esquema 23. Ciclizaçao de aminoalcenos catalisada por ácido de Bronsted
Em 2008, Ackermann e Althammer relataram o uso de ácidos fosfóricos para formaçao de pirrolidinas via hidroaminaçao, porém, o uso do catalisador enantiopuro levou à baixa discriminaçao das faces do carbocátion intermediário, levando ao produto com 17% de excesso enantiomérico.69 Apesar de diversos ácidos de Bronsted terem sido estudados, as principais reaçoes altamente enantiosseletivas que os utilizam envolvem ativaçao de uma ligaçao carbono-heteroátomo ou heteroátomo-heteroátomo, usualmente uma carbonila ou imina.70,71 A protonaçao de uma carbonila ou imina gera uma espécie que pode fazer uma ligaçao de hidrogênio com a base conjugada do ácido de Bronsted enantiopuro, mantendo a informaçao quiral próxima ao centro eletrofílico reativo e auxiliando na organizaçao molecular que favorece um dos estados de transiçao diastereoisoméricos. Por outro lado, a protonaçao de um alceno leva a um carbocátion, apesar da base conjugada quiral poder se manter próxima por interaçoes eletrostáticas, a rigidez dessa interaçao é consideravelmente menor do que no caso anterior, resultando em uma baixa discriminaçao das faces enantiotópicas. Para atacar esse desafio, Toste e colaboradores reportaram uma estratégia, em 2011, baseada no uso de dienos e ácidos ditiofosfóricos.72 Esses ácidos de Bronsted seriam fortes o suficiente para protonar o dieno e a base conjugada seria nucleofílica, podendo formar uma ligaçao C-S. Em seguida, o nitrogênio poderia realizar uma reaçao SN2' enantiosseletiva, formando a pirrolidina enantioenriquecida e liberando a base quiral. Um exemplo dessa reaçao é mostrado a seguir no Esquema 24.
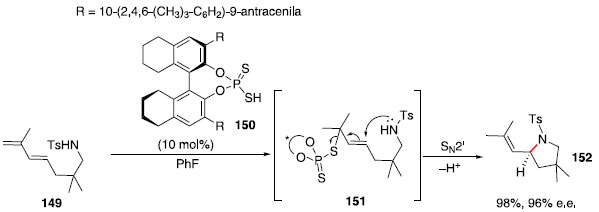 Esquema 24. Hidroaminaçao intramolecular catalisada por ácido ditiofosfórico
Construçao de alcaloides indolizidínicos A classe de alcaloides indolizidínicos ([4.3.0]-1-azabiciclos) possui grande diversidade e complexidade estrutural,73 atraindo a atençao de diversos químicos sintéticos. Assim, uma estratégia envolvendo bis-hidroaminaçao simplificaria consideravelmente o desafio sintético. Em vista disso, e da dificuldade de obter tal transformaçao com métodos conhecidos até entao, Shenvi e colaboradores estudaram um protocolo de bis-hidroaminaçao formal.74,75 A reaçao a partir da amina 153 envolve o uso de BH3•DMS e iodo molecular para realizar a hidroboraçao regiosseletiva das duplas trissubstituídas, gerando o intermediário 156. Para essa primeira etapa, foi observada ser essencial a adiçao lenta de iodo sob exclusao de luz, evitando assim a quebra da ligaçao potencialmente fotolábil B-I. Com a adiçao de NaOMe e uma segunda porçao de I2 foi possível promover uma migraçao, formando o intermediário pirrolidina 158, e após tratamento oxidativo o álcool foi instalado, fornecendo o composto 159. Para a formaçao do segundo ciclo, um protocolo baseado na reaçao de Mitsunobu foi utilizado (Esquema 25).76
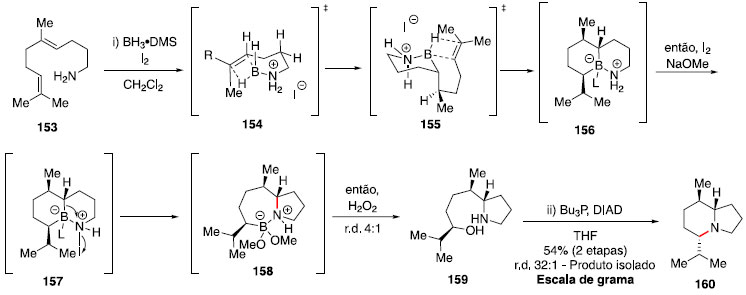 Esquema 25. Formaçao da indolizina 160 via hidroaminaçao formal
Os estados de transiçao 154 e 155, contendo ciclos de seis membros na geometria de bote, foram propostos com base na estereoquímica observada no produto final majoritário. Essa estratégia foi aplicada na síntese formal da lepadiformina (166) por Tabor e Shenvi (Esquema 26).75 Esse alcaloide de origem marinha apresenta uma estrutura tricíclica congestionada, o que torna um desafio o uso da metodologia apresentada.
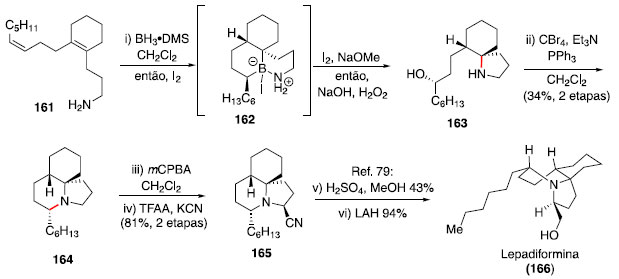 Esquema 26. Síntese do alcaloide lepadiformina via hidroaminaçao formal
Assim, a partir do dieno 161,77 foi realizado o protocolo de hidroaminaçao para formaçao do biciclo 163,78 e o último ciclo presente no produto natural foi forjado através de uma reaçao nas condiçoes de Appel. A síntese do intermediário 165 empregou uma α-cianaçao oxidativa, relatada anteriormente por Rychnovsky e colaboradores,79 que também conduziram a conclusao da síntese de 166, por meio da metanólise do grupo nitrila e reduçao completa com LiAlH4 (Esquema 26).
CONCLUSOES E PERSPECTIVAS As reaçoes de hidroaminaçao passaram por grandes avanços e contam com métodos que permitem diferentes regiosseletividades e estratégias enantiosseletivas. Métodos de hidroaminaçoes formais que permitem a utilizaçao de metais mais baratos como cobre e ferro estao sendo desenvolvidos com sucesso, tornando a síntese mais atrativa economicamente. O uso de métodos livre de metais de transiçao também é notável, utilizando conceitos estabelecidos, como ativaçao por ligaçao de hidrogênio e uso de ácidos de Bronsted. Um dos grandes desafios ainda se encontra no uso direto de amônia nesse tipo de reaçao, um importante passo para esse desenvolvimento foi o uso de catálise de Au (I) relatado por Bertrand e colaboradores.16 No entanto, usualmente essa tarefa tem sido atacada de modo indireto, por exemplo, pelo uso de Ph2C=NH com catálise de ródio31 e Bn2NOBz com catálise de cobre,51,53,54 os quais podem levar ao produto formal de uma hidroaminaçao com amônia após uma etapa extra, hidrólise da imina ou hidrogenaçao da dibenzilamina, respectivamente. Outro desafio que vem sendo discutido é a necessidade de evitar o uso de um dos parceiros (amina ou composto insaturado) em excesso para reaçoes intermoleculares, pois isso impactaria em uma perda da economia atômica, que é uma das grandes vantagens de utilizar reaçoes de hidroaminaçao. Diversos mecanismos têm sido propostos para essa classe de reaçoes, porém, há falta de estudos mecanísticos detalhados, o que pode impactar na dificuldade de propor novos sistemas catalíticos de modo racional. Nesse contexto, a utilizaçao de métodos computacionais pode auxiliar na compreensao e previsao de açao dos catalisadores.
AGRADECIMENTOS Os autores agradecem o apoio financeiro da Fundaçao de Amparo à Pesquisa do Estado de Sao Paulo (FAPESP, processos n° 2014/26378-2, 2014/25770-6, 2015/08199-6), da CAPES e do CNPq. Os autores agradecem a revisao do texto realizada pela Dra. Carolina M. A. Sant'Ana e M.a Mariana C. Frojuello.
REFERENCIAS 1. Sorribes, I.; Junge, K.; Beller, M.; J. Am. Chem. Soc. 2014, 136, 14314. 2. Ju, Y.; Varma, S.; J. Org. Chem. 2006, 71, 135. 3. Horn, M.; Mayr, H.; Lacôte, E.; Merling, E.; Deaner, J.; Wells, S.; McFadden, T.; Curran, P.; Org. Lett. 2012, 14, 82. 4. Dangerfield, M.; Plunkett, H.; Win-Mason, L.; Stocker, L.; Timmer, M.; J. Org. Chem. 2010, 75, 5470. 5. Sheehan, C.; Hess, P.; J. Am. Chem. Soc. 1955, 77, 1067. 6. Dine, M.; Erb, W.; Berhault, Y.; Roude, J.; Blanchet, J.; J. Org. Chem. 2015, 80, 4532. 7. Surry, S.; Buchwald, L.; Angew. Chem., Int. Ed. 2008, 47, 6338. 8. Hartwig, F.; Acc. Chem. Res. 2008, 41, 1534. 9. Para revisao sobre hidroaminaçao catalisada por metais de transiçao tardios, ver: Huang, L.; Arndt, M.; Gooβen, K.; Heydt, H.; Gooβen, L. J.; Chem. Rev. 2015, 115, 2596. 10. Para revisao sobre hidroaminaçao de alcenos, ver: Bernoud, E.; Lepori, C.; Mellah, M.; Schulz, E.; Hannedouche, J.; Catal. Sci. Technol. 2015, 5, 2017. 11. Anastas, T.; Warner, C.; Green Chemistry: Theory and Practice, Oxford University Press: New York, 1998. 12. Johns, M.; Sakai, N.; Ridder, A.; Hartwig, F.; J. Am. Chem. Soc. 2006, 128, 9306. 13. Os 12 princípios de química verde estao detalhados na referência 11. 14. Shen, Q.; Hartwig, F.; J. Am. Chem. Soc. 2006, 128, 10028. 15. Surry, S.; Buchwald, L.; J. Am. Chem. Soc. 2007, 129, 10354. 16. Lavallo, V.; Frey, D.; Donnadieu, B.; Soleihavoup, M.; Bertrand, G.; Angew. Chem. Int. Ed. 2008, 47, 5224. 17. Mizushima, E.; Hayashi, T.; Tanaka, M.; Org. Lett. 2003, 5, 3349. 18. Howk, W.; Little, L.; Whitman, M.; J. Am. Chem. Soc. 1954, 76, 1899. 19. Pohlki, F.; Doye, S.; Angew. Chem., Int. Ed. 2001, 40, 2305. 20. Straub, F.; Bergman, G.; Angew. Chem., Int. Ed. 2001, 40, 4768. 21. Tobisch, S.; Chem. Eur. J. 2007, 13, 4884. 22. Mujahidin, D.; Doye, S.; Eur. J. Org. Chem. 2005, 2005, 2689. 23. Composto 27 foi sintetizado em um total de 7 etapas a partir de reagentes comerciais. 24. Manna, K.; Xu, S.; Sadow, D.; Angew. Chem., Int. Ed. 2011, 50, 1865. 25. Nagano, T.; Kobayashi, S.; J. Am. Chem. Soc. 2008, 131, 4200. 26. Anderson, E.; Overman, E.; J. Am. Chem. Soc. 2003, 125, 12412. 27. Pattillo, C.; Strambeanu, I.; Calleja, P.; Vermeulen, A.; Mizuno, T.; White, C.; J. Am. Chem. Soc. 2016, 138, 1265. 28. Ngai, Y.; Barchuk, A.; Krische, J.; J. Am. Chem. Soc. 2007, 129, 12644. 29. Patil, T.; Wu, H.; Kadota, I.; Yamamoto, Y.; J. Org. Chem. 2004, 69, 8745. 30. Qiu, S.; Wei, Y.; Liu, G.; Chem. Eur. J. 2009, 15, 2751. 31. Xu, K.; Wang, H.; Khakyzadeh, V.; Breit, B.; Chem. Sci. 2016, 7, 3313. 32. Stork, G.; Landesman, K.; J. Am. Chem. Soc. 1956, 78, 5128. 33. Mukherjee, S.; Yang, W.; Hoffmann, S.; List, B.; Chem. Rev. 2007, 107, 5471. 34. Uchimaru, Y.; Chem. Commun. 1999, 1133. 35. Sakai, K.; Kochi, T.; Kakiuchi, F.; Org. Lett. 2011, 13, 3928. 36. Wakatsuki, Y.; J. Organomet. Chem. 2004, 689, 4092. 37. Siengalewicz, P.; Rinner, U.; Mulzer, J.; Chem. Soc. Rev. 2008, 37, 2676. 38. Kahsai, W.; Zhu, S.; Wardrop, J.; Lane, S.; Fenteany, G.; Chem. Biol. 2006, 13, 973. 39. Takahashi, K.; Tomita, F.; J. Antibiot. 1983, 36, 468. 40. Chiba, H.; Oishi, S.; Fujii, N.; Ohno, H.; Angew. Chem., Int. Ed. 2012, 51, 9169. 41. Composto 84 foi sintetizado em um total de 23 etapas a partir de reagentes comerciais. 42. Allan, M.; Stoltz, M.; J. Am. Chem. Soc. 2008, 130, 17270. 43. Prier, K.; Rankic, A.; MacMillan, C.; Chem. Rev. 2013, 113, 5322. 44. Musacchio, J.; Nguyen, Q.; Beard, H.; Knowles, R.; J. Am. Chem. Soc. 2014, 136, 12217. 45. Crabtree, H.; Davis, W.; J. Org. Chem. 1986, 51, 2655. 46. Para uma revisao sobre grupos diretores, ver: Rousseau, G.; Breit, B.; Angew. Chem., Int. Ed. 2011, 50, 2450. 47. Kawasaki, Y.; Ishikawa, Y.; Igawa, K.; Tomooka, K.; J. Am. Chem. Soc. 2011, 133, 20712. 48. Smith, M.; Takacs, M.; J. Am. Chem. Soc. 2010, 132, 1740. 49. Murphy, K.; Bruch, A.; Dong, M.; Angew.Chem., Int. Ed. 2014, 53, 2455. 50. Lipshutz, H.; Frieman, A.; Angew. Chem., Int. Ed. 2005, 44, 6345. 51. Xi, Y.; Butcher, W.; Zhang, J.; Hartwig, F.; Angew. Chem., Int. Ed. 2016, 55, 776. 52. A estereoquímica relativa foi determinada após conversao de 109 no cloridrato de 3-aminociclopentanol e comparaçao dos dados de RMN de 1H com amostras autênticas dos isômeros cis e trans. 53. Zhu, S.; Buchwald, L.; J. Am. Chem. Soc. 2014, 136, 15913. 54. Zhu, S.; Niljianskul, N.; Buchwald, L.; Nat. Chem. 2016, 8, 144. 55. Shi, L.; Buchwald, L.; Nat. Chem. 2015, 7, 38. 56. Lo, C.; Yabe, Y.; Baran, S.; J. Am. Chem. Soc. 2014, 136, 1304. 57. Lo, C.; Gui, J.; Yabe, Y.; Pan, M.; Baran, S.; Nature 2014, 516, 343. 58. Gui, J.; Pan, M.; Jin, Y.; Qin, T.; Lo, C.; Lee, J.; Spergel, H.; Mertzman, E.; Pitts, J.; Cruz, E.; Schmidt, A.; Darvatkar, N.; Natarajan, R.; Baran, S.; Science 2015, 348, 886. 59. Zhu, K.; Shaver, P.; Tomas, P.; Chem. Asian J. 2016, 11, 977. 60. Zhu, K.; Shaver, P.; Tomas, P.; Chem. Sci. 2016, 7, 3031. 61. Zhu, K.; Shaver, P.; Tomas, P.; Eur. J. Org. Chem. 2015, 2015, 2119. 62. Schroeder, H.; Lake, M.; Demeshko, S.; Shaver, P.; Buback, M.; Macromolecules 2015, 48, 4329. 63. Para revisoes sobre organocatálise, ver ediçao especial: Chem. Rev. 2007, 107. 64. Moran, J.; Gorelsky, I.; Dimitrijevic, E.; Lebrun, E.; Bédard, C.; Séguin, C.; Beauchemin, M.; J. Am. Chem. Soc. 2008, 130, 17893. 65. Acevedo, O.; Jorgensen, L.; J. Am. Chem. Soc. 2006, 128, 6141. 66. Brown, R.; Uyeda, C.; Brotherton, A.; Jacobsen, N.; J. Am. Chem. Soc. 2013, 135, 6747. 67. Uyeda, C.; Jacobsen, N.; J. Am. Chem. Soc. 2011, 133, 5062. 68. Schlummer, B.; Hartwig, F.; Org. Lett. 2002, 4, 1471. 69. Ackermann, L.; Althammer, A.; Synlett 2008, 995. 70. Hatano, M.; Maki, T.; Moriyama, K.; Arinobe, M.; Ishihara, K.; J. Am. Chem. Soc. 2008, 130, 16858. 71. Sai, M.; Yamamoto, H.; J. Am. Chem. Soc. 2015, 137, 7091. 72. Shapiro, D.; Rauniyar, V.; Hamilton, L.; Wu, J.; Toste, D.; Nature 2011, 470, 245. 73. Michael, P.; Nat. Prod. Rep. 2008, 25, 139. 74. Pronin, V.; Tabor, G.; Jansen, J.; Shenvi, A.; J. Am. Chem. Soc. 2012, 134, 2012. 75. Tabor, G.; Shenvi, A.; Org. Lett. 2015, 17, 5776. 76. A estereoquímica relativa de 160 foi determinada a partir da estrutura de raio-X do respectivo cloridrato. 77. Composto 161 foi sintetizado em um total de 6 etapas a partir de reagentes comerciais. 78. A estereoquímica relativa de 163 foi confirmada a partir de análise de raio-X do respectivo derivado 3,5-dinitrobenzoato-3,5-dinitrobenzamida. 79. Perry, A.; Morin, D.; Slafer, W.; Rychnovsky, D.; J. Org. Chem. 2012, 77, 3390. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access