
 |
 |
 |
 |
 |
Artigo
|
|
| Fixação de CO2 em efluente salino de indústria petroquímica Fixation of CO2 in brines of petrochemical industry |
|
Leonardo F. Santos*; André L. A. Santos; Sônia D. F. Rocha#
Departamento de Engenharia de Minas, Escola de Engenharia, Universidade Federal de Minas Gerais, Av. Antônio Carlos 6627, prédio da Escola de Engenharia - bloco II, Sala 4135, Pampulha, 31270-901 Belo Horizonte - MG, Brasil Recebido em 11/11/2016 *e-mail: leobh.fs@gmail.com In various industrial processes, such as the process of obtaining potassium by solution mining or in the oil industry, fuels are burnt concurrently to the formation of saline effluents. The opportunity to sequester carbon through capturing the CO2 from the flue gas by carbonation of these effluents and reducing the potential of scaling of the water is evident. The use of saline industrial effluent from electrodialysis for carbon sequestration by mineralization of carbon dioxide is the focus of this work. The carbonation reaction of saline effluent of reverse electrodialysis, of low ionic strength and higher CO2 solubility, was simulated through OLI®. Precipitation tests were performed by bubbling 10-20 NL h-1 CO2 in a glass reactor containing the brine and pH adjusted to 8.9. The solid was characterized and the results from the simulation and the experiments were compared. The removal of calcium and strontium in the form of carbonates reach out up to 98% and 75%. The simulation results were in line with the experimental results for the calcium behavior, but not for strontium. The OLI® simulation was powerful to represent the calcium carbonate formation, however, comparisons between predicted Sr removal and testwork results presented differences and must be investigated. INTRODUÇÃO Sequestro de carbono O principal gás de efeito estufa, devido à grande quantidade gerada e também à sua influência na intensidade do próprio aumento da temperatura global, é o dióxido de carbono ou gás carbônico (CO2).1,2 O gás carbônico compõe, aproximadamente, 0,04% da atmosfera terrestre. Entretanto, sua contribuição sobre os efeitos do aquecimento global é grande. Estimativas apontam que 60% do efeito estufa é resultado da quantidade total de CO2 presente na atmosfera.3 Por apresentar absorções vibracionais na frequência do infravermelho há o bloqueio da radiação térmica na Terra.4 Estudos têm sido desenvolvidos para reduzir a emissão de CO2 através do uso de várias tecnologias. O conjunto de medidas para capturar o dióxido de carbono emitido, mudar seu estado físico de tal forma que se possa transportá-lo e ainda as diversas formas de disposição final para este gás é identificado na literatura como Captura e Estocagem de Carbono (CEC).2,5,6 De acordo com a Agência Internacional de Energia (IEA), as tecnologias de CEC deverão ser responsáveis por uma redução de 12% nas emissões de carbono, representando um total de 94 Gton de CO2 até 2050, em um cenário necessário para a diminuição da temperatura média da atmosfera em 2 ºC.7 A captura do gás carbônico e o processo intrínseco de combustão são apresentados na literatura com destaque para três tecnologias:8
No entanto, todos os métodos discriminados anteriormente apresentam como desvantagem o uso adicional de energia comparado aos processos de combustão conhecidos, porém sem tecnologia de captura dos gases de combustão. A empresa precisa visualizar o valor econômico na redução de suas emissões de gás carbônico para que haja interesse no uso destas tecnologias ou que a legislação determine, de alguma maneira, que estes processos sejam incluídos na linha de produção.5 Assim, o uso de tais métodos ainda carecem de maiores investimentos e investigações para que possam ser amplamente aceitos no âmbito industrial e político. A fixação de CO2 na forma de carbonatos é outra forma de captura de carbono. Através deste método o dióxido de carbono reage com minerais ou soluções salinas para a produção de carbonatos. Esta reação pode ser feita in-situ ou ex-situ.9,10 No processo in-situ, o CO2 é injetado diretamente em rochas, aquíferos ou mesmo em regiões profundas do oceano, onde estão presentes minerais ou compostos de magnésio e cálcio, para a produção de novos compostos carbonatados insolúveis. No entanto, os custos de manutenção para estocagens de longo prazo e também de monitoramento podem levar a valores proibitivos.10 As vantagens do processo ex-situ sobre a estocagem geológica ou em oceanos incluem maior capacidade e tempos mais longos de estocagem, menor risco de vazamentos e até mesmo produção de energia devido à possíveis reações exotérmicas envolvidas.11 Em processo ex-situ, o sequestro de carbono pode se dar em meio aquoso ou não. O gás carbônico pode, por exemplo, ser reagido com sólidos como CaO ou MgO para finalidades específicas e assim gerar uma camada de carbonato em sua superfície.12 Outra forma pode ser a carbonatação direta de silicatos de magnésio pelo dióxido de carbono, embora altas pressões (100-150 bar) sejam exigidas para que a reação direta gás-sólido aconteça.9 A cinética de carbonatação em meio aquoso pode ser mais rápida e apresenta taxas de conversão maiores.13 A exploração da reação com o CO2 em meio aquoso rico em compostos de magnésio como forma de sequestro de dióxido de carbono é descrita na literatura como eficaz, rápida e com poucos riscos de liberação do gás carbônico após a formação dos carbonatos devido à estabilidade destes compostos.14-16 Dentre os compostos que podem ser formados se destaca o carbonato de magnésio, com estrutura da nesqueonita (MgCO3·3H2O), pela possibilidade de formação a temperatura ambiente (25 a 35 ºC) e cinética rápida de reação. O cloreto de magnésio, principal composto utilizado na reação de carbonatação e formação do MgCO3·3H2O, é abundantemente encontrado em salmouras naturais e efluentes industriais, tais como efluentes de lavras por dissolução de potássio ou mesmo água do mar.15,17 Diversos efluentes salinos, oriundos de vários processos industriais, são fontes potenciais para o sequestro de carbono ex-situ. Além disso, a geração de gás carbônico na queima de combustíveis fósseis e os efluentes salinos constituem um binômio encontrado na indústria do petróleo e outros processos industriais. Por exemplo: o processo de cristalização evaporativa para produção de KCl a partir da lavra por dissolução da carnalita envolve o consumo de energia para o processo de cristalização e também produz um efluente salino rico em MgCl2.18 Também, concentrados salinos são gerados em processos de tratamento de água como osmose reversa ou eletrodiálise reversa em processo de refino de petróleo concomitantemente a queima de combustíveis fósseis para utilização de energia no processo de refino.19 Portanto, a fixação do CO2 contido no gás de combustão nos próprios efluentes gerados na indústria é uma opção a ser considerada pelas indústrias onde esta oportunidade é presente. O sequestro de carbono e os efluentes salinos A exploração da reação de carbonatação em meio aquoso rico em compostos de magnésio e cálcio, como forma de sequestro de dióxido de carbono, está largamente descrita na literatura.14-16,20 O dióxido de carbono reage com o cátion de metais alcalinos contido em salmouras segundo a sequência de reações apresentadas pelas Equações 1 a 8:21 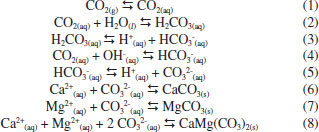 No entanto, há outra possibilidade para a reação de formação de carbonato de cálcio com estrutura de calcita (Equação 9):22 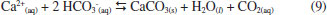 A reação representada pela Equação 9 é mais provável na faixa de pH de predominância do íon bicarbonato em meio aquoso, entre 6,3 e 10,3. Essa predominância pode variar em salmouras, dependendo da concentração e tipo de ligantes envolvidos. À medida que o meio se torna mais básico, a predominância do íon carbonato determina a reação da calcita pela reação apresentada na Equação 6, em pH acima de 10,3.22 Com relação ao magnésio, e no caso particular do carbonato de magnésio (nesqueonita), a literatura descreve uma faixa de pH ótima utilizando apenas CO2 puro e NH3 como agentes modificadores do pH.15 Observou-se que entre pH 7,8 e 8,2 a nesqueonita é formada em reações salinas onde o único componente era o MgCl2·6H2O.15 Esta faixa de pH reforça a hipótese de o mecanismo de reação envolver o íon bicarbonato, semelhante ao demonstrado na Equação 9. A hipótese mecanística para a reação de formação do carbonato de magnésio na forma de nesqueonita é apresentada por pesquisadores como sendo:17 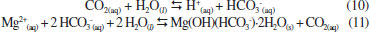 Esta hipótese deriva diretamente de cálculos termodinâmicos realizados pelos autores que apontam o pH para precipitação do carbonato de magnésio (nesqueonita) como sendo acima de 6,8.17 Portanto, a precipitação ocorre na presença do íon bicarbonato e não do carbonato. Estudos de precipitação de magnésio sob a forma de nesqueonita foram realizados também para diferentes concentrações iniciais de Mg na salmoura, a saber: 7, 16 e 32 g L-1.16 Esses estudos reportaram que o aumento da concentração de Mg favorece a precipitação de formas amorfas e hidratadas de carbonato de magnésio em detrimento da precipitação de sua forma cristalina. Entretanto, tais fases mostraram-se metaestáveis e, nas condições de temperatura avaliadas, estas fases transformam-se em nesqueonita. Em casos mais extremos a cinética de precipitação pode ser retardada de tal forma que podem ser necessários dias até que a forma cristalina do carbonato de magnésio seja detectada como produto formado.16 Por fim, como parte importante na previsão dos meios salinos estarem aptos ao sequestro de carbono, a solubilidade do CO2 influenciará na capacidade que as salmouras podem exibir para reter o gás carbônico e o tornarem disponível nas diversas possibilidades de especiação segundo a variação do pH do meio. Em pesquisa recente, um estudo, em escala de bancada, da solubilidade do CO2 em salmouras contendo NaCl e CaCl2 e a extensão das conclusões para modelos contendo Mg2+, K+ e SO42- foi comparado aos modelos computacionais disponíveis através de softwares.23 Dentre estes softwares o OLI® Studio, versão 9.0.6, apresentou desvios médios absolutos para a solubilidade do gás carbônico nos sistemas estudados entre 1,85 a 9,48% em relação aos dados experimentais. O objetivo deste trabalho é investigar o potencial da reação de carbonatação de salmouras sintéticas semelhantes às salmouras oriundas do processo de eletrodiálise reversa da indústria petroquímica, discutindo aspectos sobre a conversão obtida em escala de bancada, identificação dos sólidos formados e outros aspectos vantajosos como a redução do potencial de incrustação para reuso da água. Desta forma, avaliou-se a possibilidade do aproveitamento deste tipo efluente para mineralizar o gás carbônico.
PARTE EXPERIMENTAL Simulação Para se investigar a eficácia e possível aplicação da fixação do CO2, o concentrado salino de eletrodiálise reversa foi escolhido. Há outros efluentes possíveis, no entanto, a salmoura produzida como resíduo do processo de eletrodiálise reversa pode exibir de baixo grau de salinidade, com força iônica da ordem de 0,1 mol kg-1 ou inferior, o que poderia favorecer a simulação e o desenvolvimento da metodologia experimental.19 A reação da salmoura, com concentrações médias de elementos determinadas na literatura,19 (Tabela 1) com o dióxido de carbono foi simulada no software OLI®, versão 9.3, para previsão do comportamento dos produtos de reação em relação ao pH e temperatura. O OLI® utiliza de equações do modelo de Helgeson para estimar os coeficientes de atividade.19 Também, possui uma formulação termodinâmica para equações utilizando arranjos de Helgeson e colaboradores para os termos no estado padrão e de Bromley, Zemaitis, Pitzer, Debye-Huckel, dentre outros, para os demais termos.19
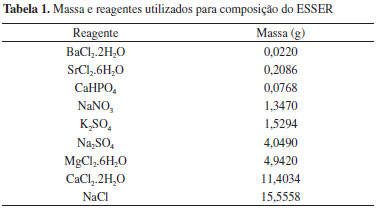
Efluente salino sintético de eletrodiálise reversa As reações de carbonatação foram realizadas com efluente salino sintético de eletrodiálise reversa (ESSER) sintético, preparado em laboratório com composição característica dos concentrados produzidos durante a eletrodiálise reversa real.19 Foi preparada uma solução sintética (10 L) do efluente, cujos sais e suas massas foram pesados em balança analítica Shimadzu AY220 e utilizados para a preparação. Os resultados são apresentados na Tabela 1. As massas expressas na Tabela 1 foram determinadas com base em valores médios apresentados na literatura para cada um dos componentes da solução (Ba, Sr, Ca, Na, K, Mg, Cl, NO3-, PO42- e SO42-).19 Foram utilizados reagentes grau P.A., porém sem purificação ou qualquer outra preparação anterior a pesagem e a solubilização dos sais em água destilada. Reação de carbonatação As reações de carbonatação foram desenvolvidas em reator de vidro com capacidade para 1 L de solução sintética de efluente. Como fonte de CO2 foi utilizado gás carbônico comercializado pela OXIPRESS, com válvula de controle de gás duplo estágio e cilindro de capacidade de 9 kg. Além da válvula duplo estágio, a vazão de gás foi controlada através de válvula tipo agulha acoplada a um rotâmetro. O reator contendo o efluente sintético foi mantido em banho termostático com circulação de água e foi mantida sob agitação de 200 rpm através de agitador mecânico com haste revestida em polipropileno. O pH e a temperatura interna do reator foram monitorados através de eletrodo de Ag/AgCl da marca ANALION e termopar conectados à um medidor de pH TECNOPON MPA-210. O gás carbônico foi solubilizado inserindo-o no reator contendo o ESSER, através de uma mangueira de TEFLON®, com vazão entre 10 e 20 NL h-1 durante o tempo necessário para que o pH se estabilizasse entre 4,2 e 4,4. Após a estabilização do pH foi adicionado hidróxido de amônio P.A. para elevação do pH a 8,9. Esta metodologia foi adaptada da literatura.14-16 A escolha da limitação do pH ao máximo de 8,9 ocorre devido à dois motivos principais: (i) para limitar ou mesmo evitar a precipitação de hidróxidos ao invés de carbonatos em pH's mais elevados, o que iria contra o objetivo de se fixar o CO2 em forma estável e definitiva. A precipitação preferencial de hidróxidos poderia manter o íon carbonato na fase aquosa; (ii) devido à Resolução 43/2011 do Conselho Nacional do Meio Ambiente (CONAMA), que estabelece o pH máximo para disposição de efluentes nos corpos hídricos igual a 9.24 Quanto à temperatura, a formação de carbonatos a partir de salmouras é descrita como viável a temperatura ambiente por vários autores.15,17,25 Os experimentos foram realizados em triplicatas, em temperaturas de 25, 30 e 35 ºC. Após a reação de carbonatação, a suspensão foi filtrada, o sólido foi seco em estufa a 60 ºC por 72 h e caracterizado por difratometria de raios-X (DRX), fluorescência de raios-X (FRX) e espectrometria no infravermelho com transformada de Fourier (FTIR). Durante o decorrer da reação foram retiradas amostras e monitorados o pH e a temperatura. As alíquotas foram caracterizadas por espectrometria de absorção atômica (EAA) GBC, modelo AVANTA para os elementos Ca, Sr, Mg, Ba, Na e K.
RESULTADOS E DISCUSSÃO Considerações sobre a solubilidade do CO2 nos efluentes sintéticos Os resultados da simulação em OLI® (v. 9.3) da solubilidade de CO2 em relação ao pH para o efluente salino sintético de eletrodiálise reversa encontra-se na Figura 1.
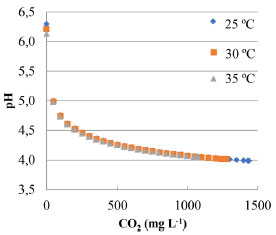 Figura 1. Solubilidade de CO2 e sua relação com o pH na composição do efluente salino sintético de eletrodiálise reversa
A Figura 1 permite observar que, se o pH fosse fixado entre 4,4 e 4,2 após o borbulhamento do CO2, a quantidade solubilizada estaria entre 300 e 600 mg L-1, respectivamente, independentemente da temperatura. Estas concentrações fornecem valores em excesso do gás, suficiente para a reação de carbonatação em relação a quantidade de íons em solução. Assim, estabeleceu-se esse intervalo como controle da quantidade de CO2 a ser solubilizada no ESSER, racionalizando-se o tempo dos experimentos e uso do gás. É importante destacar a influência da salinidade na solubilidade do CO2 salientada pela literatura.23,26,27 Segundo calculado pelo software, para uma solução com força iônica de 5,84 mol kg-1, a solubilidade do gás carbônico a 25 ºC é 454 mg L-1; já para as condições do ESSER, com força iônica calculada de 0,06 mol kg-1, a solubilidade aumenta consideravelmente, atingindo 1429 mg L-1. Simulação da reação de carbonatação do efluente salino sintético de eletrodiálise reversa Os resultados da simulação indicaram a precipitação de fases semelhantes às fases cristalinas de calcita, brucita, hidroxiapatita, estroncianita e barita. Esta predição é resultado de simulação da solução contendo a composição do ESSER, 500 mg L-1 de CO2 e variando-se o pH com adição de NH4OH P.A. A Figura 2 mostra a formação de sólidos como carbonatos de cálcio e de estrôncio, além de um hidroxifostato de cálcio. Nela, observa-se que a partir de pH 6,5 o cálcio começa a precipitar no sistema na forma de carbonato.
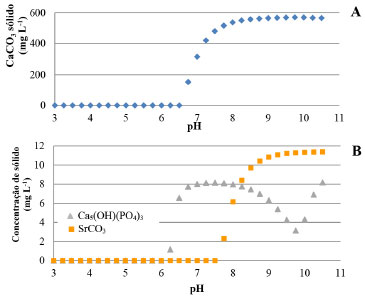 Figura 2. Predição da quantidade de (A) CaCO3 (  ); (B) Ca5(OH)(PO4)3( ); (B) Ca5(OH)(PO4)3( ) e SrCO3 ( ) e SrCO3 ( ) precipitado na reação de carbonatação para o ESSER em relação a variação do pH, 25 ºC simulado pelo OLI® ) precipitado na reação de carbonatação para o ESSER em relação a variação do pH, 25 ºC simulado pelo OLI®
A quantidade máxima de carbonato de cálcio (calcita) precipitado por volume de ESSER predita pela simulação foi de 570,2 mg L-1 em pH 9,75 (Figura 2A). Esta quantidade equivale a 228,1 mg L-1 de Ca removidos da solução na forma de CaCO3. Isto significa que a formação do carbonato de cálcio (calcita) tem um potencial de reduzir em até 98,8% a quantidade de Ca total nas condições determinadas pela simulação. SrCO3 (estroncianita) e Ca5(OH)(PO4)3 (hidroxiapatita) também podem ser formados nas mesmas condições simuladas (Figura 2B). A máxima remoção de carbonato de estrôncio por volume de ESSER ocorre em pH 10, em que há formação de 11,3 mg L-1 de precipitado. Esta quantidade equivale a 6,7 mg L-1 de Sr. A reação de carbonatação nas condições estudadas tem potencial, portanto, para remoção de até 99,6% do estrôncio total na forma de estroncianita. A precipitação do hidróxido de magnésio, sob forma de brucita (Mg(OH)2), também foi predita pela simulação, porém para pH acima de 9,88. Resultados experimentais da reação de carbonatação do ESSER A Figura 3 apresenta as triplicatas das curvas cinéticas de redução das concentrações de cálcio e estrôncio a 30 ºC. As demais curvas seguiram um padrão similar, com uma cinética relativamente rápida. Em 1 hora de reação, a concentração de cálcio sofreu uma redução média de 90% e a de estrôncio de 60%. A precipitação dos compostos de cálcio foi favorecida em relação aos compostos de estrôncio. Após as 24 horas de reação, as concentrações residuais de Ca e Sr não sofreram alteração significativa em relação ao observado com 120 minutos (2 horas).
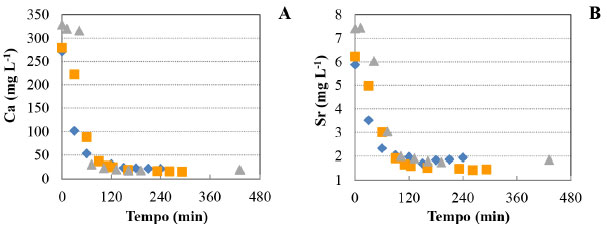 Figura 3. Variação da concentração de Ca (A) e de Sr (B) durante os experimentos de fixação de CO2 por precipitação em efluentes sintético de eletrodiálise reversa a 30 ºC
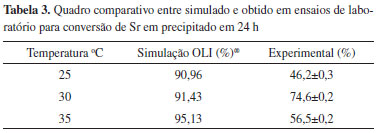
A Tabela 2 apresenta o comparativo da remoção de cálcio predita pela simulação e o obtido em ensaios de bancada em 24 h. Os resultados estão representados pelo intervalo de confiança das médias percentuais de retirada de Ca total da solução inicial, calculadas a um nível de confiança de 95%.
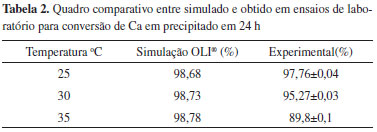
Observa-se uma discrepância entre os resultados experimentais e simulados à medida que a temperatura aumenta, mas com uma concordância boa para as temperaturas de 25 e 30 ºC. Para o estrôncio, segundo valores apresentados na Tabela 3, observa-se que o modelo não foi capaz de reproduzir os valores experimentais, inferiores ao preditos. Este aspecto merece investigação adicional.
Outros elementos, como K e Na, não apresentaram variação significativa em solução após a reação. O Mg apresentou pequena variação em solução aquosa e com resultados variáveis de um ensaio para outro, não tendo sido observado nenhum padrão. A máxima variação de magnésio foi verificada no ensaio a 35 ºC em que a redução observada foi de, aproximadamente, 16% para o efluente final. Caracterização do sólido obtido A análise por fluorescência de raios-X dos sólidos formados nas reações a 25, 30 e 35 ºC é apresentada na Tabela 4. Não foram encontradas diferenças entre os elementos contidos nos sólidos formados sob estas condições de temperatura.

A precipitação de compostos de Mg não era esperada em pH da ordem de 8,9. Conforme simulação, os compostos de Mg precipitariam a partir de pH 9,88. Também a simulação não evidenciou a presença de Cl em fase sólida. Os demais elementos, com exceção do Si, são, provavelmente, advindos de traços contidos nos reagentes. A presença de Si pode ter resultado da necessidade de raspagem do sólido para a filtração, portanto, a presença do Si deve ser oriunda do material do reator. Foram identificados sais com estrutura de calcita (CaCO3), estroncianita (SrCO3), brucita (Mg(OH)2), sulfato duplo hexahidratado de magnésio e amônio ((NH4)2Mg(SO4)2.6H2O), sulfato hidratado de cálcio (CaH2(SO4)2) e cloreto de fosfato e cálcio (Ca2PO4Cl) como fases cristalinas geradas pela reação de carbonatação à 35 ºC (Figura 4A).
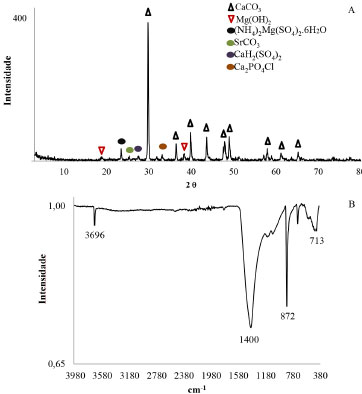 Figura 4. (A) Difratograma (DRX) e (B) Espectrograma Infra Verme lho (FTIR) do sólido da reação de carbonatação à 35 ºC
Os picos em maior número e de maior intensidade (Figura 4A) foram identificados como sendo da estrutura cristalina da calcita, confirmando a predição da simulação onde esta fase de carbonato se formaria facilmente e em maior quantidade. Uma diferença entre a simulação e as fases detectadas pela análise é que, ao invés de hidroxiapatita, a espécie de fosfato detectada pela difratometria de raio-X foi um cloreto de fosfato e cálcio, corroborando as análises de fluorescência de raios-X em que não se esperava a presença de cloretos na fase sólida, porém esses foram detectados. Nas reações a 30 e 25 ºC observou-se uma diferença na intensidade e até a ausência de alguns picos detectados a 35 ºC, mas não foram observadas fases diferentes das já citadas. A análise por infravermelho (Figura 4B) revelou a presença das bandas características do grupo carbonato em 1400 (estiramento assimétrico da ligação C-O), 872 (flexão do CO3 -2), 713 (flexão do CO3 -2) e 1081 cm-1 (estiramento simétrico da ligação C-O).28,29 A absorbância detectada em 3696 cm-1 é típica do estiramento das ligações O-H da brucita.30 As bandas de estiramento características do grupo PO43- são encontradas na região de 800-1200 cm-1, podendo estar superpostas às bandas do carbonato, sendo a banda em 872 cm-1 coincidente com a de um padrão de referência composto por fosfato de cálcio bibásico, grau P.A. Estiramentos assimétricos do grupo SO42- podem ser encontrados em 400-500 cm-1 31 e coincidem com as bandas encontradas na análise em 459 e 438 cm-1, o que também corrobora o enxofre presente na fluorescência e as fases de sulfatos no difratograma. As outras bandas podem estar superpostas com as bandas de carbonato e fosfato. Os espectros de infravermelho para os sólidos das reações a diferentes temperaturas não apresentaram diferenças.
CONCLUSÃO A reação do CO2 com o efluente salino sintético de eletrodiálise reversa revelou excelente potencial para fixar o CO2 através da formação de compostos carbonatados com cálcio e estrôncio, elementos capazes de causar incrustações em equipamentos e tubulações. Não se observou um efeito significativo da temperatura na faixa de 25 a 35 ºC, o que também revelou o potencial de ser desenvolvido um processo industrial de mais baixo custo energético uma vez que não envolveria aquecimento ou resfriamento dos efluentes. O resultado experimental para a remoção do Ca mostrou cinética rápida e resultados próximos ao simulado. Para o Sr os índices de remoção também foram elevados, mas discrepantes do simulado. Índices de remoção de cálcio entre 89 a 98% foram alcançados, enquanto para Sr, apesar de não ter sido otimizado o processo, os índices atingiram 46-75%. A fixação de CO2 sob forma de carbonatos, em concentrados de processos de eletrodiálise de efluentes de petróleo e efluentes de mineração por dissolução, pode ser viável, uma vez que o binômio efluente-gás de combustão se encontra presente nesses segmentos industriais. A reação destas soluções com o dióxido de carbono proveniente de gás de combustão, se desenvolvido estudo de viabilidade adequado, tem a possibilidade de tornar o tratamento deste tipo de efluentes salinos mais atrativo, uma vez que reduz o conteúdo de compostos formadores de incrustações e ainda atua no sentido de reduzir as emissões finais de CO2.
AGRADECIMENTOS Os autores agradecem ao CNPq, Capes, FAPEMIG, fundações fomentadoras de pesquisa no Brasil.
REFERÊNCIAS 1. Baird, C.; Em Química Ambiental. Bookman: Porto Alegre, 2002, parte I, cap. 4. 2. Figueroa, J. D.; Fout, T.; Plasynski, S.; McIlvried, H.; Srivastava, R. D.; Int. J. Greenhouse Gas Control 2008, 2, 9. 3. Yang, H; Xu, Z.; Fan, M.; Gupta, R.; Slimane, R. B.; Bland, A. E.; Wright, I.; J. Environ. Sci. (Beijing, China) 2008, 20, 14. 4. Atkins, P. W.; Shriver, D. F.; Overton, T. L.; Rourke, J. P.; Weller, M. T.; Armstrong, F. A.; Química Inorgânica, 4ª ed., Bookman: Porto Alegre, 2008. 5. Gilbbins, J.; Chalmers, H.; Energy Policy 2008, 36, 4317. 6. Pires, J. C. M.; Martins, F. G.; Alvin-Ferraz, M. C. M.; Simões, M.; Chem. Eng. Res. Des. 2011, 89, 1446. 7. http://www.iea.org/publications/freepublications/publication/technology-roadmap-carbon-capture-and-storage-2013.html, acessado em Maio 2017. 8. Olajire, A. A.; Energy 2010, 35, 2610. 9. Sanna, A.; Uibu, M.; Caramanna, G.; Kuusik, R.; Maroto-Valer, M. M.; Chem. Soc. Rev. 2014, 43, 8049. 10. Wolff-Boenisch, D.; Energy Procedia 2011, 4, 3738. 11. Hemamati, A.; Shayegan, J.; Bu, J.; Yeo, T. Y.; Sharratt, P.; Int. J. Min. Process. 2014, 130, 20. 12. Pacheco, F. G.; Dissertação de Mestrado, Universidade Federal de Minas Gerais, Brasil, 2012. 13. Fricker, K. J.; Park, A. H. A.; Chem. Eng. Sci. 2013, 100, 332. 14. De Vito, C.; Ferrini, V.; Mignardi, S.; Cagnetti, M.; Leccese, F.; Period. Mineral. 2012, 81, 333. 15. Ferrini, V.; De Vito, C.; Mignardi, S.; J. Hazard. Mater. 2009, 168, 832. 16. Mignardi, S.; De Vito, C.; Ferrini, V.; Martin, R. F.; J. Hazard. Mater. 2011, 191, 49. 17. Ma, J.; Yoon, R.; Energy Fuels 2013, 27, 4190. 18. Vale, EIA-RIMA, Estudo de Impacto Ambiental, vol I, revisão 00, 28/08/2009, 92p. 19. Becheleni, E. M. A.; Tese de Doutorado, Universidade Federal de Minas Gerais, Brasil, 2015. 20. Liu, Q.; Maroto-Valer, M. M.; Energy Procedia 2011, 4, 4503. 21. Drunkenmiller, M. L.; Maroto-Valer, M. M.; Fuel Process. Technol. 2005, 86, 1599. 22. Xiaoyan, L.; Jungang, L., Qianya, Z.; Jinlai, F.; Yingli, L.; Jingxin, S.; J. Pet. Sci. Eng. 2009, 66, 161. 23. Zhao, H.; Dilmore, R.; Allen, D. E.; Hedges, S. W.; Soong, Y.; Lvov, S. N.; Environ. Sci. Technol. 2015, 49, 1972. 24. Ministério de Minas e Energia; Resolução 430, de 13 de maio de 2011. Brasil, 2011, 9p. 25. Thi Tran, K.; Han, K. S.; Kim, S. J.; Kim, M. J.; Tran, T.; Hydrometallurgy 2016, 160, 106. 26. Carrol, J. J.; Slupsky, J. D.; Mather, A. E.; J. Phys. Chem. Ref. Data 1991, 20, 1201. 27. Gilbert, K.; Bennet, P. C.; Wolfe, W.; Zhang, T.; Komanak, K. D.; Appl. Geochem. 2016, 67, 59. 28. Andersson, M. P.; Hem, C. P.; Schultz, L. N.; Nielsen, J. W.; Pedersen, C. S.; Sand, K. K.; Okhrimenko, D. V.; Johnsson, A.; Stipp, S. L. S.; J. Phys. Chem. A 2014, 118, 10720. 29. Chen, L.; Chen, G.; Wang, X.; Fang, H.; Wu, C.; Xing, T.; Cryst. Res. Technol. 2010, 45, 254. 30. Frost, R. L.; Kloprogge, J. P.; Spectrochim. Acta, Part A 1999, 55, 2195. 31. Sandhya, S.; Sureshbabu, S.; Varna, H. K.; Komath, M. Cryst. Res. Technol. 2012, 47, 780. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access