
 |
 |
 |
 |
 |
Artigo
| Peroxidase from soybean meal: obtention, purification and application in reduction of Deoxynivalenol levels |
|
Ana Carla Penteado Feltrin; Milena Ramos Vaz Fontes; Henrique Delgado Kikumoto Gracia; Eliana Badiale-Furlong; Jaqueline Garda-Buffon*
Escola de Química e Alimentos, Universidade Federal do Rio Grande, 96203-200 Rio Grande - RS, Brasil Recebido em 28/01/2017 *e-mail: jaquelinebuffon@furg.br This study established the conditions for the extraction of the enzyme peroxidase (PO) from soybean meal (SBM). An experimental design methodology was carried out in order to evaluate the effects of stirring rate time, pH and extracting solvent volume on the enzyme extraction. By using 5 g SBM and 50 mL phosphate buffer 10 mmol L-1 pH 4.7, 60 min stirring rate at 100 rpm, an enzyme with specific activity of 100 U mg-1 for SBM was obtained. Two techniques of purification were tested and compared for purification of peroxidase from soybean meal: triphasic partition (TPP) and molecular exclusion chromatography. TPP showcased a greater efficiency with 50% recuperation and a purification factor of 13.6. Peroxidase in crude and pure forms was characterized for kinetics, thermodynamics and biochemistry. The parameter of thermal inactivation indicates high stability to exposure time and temperature increase, showing that enzyme activity is not altered by the presence of constituents of the reaction medium. Peroxidase in crude form represented a greater upkeep in activity, keeping 50% activity for 114 days at 0 °C. Peroxidase in pure form had greater affinity for substrate and reduced Deoxynivalenol levels by 80%, 20% more than the crude form. INTRODUCTION Peroxidases (PO) from vegetals (EC 1.11.1.7) catalyze redox reactions of several substrates, especially those that serve as electron donors, having hydrogen peroxide as an oxygen donor.1 PO are used in clinical diagnoses, immunoassays, pharmaceuticals, food and chemical development, synthesis, degradation and organic compounds transformation, and the removal of industrial waste and peroxides in food.2-4 The action of PO enzyme has also been related to the degradation of mycotoxin Deoxinivalenol (DON).5 In this study, the greater activity of PO enzyme generated from submerged fermentation with Rhizopus oryzae and Aspergillus oryzae was related directly to more rapid DON degradation. The PO application at an industrial scale requires the search for available sources that represent advancement in agro-industrial wastes evaluation, considering how abundant and active they are in external parts of the tissues present in grains, such as bran. Important factors must be considered when evaluating the use of the enzyme in industrial processes, including the quantity, production process, recuperation factor and purity that can be obtained from the tissue where the enzyme is located. However, despite numerous studies of their potential uses, there are few studies that consider them sources of enzymes. When the intention is to propose the use of POs from these materials, the first step is to evaluate extraction conditions and ensure efficient activity for the purpose of interest. Suitable extraction conditions require not only combinations of variables, such as pH, ionic strength and extracting solution volume, but also a way that may promote homogenization of cellular decompartmentalisation without denaturing the active protein structure. After extraction, the strategies that can be employed at the purification process are dependent on application to be given the enzyme of interest. These operations consist of several steps, which inevitably result in a significant loss of enzymatic activity abundant waste generation and difficulty recycling, and they incur a high cost.6,7 Therefore, there is a demand for efficient procedures that provide sufficient selectivity, maximum recuperation, maximum activity preservation and require acceptable costs and minimum time to obtain a desirable purification factor.8 In light of these features, this study evaluated conditions for the extraction and compared the purification techniques of PO from soybean meal (SBM), using molecular exclusion chromatography and three-phase partitioning (TPP) techniques. Afterwards, kinetic, thermodynamic and biochemical characterization of PO was performed. The study of purification and the characterization of PO allows for their application in bioprocesses, such as how decontamination of mycotoxins is associated with redox reactions.
MATERIAL AND METHODS Chemical characterization of sources of enzyme SBM were purchased in the local market in Rio Grande, RS, Brazil. Particles were larger in size than 0.500 nm. All material was stored at 4 °C until testing. Establishment of enzymatic extraction conditions The study of the extraction of PO was based on the procedure described by Cardinali et al.,9 with modifications. Fifty mL phosphate buffer at 5 mmol L-1, was added to 5 g of each source, followed by stirring for 60 min at room temperature so that the disruption of cell structure and protein solubilization could happen. Extracts were centrifuged at 3220 x g for 10 min at 4 °C and the soluble protein and specific activity of PO were determined in the filtrate and supernatant. Aiming at maximizing the extraction process, contact time and extracting solvent volume and pH were evaluated by a central composite rotatable design (CCRD) 23, whose variables and levels totalized 17 experiments. Validation was conducted by running an experiment in triplicate in the experimental condition in which high specific activity of PO was observed. After determining the extraction conditions, the effects of the ionic strength of the phosphate buffer, at concentrations ranging from 5 to 50 mmol L-1 were evaluated. Measurement of peroxidase activity Peroxidase activity was determined according to the methods described by Devaiah and Shetty,10 using an absorbance at 470 nm following the formation of tetraguaiacol. One unit of peroxidase activity was defined as the amount of enzyme catalyzing the oxidation of 1 µmol of guaiacol in 1 min. Concentration of the protein was determined by Lowry et al.,11 and the absorbance was measured at 660 nm. The standard curve was prepared with several concentrations between 0 and 1 mg mL-1 using the standard curve for bovine serum protein. Peroxidase purification In this work, PO enzyme from SBM was purified using two purification methods: one exclusion molecular chromatography and the other by TPP. The two methods were studied to compare the efficiency in preserving enzymatic activity. Exclusion molecular chromatography The crude extract was submitted to protein concentration for acetone precipitation at a ratio of 1:3 (v/v - crude extract: acetone) according to a method proposed by Boer et al.,12 adapted for this work. The mixture was allowed to rest in refrigeration for 24 h. Then it was centrifuged at 5000 rpm at 4 °C for 40 min, and the precipitate was subsequently solubilized in 5 mmol L-1 phosphate buffer pH 5. The efficiency of this step was indicated by determining the specific activity and soluble protein content. After concentration, crude enzymatic extract was subject to exclusion molecular chromatography in a column packed with Sephadex G-100 with dimensions of 1.5 cm in diameter x 30 cm of height, stabilized for 24 h with 5 mmol L-1 phosphate buffer at a pH 5, the identical buffer used in the mobile phase with 0.4 mL min-1 of flow rate. The volume of enzymatic extract corresponding to 38 mg of protein was eluted in a chromatographic column. Aliquots of 2 mL were collected for protein concentration determination in a spectrophotometer at 280 nm and measured for specific activity of PO. Three phase partitioning According to the procedure proposed by Özer et al.,13 adapted for this work, in a 15 mL centrifuge tube, 2 mL of crude enzymatic extract was added with ammonium sulfate (30%, w/v) at room temperature and vortexed for 1 min. Next, acetone (1:1.5, v/v - crude extract: solvent) was added, and the system was mixed in vortex again for 1 min and then allowed to rest for 1 h to facilitate the separation of phases. Next, it was centrifuged at 5000 rpm, 4 °C for 10 min for maximal phase separation. The top phase and bottom phase were separated by the interface formed from centrifugation. Organic and saline phases were removed, the intermediate protein precipitate was dissolved in a minimum amount of 5 mmol L-1 phosphate buffer pH 5. In all phases and purification extracts, the specific activity of PO (U mg-1) and protein concentration (mg mL-1) were determined. The adaption was performed studing some process parameters, such as the saturation percentage of ammonium sulfate adding the salt to 2 mL of crude enzymatic extract in the concentration of 30, 40, 50 and 60%, w/v), and top phase and bottom phase (1:0.5; 1:1; 1:1.5 and 1:2 proportion, v/v - crude extract: solvent) were evaluated in terms of the efficiency, purification factor (FP) and recuperation (REC) of PO, as described in Equation 1 and 2.  Purification evaluation for SDS-PAGE (Sodium Dodecyl Sulphate-Polyacrylamide Gel Electrophoresis) Enzyme purity and molecular weight were analyzed by SDS-PAGE in a vertical system. Proteins were stained with Coomassie brilliant blue.14 Characterization of enzymatic reaction conditions The greatest reaction time for PO from SBM was evaluated in reaction conditions, keeping 0.1 mol L-1 phosphate buffer pH 7 at 30 °C to evaluate activity in the range from 5 to 55 min. Both crude and purified TPP extracts were measured. With the reaction time characterized, the optimal reaction temperature was evaluated between temperatures of 0 and 80 °C. The optimal pH was determined in crude and purified extracts, employing 0.1 mol L-1 phosphate buffer in a range of pH between 4.5 and 8.5. Using time, temperature and a pH previously characterized, the ionic strength of the buffer was determined using different concentrations of phosphate buffer (5 - 50 mmol L-1). pH stability To identify the optimal pH range for PO stability from SBM, 5 mM phosphate buffer in a range of pH between 5.0 to 7.5 (with interval of 0.5 units of pH) was used. Evaluations continued until there was a total loss of activity under storage at 10 °C, in order to aim for higher efficiency purification steps. PO thermostability Thermal stability of the PO was checked in both crude and purified extracts, through storage under different temperatures ranging from 0 and 120 °C. Temperature monitoring continued until there was a total loss of enzymatic activity in pH-stable extracts previously identified. Determination of kinetic and thermodynamic parameters during thermal inactivation Kinetic parameters of thermal inactivation Data obtained during thermal stability evaluation were used for the definition of kinetic parameters relative to PO activity.15 Experimental data were calculated according to the equation below, Equation 3:  where, A0 is initial activity, A is residual activity after thermal treatment, Kd is constant thermal inactivation rate (h) and t is exposition time (h). The half-life (, h) of PO in different temperatures, was determined by the equation below, according to the period in which the enzyme maintained 50% of its activity, according Equation 4.  The value of D or the time necessary for reduction in a log 10 in the concentration of reagents species (that is, a reduction of 90% in a reagent of the concentration) was calculated by following Equation 5:  The value z (the increase in temperature necessary to reduce the value D in a log cycle) was calculated by graphing the slope of log D versus T (°C) using Equation 6:  The reaction energy of deactivating (Ed) PO was determined by linearization of the Arrhenius equation, plotted by the relation between the constants: log of denaturation speed (ln Kd) versus absolute temperature (K) using Equation 7:  Thermodynamic parameters of thermal inactivation The thermodynamic parameters were measured through changes in enthalpy (∆H°, kJ mol-1) (Equation 8), free energy (∆G°, kJ mol-1) (Equation 9) and entropy (∆S°, kJ mol-1) (Equation 10). Thermal denaturation of PO was calculated using the following equations: 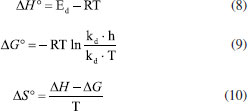 where T is absolute temperature correspondent (K), R is gas constant (8.314 J mol-1 K-1), h is Plank's constant (1.84 x 10-37 Jh), KB is Boltzmann's constant (1,38 x 10-23 J K-1) and Kd is the thermal inactivation constant (h). KM and Vmáx determination For kinetics parameters KM and Vmáx determination, the PO activity was evaluated when the reaction medium contained initially different concentrations of the substrate guaiacol (8 to 160 mmol L-1). Kinetics parameters KM and Vmáx were measured using the Lineweaver-Burk method of graphing linearization of the Michaelis-Menten equation. PO application in deoxynivalenol reduction (DON) Experiments for checking the levels of reduction were realized in the buffering medium, conducted according to optimal conditions of activity of PO from SBM in both the crude and purified forms. The experiments were realized using DON (1 µg mL-1), the cofactor of enzymatic activity (H2O2) and 1 U mL-1 of PO, completing 1 mL of reaction medium under optimal conditions for enzymatic action in crude and purified forms. At the end of the reaction time (10 min), DON residual was extracted as described by Tanaka et al.,16 where acetonitrile (30:70, v/v) was added to the reaction medium. DON present in the mixture was then solubilized through vortexing for 30 s and 3 min in an ultrasonic bath, and then sodium chlorate was added until the medium was saturated. Due to migration of DON from organic solvent following solubilization, this process was repeated three times. Lastly, DON was extracted, concentrated and quantified by high pressure liquid chromatography with ultraviolet detector (HPLC-UV). The reduction confirmation of DON levels in HPLC-UV occurred under the following conditions: Phenomenex Gemini C18 column with a particle size of 5 µm and 250 mm x 4.6 mm of internal diameter. The solvents used for elution were acetonitrile: ultrapure H2O (70:30, v/v) at a flow rate of 0.5 mL min-1, using a wavelength of 220 nm. Statistical analysis The statistical analysis of the data was performed by ANOVA, considering the averages of the triplicate tests by the Tukey's test. The "Statistic" (version 7.0, Stat Soft, Inc., Tulsa, USA) program was carried out, considering that PO specific activity (U mg-1) was response variables. The significance level was set at 95% for the mathematical model and the response surface. The significance of the regression coefficients, p (t), was determined by using the Student t test whereas the significance of the model described by the equation was determined by the Fisher's test. The variance explained by the model was given by the coefficient of multiple determination: R2.
RESULTS AND DISCUSSION Establishment of enzymatic extraction conditions Table 1 shows the results found by CCRD. It may be observed that the highest values of specific activity were obtained in the center points (trials 9, 10 and 11), 103 U mg-1 for enzyme SBM. The statistical analysis of the results displayed show Ftabulated of 3.41 in comparison to Fcalculated of 21.24 (p = 0.00003), resulted in a coded empirical model for the activity of PO in function of time of extraction, pH and extracting solvent volume. Values of R and F tests showed that the models of Equation 11 were able to represent the experimental data of the variables in the analysis for SBM. This model allow the construction of response surfaces shown in Figure 1.
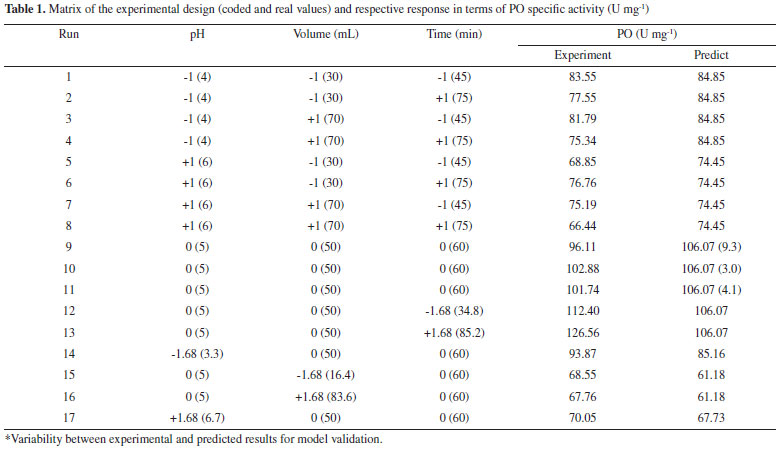
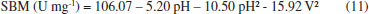 In Figure 1, the region of the highest specific activity of the enzyme from SBM corresponds to the central point evaluated in planning. The optimized conditions showed that the volume of the phosphate buffer used as extracting solvent should be 50 mL, pH 4.7, and extraction time of 60 min. Thus, extreme values of pH, volume and time, evaluated by this study, decreased the PO extraction efficiency. This possibility was mentioned by Srinivas, Barhate and Raghavarao,17 who demonstrated that the PO is not stable when pH is below 4.0. The importance of pH to the increase or maintenance of enzyme activities has also been described in the literature for enzymes from various species, such as Lentinula edodes.18 In this study, tests performed at pH 6.7 showed about 80% reduction in the specific activity from SBM, possibly due to the destabilization of the enzyme. The results indicate higher specific activity of PO from SBM (103 U mg-1).
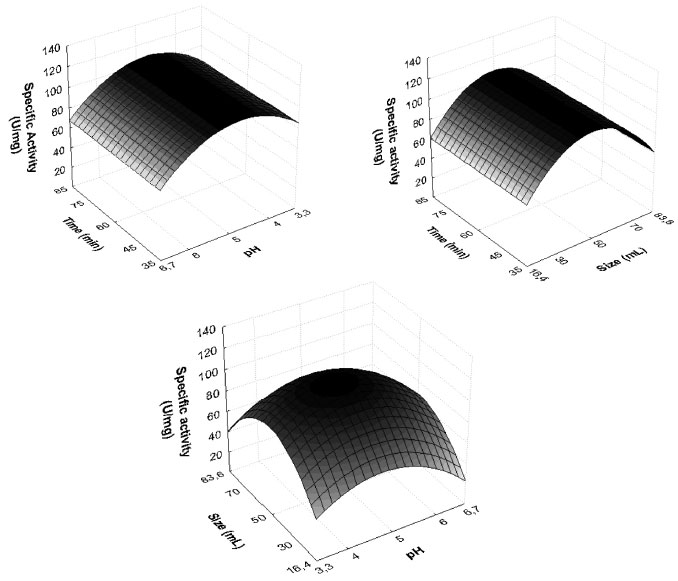 Figure 1. Reponses surfaces obtained by the experimental design CCRD 23 for PO extracted from SBM
After the definition of the conditions, the experiment was performed in triplicate to validate the extraction process. Activity of 100 U mg-1, with standard error of 3%, was achieved, a fact which shows that the use of planning established the parameters that guaranteed the desired conditions for the extraction of PO with suitable activity to be used in degradation procedures. Besides the pH, the effect of the ionic strength of the buffer on the extraction efficiency was evaluated. The highest relative activity of the enzyme extracted from SBM was obtained with ionic strengths of 10 mmol L-1. The latter was adopted due to its low cost. Exclusion molecular chromatography Chromatographic elution was searched for protein fractions with high activity of PO and the least possible protein concentration. Figure 2 shows the range 50 to 90 mL of the mobile phase contains the highest activity of PO. However, the same profile was observed at the protein level. In the interval of increased activity, aliquots were collected and pooled, and the PO action was determined by REC and FP, as shown in Table 2.
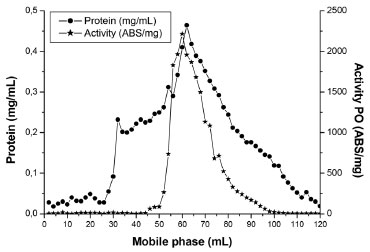 Figure 2. Elution profile obtained from exclusion molecular chromatography with Sephadex G-100, of the PO from SBM
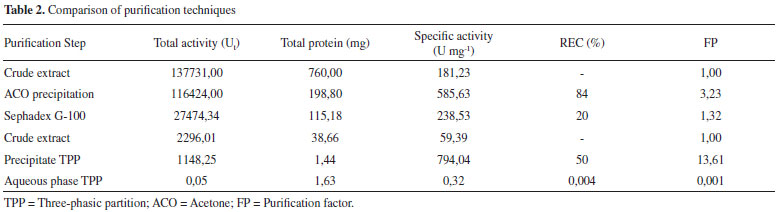
The PO purification process presented a similar chromatographic profile to that encountered for PO from palm oil by Deepa and Arumughan,19 which used three purification steps, the last step using the exclusion molecular chromatography with a Sephadex G-100 column. In study, after four purification steps, with the last step Sephadex-G-100, reached 17% of REC of PO from wheat flour.6 Based on this studies and Table 2, the percentage of REC encountered was consistent with the literature, in terms of FP, the technique did not show the necessary efficiency for application in the mycotoxicological decontamination process, which requires a high degree of purification, not just the number encountered in this work. The results show the need to search for other methodologies to improve PO purification efficiency. Three-phasic partition The original method by Özer et al.,13 describe the use of t-butanol for dehydration of the aqueous phase, however in this work, we chose to apply acetone because of its lower cost and potential increase in recuperation in the final process. The evaluation results from the parameter effects of saline saturation and extract proportions from using acetone during purification are presented in Table 3. In this work, 30% of ammonium sulfate was used to saturate the medium in PO purification. In the optimization of the relative quantity of acetone, the greatest results obtained were in a ratio of 1:1.5 crude extract: acetone, with 13.3 of FP and 50% of REC. The pH of PO in the interface in this work corresponded to 6, which is equivalent to the greatest enzyme stability, so it did not require adjustment and subsequent reduction of steps in the process.
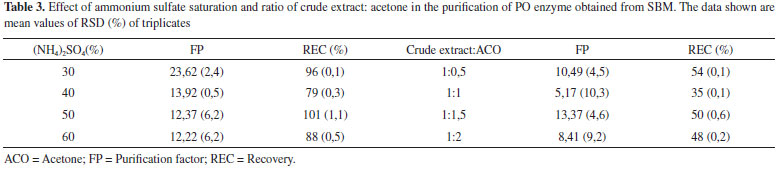
Efficiency comparison between purification techniques Comparing the two studied methods (Table 2), the parameters REC and FP favor the application of TPP over the chromatographic method. The chromatographic separation consisted of two steps, which can cause losses in enzymatic activity or denaturation. Sephadex G-100 purification had low recuperation (20%) and was disadvantageous because it required a high investment in material, provided low capacity of purification, and required high execution time (36 h) to complete the process. In contrast, the purification strategy for TPP had multiple advantages, as it consisted of only one operation step, obtained considerable recuperation (50%), was low cost, did not require much time (2 h) and has the potential to be scaled up easily. Thus, this method was faster, cheaper and more efficient for enzymatic purification, with fewer steps to decrease denaturation risk or dilution of the enzyme. This comparison was made using SDS-PAGE shown in Figure 3.
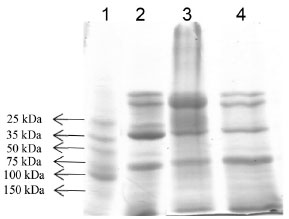 Figure 3. SDS-PAGE of the PO. The protein bands were stained by Coomassie Blue. The purification steps were: run (1) - standard molecular weight marker, run (2) - sample crude extract, run (3) - sample purified extract by Sephadex G-100 and run (4) - sample purified extract by TPP, for PO from SBM
The definition and intensity reduction of stained bands, in comparison with crude extract, confirmed the final outcome of the process, with a molecular weight of 34 kDa. The presence of more than one band in the electrophoretic profile indicates the presence of enzymatic subunits formed by denaturation or of isoenzymes present in the enzymatic extract. Enzymatic characterization Optimal reaction conditions As shown in Figure 4, the optimal conditions of time, temperature, pH and ionic force were determined for PO enzyme, both in crude and purified extracts from TPP.
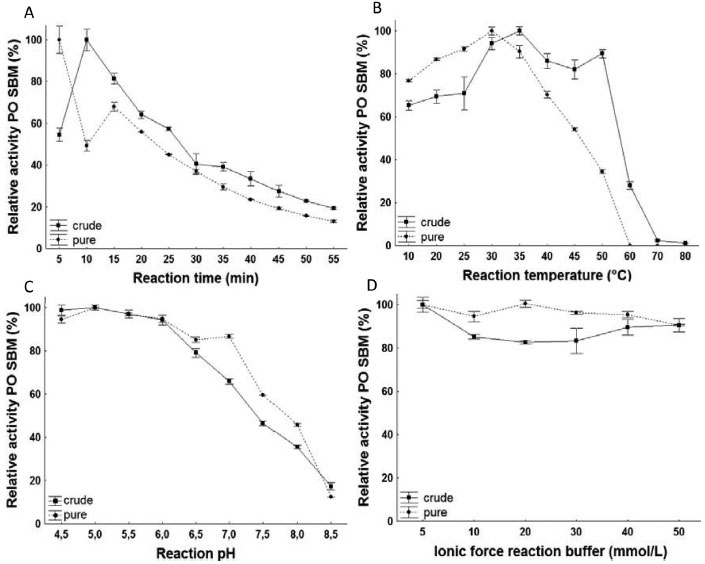 Figure 4. Profile of the effects of time (A), temperature (B), pH (C) and ionic force (D) on activity of PO purified from SBM
For PO in crude extracts, the ideal reaction time was 10 min, while in purified form, it was 5 min. The optimal temperatures were 35 and 30 °C for the crude and pure extracts, respectively. These values are consistent with previous studies, where PO was recovered from several sources with optimal temperature between 30 and 50 °C.1,20-22 For both forms of PO, the pH that presented the greatest enzymatic activity was 5, a value consistent with findings when purified from Streptomyces sp23 and Leucaena leucocephala.24 In the evaluation of pH, there weren't significant differences in reaction pH of the crude extract between 4.5 and 6 and of the pure extract between 5 and 6. The concentration of 5 mmol L-1 was identified as optimal because the low saline concentration can avoid environmental contamination and high process costs. In the purified extract by TPP, the specific activity was measured using phosphate buffer in the optimal pH, resulting in the 5 mmol L-1 concentration, the same for both extract forms. Determination of the Kinetic constants To determine affinity of the PO with their substrate guaiacol, enzyme activity was measured for each of the forms. The values of KM and Vmáx of the PO are present in Table 4. A greater affinity of PO for its substrate guaiacol was observed in purified form when compared to crude form. This is expected once the presence of the interference in the reaction medium can be correlated with inhibition. When correlated, the results obtained in this study are consistent with those described in literature, which correspond to a range from 0.28 to 18.5 mmol L-1 for different sources of the enzyme.17,22,25 Because of their high affinity, the PO from SBM shows promising results for industrial application.
pH stability pH stability in crude extract was optimized at 6. In studies referencing PO, the optimal pH value varied between 4 and 8.5, depending on the purification source.1,22,26,27 Characterization of optimal enzymatic pH stability is important when concerned with commercial exploration and industrial application.15 Thermal inactivation The residual activity rates in time incubation function for the enzyme in this study were linear, with R2 > 90%, indicating that inactivation can express first order kinetics. Half-life time calculation is accurate and reliable, especially if it is used to analyze the properties of thermal stability at different temperatures. With the increase in temperature, the values of the half-life and D are inversely proportional to Kd values, as shown in Table 5. The z values for PO in crude and purified forms were 24.71 and 22.05 °C, respectively. Higher z values indicate a greater sensitivity of the enzyme to exposure time and thermal treatment.28 This study shows that PO in crude form has a greater sensitivity to thermal treatment and time. This effect can result from other proteins that act in a reaction system for destabilization. The deactivation reaction energy was calculated to be 339.87 kJ mol-1 and 368.10 kJ mol-1 for the crude and purified forms, respectively. High values of Ed indicate that high energy is necessary to begin denaturation, which is demonstrated by the high stability of both enzymatic forms against thermal denaturation and maintenance of the structural organization of the enzyme after purification.
The denaturation enthalpy (∆H°) is the amount of heat necessary to denature the enzyme. A high enthalpy value indicates a more stable enzyme, considering that high energy quantities are necessary for the denaturation process. The calculation of denaturation entropy (∆S°), which quantifies the energy necessary for the transition of the native enzyme from the transition state to a denatured state, gave negative values. This is indicative of lower disturbance of the system and.greater.enzymatic.stability. In terms of thermal stability, the more reliable parameter to evaluate is Gibbs free energy (ΔG°).29 Negative or lower values indicate that the reaction favors thermal denaturation. The values encountered in this work confirming high enzyme stability. Ideal peroxidases for industrial application have to be abundant, purified easily, with large substrate specificity and high stability under high temperatures and extreme pH conditions. It is necessary to improve the thermal stability of enzymes for industrial application, in order to improve the integrity of the molecular structure.30 The high stability of PO demonstrated in this work shows that it has potential for use in industrial products. In comparative terms, the PO from SBM demonstrates greater thermal stability than lignin peroxidase and manganese peroxidase from Bjerkandera adusta, by Ertan et al.,31 with values of 36 and 14 kJ mol-1 for ∆H°, and 77 and 70 kJ mol-1 for ∆G°, respectively. According Zamorano et al.,32 a ∆G° of 100.32 kJ mol-1 for the purification of PO from real palm leaves (Roystonea regia). These values are lower than those in literature, demonstrating low sensitivity of the PO from SBM to thermal alterations, even when considering a variable rate of temperature. Deoxynivalenol levels reduction After the optimal conditions of purification and enzymatic reaction were defined, beyond the biochemical characterization of PO, it was evaluated for use in the reduction of DON levels. Purified extract showed advantage in DON reduction process, presenting 81% of degradation (with relative deviation of 3.4%) in relation to crude extract that showed 60% (with relative deviation of 12%), where is possible see the potential of reduction levels of DON by the PO. The affinity by substrate demonstrated for PO from SBM confirmed the necessity of purification of the enzyme by TPP when it used for application in mycotoxin degradation process. In this view, the observed decrease in the concentration of DON in HPLC-UV suggests the possibility that the enzyme is adsorbing and metabolizing DON. These results suggest the necessity of futures studies on the interaction between PO and DON and the mechanism and strategy for using PO in environmental bioremediation.
CONCLUSION When compared with TPP, the chromatographic separation with Sephadex G-100, showed a decrease in the FP of 10.3 turn. The process efficiency can be observed in the recuperation of 20% and in the chromatographic method of 50% for TPP. These results show that efficient purification and optimal characterization are necessary for enzyme application in industrial processes including environmental bioremediation.
ACKNOWLEDGEMENTS The financial support of Higher Education Personnel Improvement Coordination Coordenaçao de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), National Research Council Conselho Nacional de Pesquisa (CNPq), The Research Support Foundation in Rio Grande do Sul state Fundaçao de Amparo à pesquisa no estado do Rio Grande do Sul (FAPERGS) and Federal University of Rio Grande (FURG).
REFERENCES 1. Kalin, R.; Atasever, A.; Özdemir, H.; Food Chem. 2014, 150, 335. 2. Balasubramanian, M.; Boopathy, R.; J. Mol. Catal. B: Enzym. 2013, 90, 33. 3. Valetti, W.; Picó, G.; Sep. Purif. Technol. 2013, 119, 1. 4. Vujčić, Z.; Janović, B.; Lončar, N.; Margetić, A.; Božić, N.; Dojnov, B.; Vujčić, M. Int. Biodeterior. Biodegrad. 2015, 97, 124. 5. Garda-Buffon, J.; Kupski, L.; Badiale-Furlong, E.; Food Sci. Technol. 2011, 31, 198. 6. Manu, T.; Rao, P.; Food Chem. 2009, 114, 66. 7. Mdluli, K.; Food Chem. 2005, 92, 311. 8. Linke, D.; Berger, G.; J. Biotechnol. 2011, 152, 125. 9. Cardinali, A.; Tursi, N.; Ligorio, A.; Giuffrida, G.; Napolitano, L.; Caliandro, R.; Sergio, L.; Venere, D.; Lattanzio, V.; Sonnante, G.; Plant Physiol. Biochem. 2011, 49, 395. 10. Devaiah, P.; Shetty, S.; Pestic. Biochem. Physiol. 2009, 94, 119. 11. Lowry, H.; Rosenbrough, J.; Farr, L.; Randall, J.; J. Biol. Chem. 1951, 193, 265. 12. Boer, G.; Obici, L.; Souza, M.; Peralta, M.; Process Biochem. 2006, 41, 1203. 13. Özer, B.; Akardere, E.; Çelem, B.; Önal, S.; Biochem. Eng. J. 2010, 50, 110. 14. Laemmli, K.; Nature 1970, 227, 680. 15. Pal, A.; Khanum, F.; Process Biochem. 2011, 46, 879. 16. Tanaka, T.; Yoneda, A.; Inoue, S.; Sugiura, Y.; Ueno, Y.; J. Chromatogr. A 2000, 882, 23. 17. Srinivas, D.; Barhate, S.; Raghavarao, S.; J. Food Eng. 2002, 54, 1. 18. Silva, B.; Souza, M.; da Costa, A. M. de S.; Jorge, L. M. de M.; Paraíso, R.; Powder Technol. 2013, 229, 61. 19. Deepa, S.; Arumughan, C.; Phytochemistry 2002, 61, 503. 20. Al-Senaidy, A.; Ismael, A.; Saudi J. Biol. Sci. 2011, 18, 293. 21. Kumar, R.; Singh, A.; Singh, K.; Jagannadham, V.; Process Biochem. 2011, 46, 1350. 22. Singh, P.; Prakash, R.; Shah, K.; Talanta 2012, 97, 204. 23. Fodil, D.; Badis, A.; Jaouadi, B.; Zaraî, N.; Ferradji, Z.; Boutoumi, H.; Int. Biodeterior. Biodegrad. 2012, 65, 470. 24. Pandey, P.; Dwivedi, N.; J. Mol. Catal. B: Enzym. 2011, 68, 168. 25. Suzuki, T.; Honda, Y.; Mukasa, Y.; Kim, S.; Phytochemistry 2006, 67, 219. 26. Motamed, S.; Ghaemmaghami, F.; Alemzadeh, I.; Ind. Eng. Chem. Res. 2009, 48, 10614. 27. Srinivas, D.; Rashmi, R.; Raghavarao, S.; Process Biochem. 1999, 35, 43. 28. Tayefi-Nasrabadi, H.; Asadpour, R.; J. Biol. Sci. 2008, 8, 1310. 29. Saqib, N.; Hassan, M.; Khan, F.; Baig, S.; Process Biochem. 2010, 45, 641. 30. Kagliwal, D.; Singhal, S.; Int. J. Biol. Macromol. 2014, 69, 329. 31. Ertan, H.; Siddiqui, S.; Muenchhoff, J.; Charlton, T.; Cavicchioli, R.; Biochimie 2012, 94, 1221. 32. Zamorano, S.; Pina, G.; Arellano, B.; Bursakov, A.; Zhadan, P.; Calvete, J.; Sanz, L.; Nielsen, R.; Villar, E.; Gavel, O.; Roig, G.; Watanabe, L.; Polikarpov, I.; Shnyrov, L.; Biochimie 2008, 90, 1737. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access