
 |
 |
 |
 |
 |
Artigo
| Facile synthesis, characterization and DFT calculations of 2-acetyl pyridine derivatives |
|
Haroon ur RashidI; Adnan ShahzadII,IV; Zarif GulIII; Ezzat KhanIII,*; Muhammad Naveed UmarIII,#; Muhammad Raza ShahIV; Awal NoorV; Sher Wali KhanVI
IDepartment of Chemistry, Sarhad University of Science & Information Technology, Peshawar, Khyber Pakhtunkhwa, Pakistan Recebido em 29/12/2016 *e-mail: ekhan@uom.edu.pk Deprotonation of 2-acetylpyridine was carried out in the presence of NaH as a phase transfer catalyst instead of NaOH to minimize side products formation. The products were then treated with two equivalents of alkyl/aryl halide to synthesize a variety of acetyl pyridine derivatives. The compounds were structurally characterized by 1H- and 13C-NMR spectroscopy in solution state. Structure of compound 2 was also confirmed in solid state by X-ray diffraction. Electronic properties such as Mulliken charges, frontier molecular orbitals (HOMO-LUMO), ionization energy (IE), electron affinity (EA), global hardness (η), Chemical Potential (µ) and Global electrophilicity (ω) of some compounds were investigated by the same theory using B3LYP-6-311G basis set. INTRODUCTION The pyridine as nucleus can be found in numerous natural products as well as in synthetic compounds and play a key role in chemistry of biological and non-biological systems.1Nitrogen containing heterocyclic compounds have anti-allergic,2 anti-microbial,3-10 anti-bacterial, anti-fungal,11,12 anti-inflammatory,13,14 and anti-cancer15,16 activities. Many ligands derived from pyridine containing precursors are capable to couple with metals, the resulted complexes exhibit anti-tumor activities.17 Among metals, Cu (II) and Co(III) complexes with pyridine derivatives efficiently interact with DNA18 and can bind the two strands of DNA in an effective manner. Pyridine containing compounds and its metal complexes get much attention in the field of catalysis i.e. asymmetric synthesis19 atom transfer radical polymerization20 oligomerization21 polymerization22 and transfer hydrogenation of carbonyl compounds.23 In this work an easy and facile substitution on -CH3 group of the 2-acetylpyridine was carried out. The reaction was found to be equally effective with aryl and alkyl groups, formation of ether as byproduct or other side reactions were not observed. NaH instead of KOH/NaOH was used for the deprotonation of CH3 group of 2-acetyl pyridine. In case of KOH/NaOH unexpectedly ether is formed as major product exactly as in Williamson's synthesis.24 All the desired compounds were obtained in very good quantity, they were purified by using purification techniques and were structurally characterized with the help of 1H- and 13C-NMR spectroscopy. Structure of compound 1 was confirmed in solid state by X-ray diffraction. Structural data of compound 2 and electronic properties of compounds 1-3 were calculated by DFT. Experimental and theoretical data of compounds are discussed.
EXPERIMENTAL General All manipulations were performed in Schlenk-type glassware. Carefully dried toluene and oven-dried glassware were used throughout. The 2-acetyl pyridine (Sigma Aldrich), 18-Crown-6-ether (TCI Japan), Sodium hydride (Alpha Aesar), 1-(bromomethyl)-4-methylbenzene (TCI, Japan), 2-(bromomethyl)naphthalene (TCI, Japan), iodomethane (Alpha Aesar) were commercial products and were used as received. The NMR spectra for compounds 1-3 were recorded in CDCl3 using AVANCE AV-300 spectrophotometer, chemical shifts are given relative to Me4Si [δ1H (CHCl3) = 7.23; δ 13C(CDCl3) = 77.0; Chemical shifts are given to ±0.1 for both 1H and 13C-NMR. Crystal structure of compound 2 was determined by STOE-IPDS II equipped with low temperature unit. Structural solutions and refinements were accomplished by using SIR 97,19 SHELXL97,20 and WinGX.21 Melting point uncorrected was determined by melting point apparatus model SMP 10, England. All quantum-chemical calculations were carried out using the Gaussian 03 program package. Optimized geometries at the B3LYP/6-311+G level of theory were found to be minima by the absence of imaginary frequencies.25-30 Synthesis of 2-Methyl-1-(pyridine-2-yl)propane-1-one, 1 Following the literature procedure31,32 with modification, a 250 mL round bottom flask was charged with 29 mmol L-1 (3.53 g, 2 mL) of 2-Acetylpyridine in 58 mL dry toluene. After some time 3 equivalents, 2.1 g of Sodium hydride and 0.2 mol% of 18-crown-6-ether were added and the mixture was stirred for 20 minutes followed by addition of 2 equivalents (3.5 mL) of iodomethane. The reaction mixture was kept stirring for 3 h at 23±1 °C. The reaction mixture was washed with ethyl acetate several times. The solvent and other readily volatile materials were evaporated. The final product was purified with the help of column chromatography and was obtained as viscous oils. 1. R = Me; Yield = 70%; 1H-NMR (400 MHz, CDCl3): δ (ppm) = 1.20 (d, 6H, Me),4.10(sep, 1H, CH),7.44, 7.82, 8.03, 8.67 (m, m, m, m, 4H, Aromatic); 13C-NMR (100 MHz): δ (ppm) = 18.38 (Me), 36.31, (CH), 126.38, 129.66, 143.44, 145.87, 146.06 (Aromatic Carbons not assigned), 201.52 (C=O). Synthesis of 2-p-Methylbenzyl-1-(pyridine-2-yl)-3-p-tolylpropane-1-one, 2 Compound 2 was prepared exactly in the same way as compound 1 by the reaction of 2-Acetylpyridine (29 mmol, 3.53 g, 2 mL) Sodium hydride (2.1 g), 18-crown-6-ether (0.2 mol%) and 1-(bromomethyl)-4-methylbenzene (2 equivalents, 10.61 g). Crystals of compound 2 were isolated from hexane solution. R = CH3-C6H4-CH2;Yield = 70%; 1H-NMR (400 MHz, CDCl3): δ (ppm) = 2.24 (s, 6H, Me), 2.89 (m, 4H, CH2), 4.77 (Sep. 1H, CH), 6.99, 7.06, 7.37, 7.73, 7.93, 8.63, (m, m, m, m, m, m, 12H, Aromatic); 13C-NMR (100 MHz, CDCl3): δ (ppm) =20.98 (Me), 36.78 (CH2), 48.36 (CH), 122.50, 126.84, 129.04, 135.40, 136.71, 137.04, 148.57, 152.86 (Aromatic Carbons), 203.46 (C=O). Synthesis of naphthalene substituted derivative 3 Compound was prepared according to the procedure described above by using 2-(bromomethyl)naphthalene (12.88 g, 58 mmol), 2-Acetylpyridine (29 mmol, 3.53 g, 2 mL) Sodium hydride (2.1 g) and 18-crown-6-ether (0.2 mol%). R= 2-CH2-C10H6;Yield = 70%; 1H-NMR (400 MHz, CDCl3): δ (ppm) =2.95 (dd, 2H, CH2), 3.39 (dd, 2H, CH2), 5.04 (pent, 1H, CH), 7.38, 7.40, 7.63-7.69, 7.73, 8.61 (m, m, m, m, m, 18H, aromatic protons); 13C-NMR (100 MHz): δ (ppm) = 37.45 (CH2), 48.00 (CH), 125.18, 125.78, 126.88, 127.48, 127.53, 127.69, 127.80, 136.67, 137.39, 148.83, 152.91 (Aromatic Carbons not assigned), 203.50 (C=O).
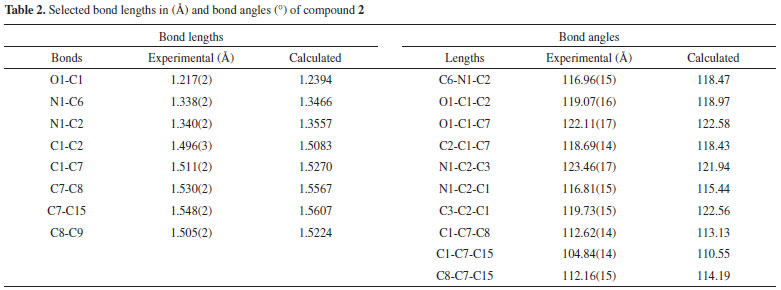
X-RAY CRYSTALLOGRAPHY A crystal of appropriate size and suitable dimensions was selected for X-Ray structure determination. The diffraction intensity data were collected on a IPDS II diffractometer Mo-Kα radiation (λ = 0.71073 Å) at 133±1 °C. Final refinement on F2 was carried out by full-matrix least-squares techniques. Structure solution and refinements were accomplished with SIR97, SHELXL-97 19-21 and WinGX.33 DFT calculations The structure of compound (2) was obtained from X-ray crystallographic data. The molecular structure of (2) (C23H23NO) in ground state was optimized by DFT method including correlation correction using B3LYP/6-311G model of theory.25-30,34-36 Optimized structure of the compound is given in Figure 2. Frontier molecular orbitals (HOMO-LUMO) energies, optimized geometries gaps, global hardness, ionization potential, electron affinity and global electrophilicity was calculated for compounds 1-3, using the same basis set. All quantum-chemical calculations were carried out using the Gaussian 03 program package.37,38 Optimized geometries at the B3LYP/6-311+G level of theory were found to be minima by the absence of imaginary frequencies.25-30
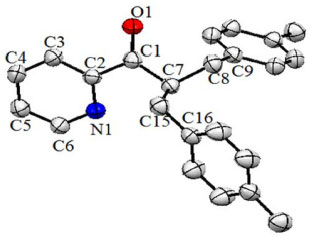 Figure 1. Crystal of compound 2 with partial labelling scheme ellipsoids drawn at 50% probability, hydrogen atoms are omitted for clarity
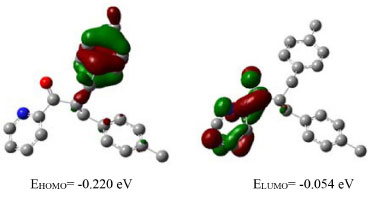 Figure 2. HOMO-LUMO representation of compound 2. Related energy values are also given
NMR SPECTROSCOPY The NMR data of compounds 1-3 are given in experimental section. The data is straightforward and show certain characteristic peaks in both 1H and 13C-NMR spectra. The R2CH proton appeared in its characteristic aliphatic region (≈ 2.9-4.0 ppm) and can easily be identified from its multiplicity. Other protons corresponding to R group and pyridyl group appear in the expected region. The 13C-NMR spectra of all compounds so obtained contain a signal at 201.52, 203.46 and 203.50 ppm corresponding to CO carbon in compound 1, 2 and 3, respectively. The R2CH in compound 1 appears at 36.31 ppm while the same carbon in compound 2 and 3 appears at 48.36 and 48.00 ppm, respectively.
RESULTS AND DISCUSSION Treatment of 2-acetyl pyridine with twofold excess of benzyl bromide in the presence of NaOH (as base) normally afford α-substituted acetyl pyridine (desired products). Analyzing reactions with the help of GC and HPLC show considerable amount of unreacted 2-acetyl pyridine and some peaks corresponding to unknown side products. The same reaction was repeated without adding 2-acetyl pyridine under the same reaction conditions and the same peak was observed as in previous reaction. The compound was characterized by NMR which shows 4 aliphatic and 10 aromatic hydrogen atoms. The data so obtained show that during reactions dibenzyl ether is formed as side product instead of the desired products. The same compound can be obtained as major product in Williamson's synthesis24 as shown in Scheme 2.
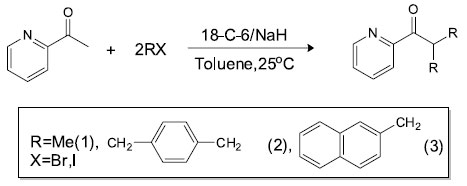 Scheme 1. Reaction of 2-acetyl pyridine with alkyl/aryl halides: A novel method towards syntheses of 2-acetyl pyridine derivatives
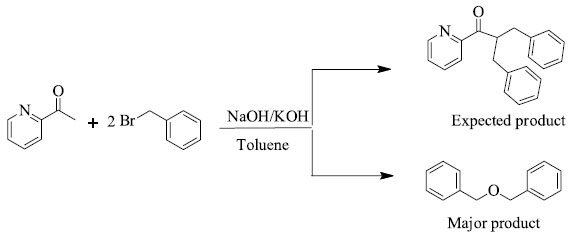 Scheme 2. Reaction of 2-acetyl pyridine with benzyl bromide in the presence of NaOH/KOH, Yield = 72%; 1H-NMR: 4.99 (CH2), 7.25 (aromatic)
Proposed mechanism A detailed mechanism of the reaction is given in Scheme 3. In first step, Br-from the corresponding Ar-Br substrate is replaced by HO- followed by water elimination and oxide ion formation. Since Aryl halide is present in excess and another equivalent reacts with the oxide radical to give dibenzyl ether (side product).
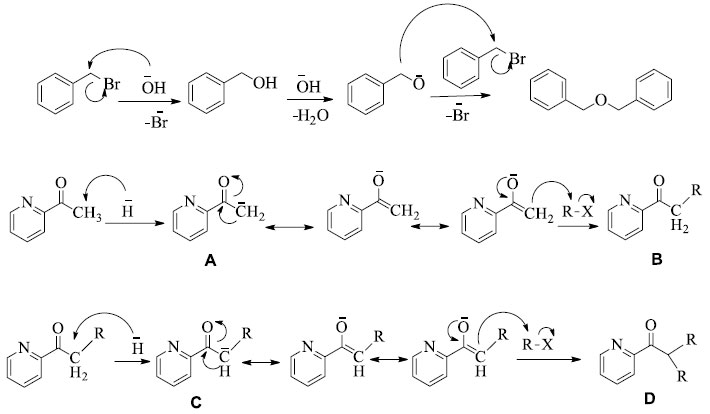 Scheme 3. Proposed mechanism for the formation of the expected product obtained in this study
To avoid the formation of side products, the strategy was modified by using NaH as a base instead of NaOH as shown in Scheme 1. In first step, H abstraction takes place to afford radical A which then reacts with R-X to give mono-substituted precursor B. The precursor B is still sensitive towards hydride attack and another H is abstracted since NaH is being used in excess (> two equivalents) shown by C and the reaction of R-X gives the final product D in the same way as in previous step. During the course of these reactions, side products were not detected as discussed above. Single crystal X-ray diffraction analysis Compound 2 was synthesized and subsequently purified as discussed in experimental section. The solid was dissolved in n-hexane and kept undisturbed. After few days needle-like crystals appeared in the solution. Crystals were separated from the solution and the structure was determined for a single crystal. The compound crystallizes in monoclinic system (α = γ = 90, β ≠ 90) with the space group P21/c. Crystallographic data pertinent to compound 2 are given in Table 1. The experimental and calculated data (for gaseous molecule), DFT (B3LYP/6-311G), (bond lengths and bond angles) are given in Table 2. The molecular structure of compound 2 is presented in Figure 1. There is negligible effect of substituents (p-Me) on the geometry around C1. The sum of angles around C1, ∠O1-C1-C2 119.07, ∠C7-C1-C2 118.69 and ∠O1-C1-C7 122.11 is approximately 360° and geometry around C1 is trigonal planer. The atoms C1-O1-C7-C2 are co-planar and are even in the same plane with the principal pyridine ring. This data show the same structural features as already described in literature.31,32 The paraxylyl rings are twisted by 83.76° and 35.03° with respect to the principal pyridine ring of the molecule.
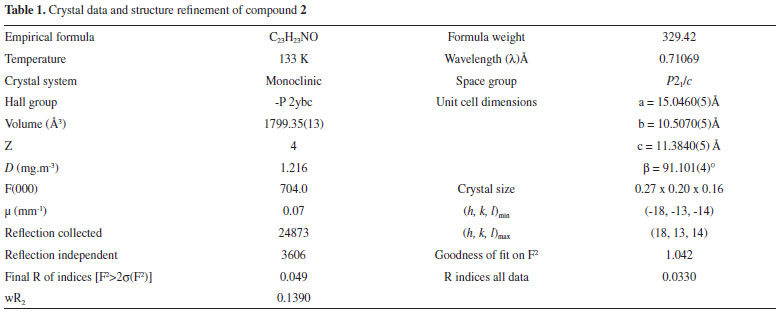
Frontier Orbitals (HOMO-LUMO) In general, electrons occupy the atomic orbitals which combine to form molecular orbitals. These orbitals are either HOMO (Highest occupied molecular orbital) or LUMO (Lowest unoccupied molecular orbital) also called frontier orbitals and the gap between these orbitals is known as the energy gap or band-gap. For explanation of different types of reactions and determination of different reactive sites in conjugated molecules, frontier orbital and their properties like band-gap is very important. The study of these orbitals and their energy gaps is helpful to get information about reactivity and stability of molecules.30 The HOMO-LUMO gaps are strongly dependent upon the substituent attached to a molecule and may cause an increase or decrease in overall energy of the system.39 The band-gap is sometimes used as a measure of excitability of molecules. The smaller the energy gap the more easily electron will be excited. It helps to evaluate a system for luminescence properties to be exhibited by that system.28 The study of energy related to frontier orbitals of molecules provides quantitative data.40 The HOMO are outer orbitals containing electrons and tend to donate these electrons so the energy of HOMO is associated with ionization potential. The LUMO tend to accept electrons so the energy of LUMO is associated with electron affinity.30 The HOMO/LUMO energy gap of compound 1-3 and starting material i.e., 2-acetylpyridine has been calculated by using B3LYP 6-311G and pop = Reg bases sets of theory (Table 3 and Figures 1S-4S in supporting information). The HOMO of starting material (2-acetyl pyridine) and compound 1 are slightly delocalized on pyridine ring and carbonyl group and in both cases the LUMO are localized on carbonyl moiety with band-gap of 0.176 eV and 0.183 eV respectively. The HOMO of compound 2 is localized over the C6H4 moiety of the 4-Me-C6H4-CH2 group and LUMO are localized over pyridine and carbonyl carbon with band-gap of 0.165 eV. And the HOMO of compound 3 are localized on nepthyl moiety and LUMO are localized on pyridine ring with band-gap of 0.132 eV given in Figures 2S-4S in supporting information. The data obtained regarding HOMO-LUMO energy and other selected electronic properties and summarized in Table 3.
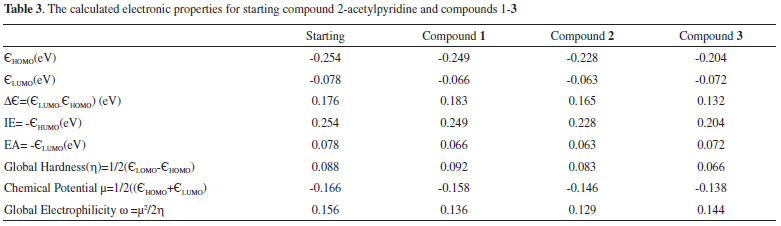
Global hardness is hypothetical feature of a molecule and cannot be estimated experimentally. It is an opposition towards polarization of electron cloud of an ion, molecule or atom under small perturbation of chemical response. The global hardness is less for alkali metals and high for noble gases.41 It is a measure of stability and reactivity of molecules. If the molecules are hard it defines that the molecules tend to be more stable and un-reactive because they cannot undergo change in electronic charge much easily.42 The value observed for compound 1 is 0.083 (see Table 3) which is very low and shows that the molecule has considerable hard characters. The concept of electrophilicity helps in understanding the mechanism of organic reactions.43 Ingold proposed the first global electrophilicity scale to describe electrophile and nucloephile specie based on valence electron theory.44 The global electrophilicity is a useful structural predicator of reactivity and is used in investigation of chemical reactivity of molecules.45 The concerned data is relatively high and indicates that the molecule can show moderate reactivity.
CONCLUSION A comprehensive literature survey shows that very few is known about the analogous compounds.46,47 As far as our results are concerned all our compounds are novel and the structural data related to these compounds are reported for the first time. The method is quite efficient and no side reaction/products were observed during this work. Reaction of 2-acetylpyridine with NaH in the presence of phase transfer catalyst affords substituted products. The synthetic method is very efficient and is the only access to such type of derivatives. The solid state and gaseous phase data of one derivative can be well compared. The energy gap between HOMO-LUMO of the molecules is considerably low, indicating high reactivity of the molecule to be effective in secondary reactivity.
SUPPLEMENTARY MATERIAL Figures 1S to 7S are available for download at http://quimicanova.sbq.org.br in pdf format with free access.
ACKNOWLEDGEMENTS The financial support from Higher Education Commission (HEC) Pakistan under National Research Program for Universities (NRPU) No. 20-1488/R&D is highly acknowledged. We are thankful to Professor Dr. Rhett Kempe, Anorganische Chemie II, University of Bayreuth, Germany, for his kind assistance in the X-ray laboratory.
REFERENCES 1. Bringmann, G.; Reichert, Y.; Kane, V.; Tetrahedron 2004, 60, 3539. 2. Hwang, C.; Tu, H.; Wang, H.; Lee, H.; Molecules 2006, 11, 169. 3. Hiremath, P.; Ullagaddi, A.; Shivaramayya, K.; Purohit, G.; Indian J. Heterocycl. Chem. 1999, 3, 145. 4. Savchenko, V.; Dorokhov, V.; Yakushchenko, I.; Zyuzin, I.; Aldoshin, S.; Herald Russ. Acad. Sci. 2010, 80, 149. 5. Zavyalova, V.; Zubarev, A.; Shestopalov, A.; Rus. Chem. Bull. 2009, 58, 1939. 6. Naik, T.; Chikhalia, K.; E-J. Chem. 2007, 4, 60. 7. Mashaly, M.; Abd-Elwahab, H.; Faheim, A.; J.-Chin. Chem. Soc. Taipei 2004, 51, 901. 8. Sabet, R.; Fassihi, A.; Moeinifard, B.; Res. Pharma. Sci. 2009, 2, 103. 9. Patel, B.; Agrava, N.; Shaikh, M.; Med. Chem. Res. 2011, 20, 1033. 10. Al-Salahi, A.; Al-Omar, A.; Amr, E.; Molecules 2010, 15, 6588. 11. Zheng, Y.; Ma, Z.; Zhang, X.; Yang, N.; Yang, G.; Int. J. Chem. 2011, 3, 42. 12. Rajput, S.; Sharma, S.; Int. J. Pharm. Bio. Sci. 2011, 2, 200. 13. Muthal, N.; Ahirwar, J.; Ahriwar, D.; Masih, P.; Mahmdapure, T.; Siva-kumar, T.; Synthesis 2010, 2, 2450. 14. Márquez-Flores, K.; Campos-Aldrete, E.; Salgado-Zamora, H.; Correa-Basurto, J.; Meléndez-Camargo, E.; Med. Chem. Res. 2012, 21, 775. 15. Nassar, E.; J. Am. Sci. 2010, 6, 338. 16. Bassyouni, A.; Tawfik, A.; Soliman, M.; Rehim, A.; Res. Chem. Intermed. 2011, 38, 1291. 17. Yang, T.; Wu, H.; Ma, J.; Hu, J.; Wang, Y.; Trans. Met. Chem. 2011, 36, 403. 18. Nagababu, P.; Kumar, A.; Reddy, L.; Kumar, A.; Mustafa, B.; Shilpa, M.; Satyanarayana, S.; Met.-Based Drugs 2008, 24, 1. 19. Busto, E.; Gotor-Fernández, V.; Gotor, V.; Tetrahedron: Asymmetry 2005, 16, 3427. 20. Abu-Surrah, A.; Ibrahim, A.; Abdalla, Y.; Issa, A.; J. Polym. Res. 2011, 18, 59. 21. Seitz, M.; Görl, C.; Milius, W.; Alt, G.; Jordan J. Chem. 2008, 3, 109. 22. Abu-Surrah, S.; Lappalainen, K.; Piironen, U.; Lehmus, P.; Repo, T.; Leskelä, M.; J. Organomet. Chem. 2002, 648, 55. 23. Baratta, W.; Ballico, M.; Baldino, S.; Chelucci, G.; Herdtweck, E.; Siega, K.; Magnolia. S.; Rigo, P.; hem. - Eur. J. 2008, 14, 9148. 24. Peng, Y.; Song, G.; Green Chemistry 2002, 4, 349. 25. Ahmadi, S.; Seif, A.; Rozbani, M.; Arabian J. Chem. 2010, 3, 121. 26. Jacquemin, D.; Perpete, A.; Scueseria, E.; Ciofini, I.; Adamo, C.; J. Chem. Theory Comput. 2008, 4, 123. 27. Schlegl, B.; Millam, M.; Lyenge, S.; Voth, A.; Daniels, E.; Chem. Phys. 2001, 114, 9758. 28. Zheng, G.; Musgrave, B.; J. Phys. Chem. A 2007, 111, 1554. 29. Altaf, A.; Shahzad, A.; Gul, Z.; Khan, A.; Badshah, A.; Tahir, N.; Zafar, I.; Khan, E.; J. Chem. 2015, 1-5, Article ID 913435. 30. Taghki, I.; Larif, M.; Hmammouchi, R.; Adad, A.; Bouachrine, M.; Lakhlifi, T.; J. Mater. Env. Sci. 2012, 3, 870. 31. Umar, N.; Shoaib, M.; Ng, W.; Acta Crystallogr., Sect. E: Struct. Rep. Online 2012, 68, o575. 32. Umar, N.; Shoaib, M.; Ng, W.; Acta Crystallogr., Sect. E: Struct. Rep. Online 2012, 68, o576. 33. Sheldrick, M.; Acta Crystallogr., Sect. A: Found. Adv. 2008, 64, 112. 34. Zheng, G.; Musgrave, B.; J. Phys. Chem. A 2007, 111, 1554. 35. Weng, H.; Weng, X.; Wang, H.; Wang, L.; Liu, A.; J. Mol. Model. 2007, 13, 147. 36. Naei, M.; Hollosadi, M.; Anaraki-Ardakani, H.; Arabian J. Chem. (2013), DOI: 10.1016/j.arabjc.2013.07.021. DOI: http://dx.doi.org/10.1016/j.arabjc.2013.07.021 37. Noel, M; Tenderholt, L; Langner. M; J. Comput. Chem. 2008, 29, 839. 38. Hong, J; Kai, Z; Li, Y; Corros. Sci. 2008, 50, 865. 39. Kaur, I.; Jia, W.; Kopreski, P.; Selvarasah, S.; Dokmeci, R.; Pramanik, C.; McGruer, E.; Miller, P.; J. Am. Chem. Soc. 2008, 130, 16274. 40. Joseph, T.; Vaghese, T.; Panicker, Y.; Viswanthan, K.; Dolezal, M.; Alsenoy, V.; Arabian J. Chem. 2013,113, 203. 41. Chattaraj, K.; Matti, B.; Sarkar U.; J. Phys. Chem. 2003, A107, 4973. 42. Islam, N.; Ghosh, C.; J. Quantum. Inf. Sci. 2011, 1, 135. 43. Chattaraj, K.; Matti, B.; Sarkar, U.; J. Phys. Chem. A 2003, 107, 4973. 44. Ingold, K.; Chem. Rev.1935, 15, 225. 45. Soto-Delgado, J.; Domigo, R.; Contreras, R.; Org. Biomol. Chem. 2010, 8, 3678. 46. Umar, N.; Shoaib, M; Ng, W.; Acta Crystallogr., Sect. E: Struct. Rep. Online 2012, 68, o575. 47. Umar, N.; Shoaib, M; Ng, W.; Acta Crystallogr., Sect. E: Struct. Rep. Online 2012, 68, o576. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access