
 |
 |
 |
 |
 |
Revisão
|
|
| Células solares de perovskitas: uma nova tecnologia emergente Perovskites solar cells: a new emerging technology |
|
Ellen RaphaelI; Mariana Nascimento SilvaI; Rodrigo SzostakII; Marco Antônio SchiavonI,II,*; Ana Flávia NogueiraII
IDepartamento de Ciências Naturais, Universidade Federal de São João del-Rei, Campus Dom Bosco, 36301-160 São João del-Rei - MG, Brasil Recebido em 31/01/2017 *e-mail: schiavon@ufsj.edu.br Solar energy has been considered as an important source of clean and safe energy to overcome the problems associate to the burn of fossil fuels (e.g. climate changes, pollution, health problems, etc.). In the area of photovoltaics, devices that convert solar energy into electricity, perovskites solar cells (PSC) have attracted great attention due to their rapid development, high energy conversion efficiency, diversification of the processing methods and different materials. The fantastic properties of the hybrid perovskite materials, such as high absorption coefficient, direct and tunable bandgap, ambipolar charge carrier, simple preparation, etc., have quoted this technology as one the most important in this century. The rapid development of PSC has provided a significant increase in energy conversion efficiency, which was first reported, in 2009, as 3.8% and now reaches up to 22.10%, according to the National Laboratory of Renewable Energy (NREL). Many studies have been carried out to further increase the efficiency of devices and solve problems as the instability of the material, and the presence of lead, so that the PSC can be applied commercially. This paper presents a review on PSC and the major advances reported in device's architecture, preparation methods, novel materials and interface engineering. INTRODUÇÃO Há muitos anos a humanidade tem feito uso de combustíveis fósseis para satisfazer grande parte da demanda de energia. Porém, com a diminuição progressiva das reservas de combustíveis fósseis, deterioração do meio ambiente (devido ao efeito estufa), poluição do ar e água e, consequentemente, com os problemas de saúde humana associados ao aquecimento global, além das questões econômicas e políticas relacionadas à saúde e preservação do meio ambiente, o desenvolvimento de fontes alternativas de energia tem se tornado cada vez mais importante.1,2 Tendo em vista essas preocupações, a energia solar fotovoltaica pode ser considerada umas das fontes de energia alternativa mais promissora, pois provém de uma fonte abundante, limpa e segura, não gera ruídos e ainda permite a geração de energia elétrica em áreas remotas.3 A energia solar é, de uma forma ou de outra, a fonte de quase toda a energia na Terra.4 O sol fornece ao nosso planeta cerca de 10.000 vezes mais energia que o nosso consumo diário global. A energia solar pode ser convertida em calor, o qual pode ser utilizado diretamente para o abastecimento de água quente, ou em eletricidade, através do efeito fotovoltaico, nas células solares.5,6 A constituição básica de uma célula solar é baseada em duas camadas de semicondutores (junção), sendo uma camada contendo carga positiva (p) e outra contendo carga negativa (n), que geram corrente elétrica quando expostas à radiação solar.7 Atualmente, as células solares são classificadas em três tipos, a saber: Células Solares de primeira, segunda e terceira gerações, as quais são diferenciadas basicamente pelos materiais e tecnologias de processamento utilizados em sua fabricação. As células solares de primeira geração, também conhecidas como células de Wafer (Figura 1),8 são baseadas na junção pn, cujo principal exemplo são as células solares de silício cristalino, tecnologia dominante no mercado atual, que apresentam eficiências entre 15-20%, e que, apesar da queda acentuada dos preços nos últimos anos, ainda apresentam alto custo de produção e instalação.9 As células solares de segunda geração, ou de filmes finos comerciais à base de silício amorfo, CIGS (seleneto de cobre, índio, gálio), CdTe (telureto de cádmio) ou CZTS (sulfeto de cobre, zinco, estanho), têm custo mais baixo, se comparado com as de silício cristalino, embora ainda exijam processos de produção envolvendo vácuo e tratamentos térmicos a altas temperaturas -, porém apresentam uma eficiência mais baixa que as anteriores. Já as células de terceira geração, ou de filmes finos emergentes, representam as tecnologias atuais mais pesquisadas no meio científico. São à base de materiais orgânicos, corantes, pontos quânticos ou perovskitas, e também englobam estruturas mais complexas como as células tandem (ou multi-junção), células de portadores quentes e tecnologias de upconversion. Essas células envolvem a geração de múltiplos éxcitons, oferecendo a possibilidade de ultrapassar os limites teóricos de eficiência de Shockley e Queisser (S-Q), associando elevada eficiência e baixo custo.7,8,10
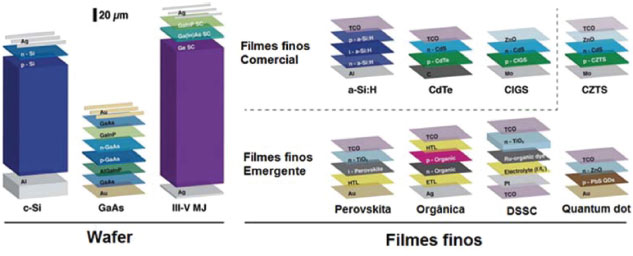 Figura 1. Representação das células solares que compõem as três gerações de células solares. Primeira geração (Wafer), segunda geração (Filmes finos comerciais) e terceira geração (Filmes finos emergentes) (Adaptado com permissão da referência 8)
Os últimos anos trouxeram uma rápida, e sem precedentes, ascensão de uma nova classe de células solares, dentro das células de terceira geração, baseadas em perovskitas.11-18 Atualmente, a aplicação desses materiais em dispositivos fotovoltaicos tem se tornado objeto de estudo de vários grupos de pesquisa, apresentando considerável evolução na arquitetura das células solares, bem como no valor de eficiência de conversão (PCE, do inglês Power Conversion Efficiency), a qual atualmente é certificada com valor de 22,1%, de acordo com o Laboratório Nacional de Energia Renovável (NREL) dos Estados Unidos.19
PRINCIPAIS TIPOS DE CÉLULAS SOLARES A história das células solares iniciou-se em 1839, quando Edmond Becquerel observou que duas placas de latão imersas em um eletrólito líquido produziam eletricidade quando expostas à luz solar, fenômeno que recebeu o nome de efeito fotovoltaico.10 Mais tarde, na década de 1870, Willoughby Smith, W. G. Adams e R. E. Day observaram o efeito fotovoltaico usando selênio.20 Esse fato possibilitou que, em 1883, Charles Fritts construísse a primeira bateria solar com folhas de selênio, com uma eficiência de apenas 1%, mas que gerou grande repercussão pelo fato de que naquela época não havia geração de energia sem a queima de combustíveis.10 Foi então que, em 1954, surgiu a primeira célula solar à base de silício, com eficiência de 6%, desenvolvida por Chapin e colaboradores da Bell Labs.7 Devido ao custo desses dispositivos ser bastante elevado, somente em 1973, com a crise do petróleo, o interesse nas células solares foi retomado, com o intuito de torná-las uma fonte de energia substituta e mais acessível; o foco principal das pesquisas nessa época foram as células de silício cristalino, propiciando um grande avanço e desenvolvimento de diversas tecnologias de fabricação, o que é refletido atualmente no domínio do mercado mundial dessa classe de células solares.21 Estudos mais aprofundados, envolvendo novos tipos de materiais para confecção de células solares e novas tecnologias, começaram depois que Grätzel e O'Regan, em 1991, produziram a primeira célula solar sensibilizada por corante.22 Na avaliação de células solares, a eficiência e o custo são os parâmetros mais importantes a serem considerados; dentro desse contexto, os tipos de materiais utilizados na confecção, a geometria e a estrutura da célula são fatores-chave para obtenção de um elevado desempenho com redução de custos, fato que pode ser observado na evolução das tecnologia das células solares.23 Nas células solares convencionais, também conhecidas como células solares de primeira geração, o semicondutor utilizado é o silício, um elemento tetravalente em rede cristalina, sendo um semicondutor intrínseco.24 Diferentemente de um átomo isolado no material sólido, com a aproximação de diversos átomos, as funções de onda que descrevem os elétrons individuais se superpõem e somam-se de modo que resultam em um desdobramento dos níveis de energia, constituindo praticamente uma banda contínua de energia, na qual as energias possíveis dos elétrons estão agrupadas em bandas permitidas (bandas de valência e condução) separadas por bandas proibidas de energia (bandgap). Na construção de uma célula solar de silício é necessária uma junção pn.14 Na dopagem para obtenção da camada de silício tipo n, um átomo de silício é substituído por um átomo do grupo V, o qual tem 5 elétrons de valência; nesse caso, o átomo dopante é um doador de elétrons e o silício é considerado um semicondutor tipo n. Porém, quando é adicionado um átomo dopante do grupo III, com 3 elétrons de valência, ocorre a falta de um elétron no retículo cristalino para cada átomo de impureza, ou seja, aparecem buracos, que podem ser completados por elétrons vizinhos, fazendo com que o buraco se desloque. Sendo assim, o átomo dopante é um aceptor de elétrons e o silício é considerado um semicondutor tipo p.7 Uma vez que a região tipo n apresenta uma concentração elevada de elétrons e a de tipo p uma concentração elevada de buracos, os elétrons livres do lado n passam para o lado p, onde encontram buracos que os anulam. Ocorre, então, um acúmulo de elétrons no lado p, que passa a ficar negativamente carregado. O mesmo processo ocorre com o lado n, porém, este fica positivamente carregado.24 Esse acúmulo de cargas na interface de contato cria um campo elétrico que eventualmente leva a um equilíbrio da passagem de cargas de um lado para o outro. Sendo assim, quando essa junção pn for exposta à luz, com energia maior que a banda proibida de energia ou bandgap (BG), ocorrerá a formação de elétrons e buracos livres. Devido ao campo elétrico formado na junção pn, os elétrons irão para o silício tipo n e os buracos para o tipo p. Assim, uma corrente é produzida através da junção, fazendo com que surja uma diferença de potencial, no chamado efeito fotovoltaico, e o dispositivo passa a operar como uma célula de silício ao se fechar o circuito externo.24 Diferentes tipos de células solares, preparadas com diversos materiais além do silício e em diferentes configurações, vêm se tornando destaque na pesquisa atualmente, fazendo parte da nova geração de dispositivos fotovoltaicos. São elas: as células solares sensibilizadas por corante (DSSC, do inglês Dye-Sensitized Solar Cells), as células solares sensibilizadas por pontos quânticos (QDSSC, do inglês Quantum Dot-Sensitized Solar Cells) e as células solares de perovskita (PSC, do inglês Perovskite Solar Cells). A história das DSSCs começou em 1887, quando o primeiro fenômeno de fotossensibilização por corantes orgânicos foi relatado; porém, somente a partir de 1991, com o trabalho de Gratzel e O´Regan reportando uma célula com eficiência de 7,1%, as DSSCs receberam maior atenção da comunidade científica. O recorde de eficiência para esse tipo de célula tem certificação de 11,9% pelo NREL,19,22 e 14% sem certificação, reportado recentemente por Kakiage et al.25 Esse tipo de célula é constituído basicamente por um fotoanodo composto por um substrato de vidro condutor transparente, formado de óxido de estanho dopado com flúor (do inglês: Fluorine doped Tin Oxide - FTO) ou índio (do inglês: Indium doped Tin Oxide - ITO), sobre o qual é depositada uma camada de TiO2 nanocristalino sensibilizado por um corante, um eletrólito (par redox) e um contra eletrodo, montados em um arranjo tipo sanduíche.26,27 Quando a célula é exposta à luz solar, o sensibilizador absorve os fótons incidentes, fazendo com que os elétrons passem do seu orbital molecular ocupado de maior energia (HOMO, do inglês Highest Occupied Molecular Orbital) para o orbital molecular desocupado de menor energia (LUMO, do inglês Lowest Unccupied Molecular Orbital). Os elétrons excitados são, então, injetados na banda de condução do TiO2, sendo transportados externamente até o contra eletrodo, e com isso ocorre a oxidação do corante. Já o eletrólito promove a regeneração do corante a partir dos elétrons recebidos do contraeletrodo.28-30 As QDSSCs podem ser consideradas derivadas das DSSCs, em que pontos quânticos (PQs) semicondutores têm sido utilizados como sensibilizadores no lugar de corantes, com o intuito de aumentar a eficiência das células solares devido às suas propriedades optoeletrônicas excepcionais, incluindo: bandgap que varia em função do tamanho da nanopartícula, elevado coeficiente de absorção, maior estabilidade e emissão de múltiplos éxcitons.31-33 Quando o dispositivo é exposto à luz solar, são criados pares elétron-buraco nos PQs, já que os elétrons da banda de valência adquirem energia suficiente para alcançar a banda de condução e, na sequência, esses elétrons da banda de condução do PQ são injetados no filme de TiO2, resultando na formação de buracos no sensibilizador, que será regenerado através da doação de elétrons do par redox do eletrólito, este, por sua vez, recebe elétrons do contraeletrodo, de modo semelhante ao que ocorre nas DSSCs.31,32,34
CÉLULAS SOLARES DE PEROVSKITAS (PSCS) Células solares de perovskitas (PSCs) baseadas em haletos orgânico-inorgânicos têm chamado a atenção da comunidade científica nos últimos anos por chegar à eficiência superior a 20% com apenas meia década de estudos.11,12,16,35 Além de terem alta eficiência, as PSCs podem ser preparadas com materiais e técnicas de baixo custo, tornando-se extremamente interessantes para comercialização em larga escala.36,37 As propriedades únicas desses materiais absorventes fornecem numerosas vantagens em aplicações optoeletrônicas, que em muitos aspectos se originam a partir da natureza da estrutura da perovskita.38 Perovskitas de haleto de metilamônio de chumbo surgiram como materiais fotovoltaicos promissores devido às suas propriedades, tais como: elevado coeficiente de absorção na região visível do espectro solar;39 bandgap direto (devido ao alinhamento das bandas de valência e condução) e tunelável (em que elétrons podem escapar de regiões cercadas por barreiras de potencial, mesmo quando sua energia cinética é menor que a energia potencial da barreira), com a alteração dos cátions e ânions;40,41 transporte ambipolar de carga;42,43 alta mobilidade de elétrons e buracos comparados com semicondutores orgânicos;44 e comprimento de difusão que excede micrometros em monocristais.45-48 Em uma típica PSC, a camada ativa é composta pela perovskita (sendo a responsável por absorver a radiação), e é colocada entre uma camada transportadora de elétrons (ETL, do inglês Electron Transport Layer), geralmente TiO2 ou fulereno, e uma camada transportadora de buracos (HTL, do inglês Hole Transport Layer), geralmente SPIRO-OMeTAD ou PEDOT:PSS.49-51 Uma das camadas, HTL ou ETL, é depositada sobre um eletrodo condutor transparente, sendo FTO ou ITO, e sobre a outra camada é depositado um metal (Au, Ag ou Al), completando o dispositivo. Com esses materiais é possível preparar células solares de perovskitas na configuração regular e invertida. A configuração regular teve origem nas DSSCs de estado sólido e se caracteriza pelas camadas na ordem FTO/ETL/perovskita/HTL/metal, em que a iluminação é feita pelo lado da camada transportadora de elétrons. Já a configuração invertida tem origem nas células solares orgânicas regulares com as camadas ETL e HTL invertidas, em que o eletrodo condutor transparente utilizado geralmente é o ITO.52 A Figura 2 resume a origem das diferentes configurações das células solares de perovskitas.
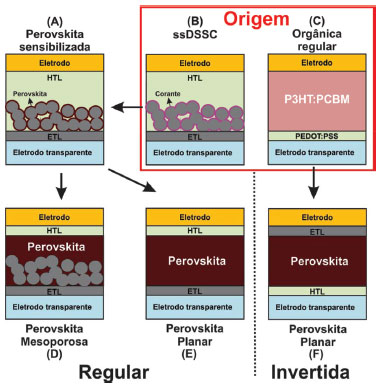 Figura 2. Diferentes configurações de células solares de perovskitas. (A) Célula solar de perovskita sensibilizada, (B) célula solar sensibilizada com corantes de estado sólido (ssDSSC, do inglês solid state dye sensitized solar cell), (C) célula solar orgânica regular, (D) célula solar de perovskita mesoporosa, (E) célula solar de perovskita planar e (F) célula solar de perovskita invertida
PEROVSKITAS Descobertas por Gustav Rose, em 1839, as perovskitas representam uma classe de materiais com características únicas que hoje estão revelando inúmeras e versáteis aplicações em uma ampla gama de dispositivos tecnológicos.53 O termo perovskita é usado para se referir a um grande grupo de compostos com uma estrutura cristalina que se assemelha à estrutura da perovskita mineral titanato de cálcio (CaTiO3). Sua fórmula geral é ABC3, em que A e B são cátions e C é um ânion, o qual geralmente é o íon óxido O2-, ou íons haleto (Cl-, Br-, F-). A estrutura cristalina da perovskita descrita na Figura 3(a) é idealmente cúbica. Apesar da estrutura básica ser bem simples, existe ainda um grande número de variações estruturais entre elas, pois as posições A e B podem acomodar diversos elementos da tabela periódica, como ilustrado na Figura 3(b).54,55
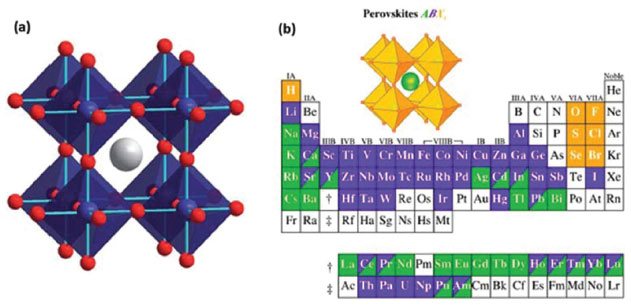 Figura 3. (a) Estrutura da perovskita cúbica ABC3, em que a esfera cinza é o cátion A e as esferas azuis e vermelhas representam os íons B e C, respectivamente; (b) possível combinação de elementos que podem ocupar sítios na grade cristalina de perovskita (Adaptado com permissão das referências 54 e 55)
Dada a compatibilidade química e estrutural entre as perovskitas, seu alojamento estrutural maleável oferece uma oportunidade para personalizar propriedades eletrônicas, magnéticas e ópticas em filmes finos, de maneira que não são possíveis com semicondutores convencionais.55 Ferroeletricidade, ferromagnetismo, magnetoeletricidade, luminescência e supercondutividade são algumas de suas características.49,56 A maior classe de perovskitas é aquela para a qual o ânion é o íon óxido, sendo que sua abrangência é ainda aumentada ao se observar que soluções sólidas e não-estequiometria também são comuns da estrutura da perovskita, como em Ba1-xSrxTiO3 e SrFeO3-y.57 Um tipo de perovskita muito utilizado em células solares é a baseada em haletos orgânico-inorgânicos, cuja fórmula geral é ABX3, em que A e B são cátions e X é um ânion, geralmente íons haleto (X = I-, Br-, Cl-). O cátion A é orgânico (tipicamente CH3NH3+, C2H5NH3+ ou HC(NH2)2+), geralmente maior e mais eletropositivo em comparação aos cátions B, os quais são tipicamente íons metálicos bivalentes (Pb2+ ou Sn2+). O maior cátion, o cátion A, ocupa um sítio cubo-octaédrico compartilhado com doze ânions X, localizado entre oito unidades octaédricas BX6, enquanto o menor cátion, o cátion B, está estabilizado em um sítio octaédrico compartilhado com seis ânions X.16,17,36,37 A Figura 4 representa a estrutura cristalina da perovskita e sua célula unitária.
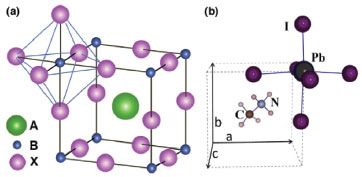 Figura 4. Estrutura da perovskita ABX3 mostrando o sítio octaédrico BX6 e o maior cátion A ocupando o sítio cubo-octaédrico, (b) Célula unitária da perovskita cúbica CH3NH3PbI3 (Adaptado com permissão das referências 37 e 58)
O tamanho dos íons A e B influenciam diretamente na estrutura cristalina, sendo a estabilidade cristalográfica da perovskita estimada pelo fator octaédrico µ (µ=rB/rX) e pelo fator de tolerância τ (τ=(rA+rX)/[21/2(rB+rX)]), em que rA, rB e rX são os raios iônicos de A, B e X, respectivamente.1,10,12 A estrutura cúbica ideal ocorre quando τ = 1, enquanto τ < 1 indica que o cátion A é pequeno demais e τ > 1 indica que A é grande demais para caber nas cavidades entre as unidades octaédricas BX6. Quando τ < 1, ocorre a chamada distorção octaédrica, em que o tamanho muito pequeno do cátion A resulta na diminuição da simetria do sistema. Com o intuito de manter a ligação, cada octaedro deve girar sempre que um deles mudar sua posição em torno do eixo normal da camada, podendo resultar, assim, nas chamadas inclinações out of phase (no sentido oposto da fase, quando a direção da rotação se alterna entre diferentes camadas) ou inclinações in phase (quando as rotações ocorrem na mesma direção). A distorção Jahn-Teller ocorre quando a configuração eletrônica do cátion é degenerada orbitalmente, produzindo estado de menor energia instável. Esse efeito ocorre como resultado de um declínio linear da energia, seguido de desarranjos no cristal, enquanto, ao mesmo tempo, a energia elástica do sistema aumenta. Rearranjos distorcidos podem se manifestar tanto pelo alongamento quanto pelo encolhimento das ligações octaédricas, causando uma queda da energia ao mínimo. Assim, uma série de elementos podem servir como substituintes para os materiais de perovskitas absorvedores de luz.1 A estrutura cúbica da perovskita ocorre geralmente em 0,81 < τ < 1,11 e 0,44 < µ < 0,90.1,17,53 Os valores mais baixos de τ resultam em uma estrutura menos simétrica tetragonal ou ortorrômbica.16 Tomando o CH3NH3PbX3 como exemplo, de acordo com os valores dos raios dos respectivos íons, os fatores τ calculados são 0,85, 0,84 e 0,83 e os fatores µ são 0,66, 0,61 e 0,54, para X = Cl-, Br- e I-, respectivamente. Desta forma, espera-se que perovskitas de haletos de metilamônio de chumbo apresentem estrutura cúbica. Apesar dessas limitações, transições de fases de CH3NH3PbX3 ocorrem com a variação da temperatura, de tal forma que em temperaturas mais baixas ocorre a fase ortorrômbica, que se transforma em tetragonal e depois em fase cúbica à medida que se aumenta a temperatura.59 À temperatura ambiente, as perovskitas com íons Cl- e Br- são cúbicas, enquanto que aquela com íons I- é tetragonal, como mostrado na Tabela 1, que lista os fatores estruturais em função da temperatura.
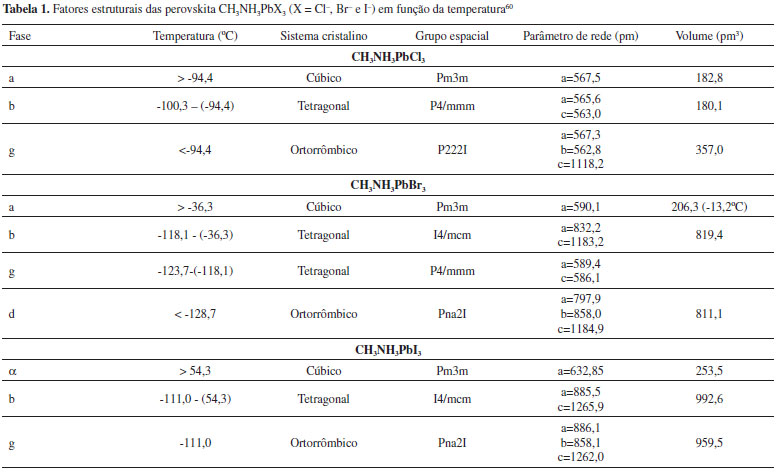
Para os haletos de perovskitas orgânico-inorgânicos de presente interesse em células solares, o cátion A, geralmente, é o metilamônio (MA (CH3NH3+)), com rA = 1,80 Å; porém, o etilamônio (EA (CH3CH2NH3+)), com rA = 2,30 Å, e o formamidínio (FA (NH2CH=NH2+)), com rA estimado entre 1,90 e 2,20 Å, também têm apresentado bons resultados. O ânion X mais utilizado é o halogênio iodeto, I-, (rX = 2,20 Å), sendo o Br- (rX = 1,96 Å) e Cl- (rX = 1,81 Å) também utilizados em um material de haletos mistos. Com o intuito de se obter uma maior eficiência, o cátion B mais utilizado universalmente é o Pb, com rB = 1,19 Å. O cátion Sn (rB = 1,10 Å) forma compostos similares, porém com uma menor estabilidade, devido à facilidade de oxidação do Sn em SnI4 em perovskitas iodadas, enquanto o Pb apresenta uma maior proteção contra a oxidação. Desse modo, os materiais de perovskita mais bem sucedidos até hoje são as variações sobre o CH3NH3PbX3, como o iodeto de metilamônio de chumbo (CH3NH3PbI3)61-63 e os haletos mistos CH3NH3PbI3-xBrx ,64 CH3NH3PbI3-xClx,65,66 FAPbI367 e FA0,83MA0,17Pb(I0,83Br0,17)3.16,55,68
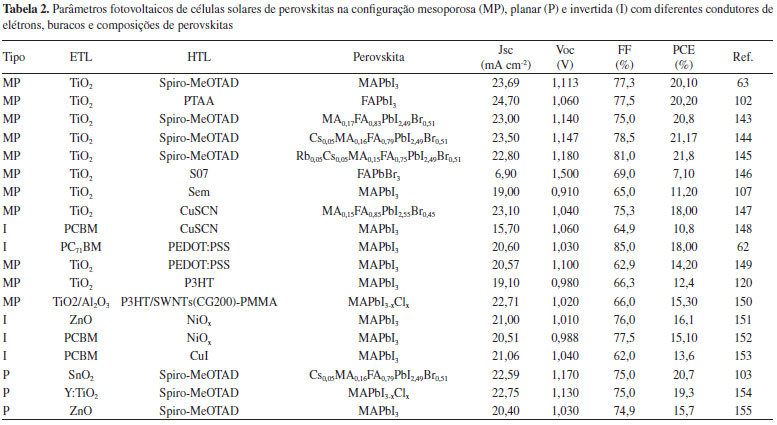
FORMAÇÃO DO FILME DE PEROVSKITA A formação do filme de perovskita, geralmente segue a seguinte reação: 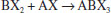 em que BX2 (M = Pb, Sn; X = I, Br, Cl) e AX (A = MA, EA, FA; X = I, Br, Cl). A cinética da reação de formação da perovskita é impressionantemente rápida, e existem diversos métodos de formação do filme que confirmam essa cinética. A qualidade do filme desempenha um papel fundamental para alcançar alta eficiência em células solares de perovskitas, necessitando, assim, de uma maior compreensão dos mecanismos de formação do filme.38,69-72 A morfologia de um filme, o qual conta com dois ou mais precursores da perovskita, está diretamente relacionada à eficiência, a qual depende da otimização de parâmetros relacionados à concentração dos precursores, solvente, temperatura, velocidade de rotação, tempo, entre outros, e até dos processos de pós-deposição.73 Os precursores podem ser depositados de forma simultânea ou independente, resultando em uma variedade de técnicas de deposição.74 Para perovskitas simples (CH3NH3PbI3, CH3NH3PbBr3, FAPbI3), a maioria dos métodos de deposição baseia-se no mesmo princípio: a combinação de um componente orgânico, iodeto ou brometo de metilamônio (MAI ou MABr), por exemplo, com um componente inorgânico, como o iodeto, cloreto ou brometo de chumbo (PbI2, PbCl2 ou PbBr2), para formar a perovskita (MAPbI3, MAPbI3-xClx ou MAPbBr3, respectivamente).75 Dentre os métodos de preparação de perovskitas, cinco se destacam: deposição de solução em uma etapa (One-step solution deposition), deposição sequencial de soluções em duas etapas (Two-step sequential solution deposition), deposição a vácuo, processo de deposição assistida por vapor e engenharia de solvente.59 Deposição de solução em uma etapa Nesta técnica, geralmente, a solução precursora é preparada a partir da dissolução de uma mistura de BX2 (B = Pb, Sn; X = I, Br, Cl) e AX (A = MA, FA; X = I, Br, Cl), ambos em estado sólido, em um solvente orgânico apropriado, como N,N-dimetilformamida (DMF), gama-butirolactona (GBL) ou dimetilsulfóxido (DMSO).38,41,50,76-78 Em seguida, a solução é depositada sobre o ETL ou HTL - dependendo da configuração da célula solar - e, finalmente, passa por um tratamento térmico, produzindo a fase final da perovskita.38,58 Entretanto, esse método de deposição ainda enfrenta o desafio da formação de uma camada de perovskita homogênea, de modo que requer a otimização de múltiplos parâmetros incluindo processo de pós deposição.52,76,79,80 Deposição sequencial de soluções em duas etapas No processo de deposição sequencial,71,72 inicialmente, uma camada de BX2 (B = Pb, Sn; X = I, Br, Cl) é depositada, sob condições apropriadas para cada caso, e a camada de perovskita é então finalizada, tanto pela imersão do filme de MX2 em uma solução de AX (A = MA, FA; X = I, Br, Cl) em isopropanol, quanto também pelo spin-coating da solução de AX no filme de MX2. 55,81 Um controle adicional sobre a morfologia do filme pode ser obtido com esse método de deposição, visto que é possível se obter cristais tipo cuboides uniformes, enquanto o método em uma etapa apresenta uma morfologia irregular.75,76 Deposição de vapor de fonte dupla O método de deposição de vapor à vácuo conta com a utilização dos compostos BX2 e AX como precursores inorgânico e orgânico, respectivamente. Esses precursores são evaporados de forma simultânea a partir de fontes separadas com temperaturas de aquecimento diferentes, sobre um substrato de FTO revestido com TiO2, em rotação, sob vácuo.38,59,66,76,82,83 Imediatamente após a formação do filme, é realizado um processo de aquecimento em atmosfera de N2 para a cristalização da perovskita. A utilização desse método resulta em filmes extremamente uniformes e nanocristalinos, o que é dificilmente observado em processamentos de soluções.59 Entretanto, essa evaporação térmica requer um vácuo elevado, limitando, assim, a eficiência de custo e a produção em massa dos filmes de perovskitas.73,76 Processo de deposição assistida por vapor O processo de deposição assistida por vapor pode ser considerado como uma modificação da deposição sequencial de soluções em duas etapas, sendo a segunda etapa substituída por uma deposição de vapor.59,73,82 Inicialmente, o filme de MX2 é depositado por spin-coating sobre o substrato, seguido da exposição a um ambiente com atmosfera de N2, rico em vapor de AX, sob elevada temperatura.38,59,74,83 O filme formado pelo processo de solução assistida por vapor é mais estável, apresentando cobertura completa da superfície, com grãos bem definidos e uniformes, e baixa rugosidade da superfície em comparação aos demais métodos também utilizados.59,74,83 Os esquemas dos diferentes métodos utilizados são apresentados na Figura 5.
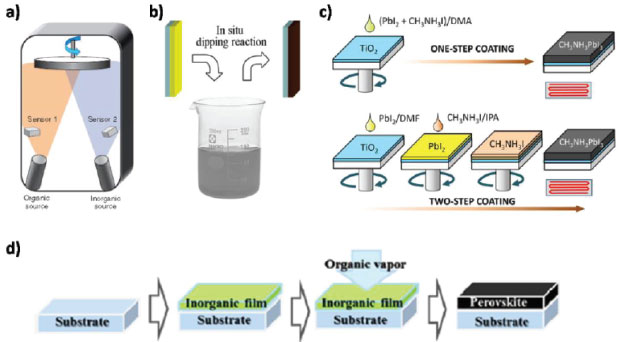 Figura 5. Preparação de filme de perovskita por diferentes métodos de deposição: (a) coevaporação de fonte dupla utilizando PbCl2 e MAI; (b) deposição sequencial de PbI2 em solução de MAI; (c) deposição em solução de etapa única utilizando a mistura PbI2 e MAI, e deposição sequencial de PbI2 e MAI; (d) solução assistida por vapor utilizando PbI2 e vapor de MAI (Adaptado com permissão das referências 66, 83, 84 e 85)
Método de engenharia de solventes Atualmente, o método que tem trazido melhores resultados nas PSCs é o método de engenharia de solventes. Esse método consiste na preparação de uma solução de perovskita em uma mistura de solventes (DMSO/GBL ou DMSO/DMF), e esta é depositada por spin coating. Ainda durante os últimos segundos de rotação do spin coating, um não-solvente da perovskita (tolueno, clorobenzeno, diclorobenzeno ou dietil eter) é colocado sobre o filme de perovskita, levando à formação do intermediário MA2Pb3I8.2DMSO. As moléculas de DMSO não deixam ocorrer a formação direta da perovskita, formando, assim, um intermediário, que com tratamento térmico perde as moléculas de DMSO, formando a perovskita. Com esse método, eficiências acima de 20%63,68,86,87 têm sido obtidas, com máximo de 21,6%, reportado recentemente por Saliba et al.88 Pontos quânticos de perovskita As perovskitas híbridas orgânica-inorgânica (CH3NH3PbX3, X = I, Br, Cl) têm ganhado destaque como os mais promissores materiais fotovoltaicos, no entanto, são extremamente sensíveis ao oxigênio e umidade, o que dificulta sua aplicação prática. Como uma alternativa para melhorar a estabilidade, uma nova classe de perovskitas completamente inorgânicas, à base de haletos de césio e chumbo, têm sido preparadas na forma de pontos quânticos coloidais (CsPbX3, X = Cl, Br, I).89,90 Esses pontos quânticos de perovskitas inorgânicas (PQPIs) foram reportados pela primeira vez por Kovalenko et al.,81 e foram incorporados em células solares de perovskitas por Kulbak et al.,91 apresentando estabilidade e eficiência de conversão de 6,6%. Desde então têm sido foco das pesquisas atuais, como materiais de alta eficiência de luminescência, com alta absorção de seção transversal, elevado rendimento quântico de emissão (~90%), dinâmica de transferência de carga ultrarrápida, além de que o comprimento de onda de emissão pode ser sintonizado em todo o espectro visível através do controle de tamanho dos PQPIs, ou de sua constituição estequiométrica.81,92-95 No que tange à área de células solares, a princípio, um elevado rendimento quântico não é interessante, pois, pela definição do conceito, indica que a probabilidade da desativação do estado excitado pela emissão de fótons é grande. No entanto, um alto rendimento quântico indica baixa densidade de estados de armadilha presente no material, ou seja, o material apresenta uma alta qualidade óptica. Estados de armadilha são a principal fonte de desativação do estado excitado de pontos quânticos, por ser um processo mais rápido (10-10 s) do que a fluorescência (10-8 s). Como o processo de transferência de carga também é da ordem de 10-10 s, uma alta densidade de estados de armadilha diminui a taxa da transferência de carga. Portanto, pontos quânticos com alta eficiência de luminescência e elevado rendimento quânticos são também desejados para aplicação em células solares.
DESENVOLVIMENTO DAS CÉLULAS SOLARES DE PEROVSKITA Progressos nos dispositivos e eficiências de conversão de energia O interesse em haletos de perovskita já existe há mais de um século, porém, foi na década de 1990 que o grupo de Mitzi se concentrou em haletos de perovskitas orgânico-inorgânico em camadas, que apresentavam fortes características excitônicas, e descobriu sua aplicação em transístores de película fina (TFT, do inglês Thin Film Transistors) e diodos emissores de luz (LEDs, do inglês Light-Emitting Diodes).96,97 Embora esses LEDs exibam propriedades fotovoltaicas inegáveis e seu uso em células solares já tenha sido realizado anteriormente, esse assunto não foi estudado na época, devido à toxicidade do Pb, e pelo fato de materiais menos nocivos, como Sn, não terem sido considerados suficientemente robustos. Assim, somente recentemente é que esses materiais foram novamente preparados.16,98,99 As células solares de perovskitas se derivaram das DSSCs, quando, em 2009, Kojima et. al.100 substituíram o corante das DSSCs pelas perovskitas MAPbI3 e MAPbBr3, que foram depositadas sobre o semicondutor TiO2 por processo de auto-organização com deposição de cada uma das soluções precursoras contendo quantidades estequiométricas de CH3NH3X e PbX2, por gotejamento através de um spin-coater, e utilizando um eletrólito à base de LiI/I2 dissolvido em metoxiacetonitrila, obtendo eficiências em torno de 3%. A injeção de elétrons de MAPbX3 (X = I, Br) na camada de TiO2 com os buracos reduzidos pelo par redox é considerada possível de acordo com os níveis de energia correspondentes, os quais são apresentados na Figura 6.59
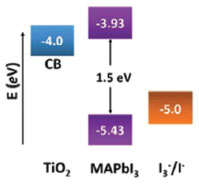 Figura 6. Diagrama de nível de energia do TiO2, CH3NH3PbI3 e do par redox I3-/I- (Adaptado com permissão da referência 59)
Com a mesma configuração, porém utilizando um eletrólito a base de LiI/I2 dissolvido em acetato de etila, dois anos depois, Im et al. atingiram a eficiência de 6,5% com a perovskita MAPbI3, que foi formada in situ sobre o TiO2 através da deposiçao de um solução preparada previamente com a mistura dos precursores CH3NH3I e PbI2 em proporção molar 1:1 em γ-butirolactona. Os autores observaram, porém, que a perovskita se dissolvia no eletrólito de I-/I3- rapidamente, reduzindo a eficiência das células solares.101 Esses resultados não superaram a eficiência das DSSCs naquele momento, porém um grande marco se deu em 2012, quando simultaneamente os grupos de Snaith de Oxford e Gratzel da EPFL, motivados por resolver o problema de dissolução da perovskita pelo eletrólito, substituíram o eletrólito líquido pelo HTL 2,2',7,7'-tetrakis (N,N-di-p-methoxyphenylamine) 9,9'-spirobifluorene (spiro-OMeTAD), chegando, assim, à eficiência de 10%.42,77 A Figura 7 apresenta a estrutura de uma célula solar mesoporosa (a) e o esquema do alinhamento das bandas dos materiais utilizados na construção dessa célula (b). A partir de então, as PSCs receberam atenção de cientistas da área do mundo inteiro, superando rapidamente a barreira dos 20% de eficiência em 2015,102 quando utilizaram perovskitas de iodeto de formamidínio de chumbo (FAPbI3) depositada pelo método de duas etapas e atingiram 22,1% em 2016, conforme certificação NREL.19
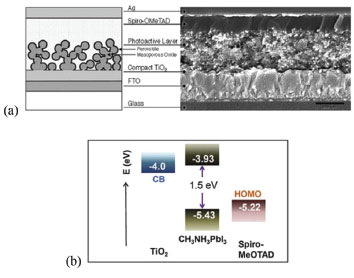 Figura 7. (a) Diagrama esquemático (esquerda) e microscopia da sessão transversal da célula solar mesoporosa sensibilizada por perovskita (direita). (b) Diagrama de nível de energia de TiO2, MAPbI3 e spiro-MeOTAD (Adaptado com permissão das referências 37, 38 e 66)
As primeiras PSCs utilizavam a perovskita como pontos quânticos ou como uma camada fina da perovskita sobre o eletrodo poroso de TiO2. Porém, em 2013 Bruschka et al.72 prepararam PSCs com uma camada espessa de perovskita, preenchendo totalmente os poros e formando uma camada de perovskita sobre o TiO2 poroso, conhecida como capping layer. Com isso a eficiência das células solares chegou a 15%, e hoje todas as células solares de perovskitas com TiO2 poroso apresentam capping layer, o que melhora a separação entre a ETL e HTL, reduzindo a recombinação entre os elétrons e buracos das duas camadas. Ainda em 2013, foram preparadas PSCs sem a presença de camada porosa de TiO2 e com apenas uma camada compacta de TiO2. Ball et al.50 prepararam células solares com 5% de eficiência, sendo estas as primeiras células solares de perovskitas planares, semelhantes às células de filmes finos de silício amorfo e CdTe. Desenvolvendo mais essa ideia, ainda no mesmo ano, Liu et al.66 prepararam células solares com filmes finos de peroviskitas de haletos mistos de metilamônio e chumbo (CH3NH3PbI3-xClx) depositado por fonte de vapor, obtendo 15% de eficiência, demonstrando definitivamente o potencial da arquitetura planar. Recentemente, Anaraki et al. substituíram o TiO2 por SnO2 na camada planar e chegaram à eficiência de 20,7%,103 que é o atual recorde de células solares de perovskitas planares. A falta de uma camada mesoporosa na estrutura contribui para a capacidade de processamento a baixas temperaturas, custos reduzidos de materiais e ainda oferece uma versatilidade considerável em técnicas de deposição dos filmes de perovskitas, o que aumenta ainda mais as opções de materiais transportadores de elétrons e buracos.74,104 Uma das vantagens que as perovskitas têm sobre os corantes é que elas apresentam um alto coeficiente de absorção da luz, sendo necessária uma fina camada de perovskita para uma completa absorção.105 Além disso, o longo caminho de difusão de cargas, que reduz a recombinação na perovskita, e o transporte ambipolar de cargas são fatores importante para o alto desempenho das células solares. Esse transporte ambipolar foi descoberto em 2012, quando Lee et al.42 prepararam células sem camada mesoporosa de TiO2 mas com uma camada mesoporosa de Al2O3. Nesse caso, a camada de Al2O3 isolante é depositada sobre a camada compacta de TiO2, servindo como um suporte para as partículas da perovskita, e não como um receptor de elétrons, uma vez que a injeção de elétrons é impossível devido a uma maior banda de condução (BC) do Al2O3 em relação à da perovskita, como mostrado na Figura 8. Desse modo, o transporte de elétrons ocorre na própria camada da perovskita até que sejam coletadas pelo TiO2 compacto sobre o eletrodo de FTO. Uma PCE de 10,9% foi obtida juntamente com Jsc = 17,8 mA cm-2, Voc = 0,98 V e FF = 0,63. O resultado também demonstrou que as perovskitas, além de sensibilizadores, são capazes de transportar elétrons, e também que a perovskita dentro da camada mesoporosa é contínua, em contraste com estudos anteriores que as consideravam pontos quânticos isolados.100,101 Devido à estrutura mesoporosa e à falta de injeção de elétrons, esse dispositivo foi denominado como "célula solar meso-superestruturada" (CSMS, do inglês meso-superstructured solar cells - MSSC).36,37,59
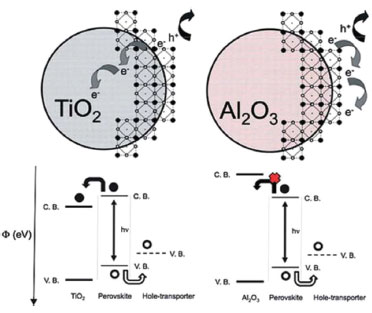 Figura 8. Esquema ilustrando a transferência e o transporte de carga na célula solar de perovskita de TiO2 e uma célula solar não-injetora baseada em Al2O3 (Reimpresso com permissão da referência 105)
Surpreendentemente, no mesmo ano, Etgar et al.43 demonstraram que essas células solares de TiO2 mesoporoso não necessitam de materiais HTL para funcionar - nesse caso, o transporte de buracos ocorre simplesmente através da camada de perovskita. O estudo inicial foi a partir da deposição da perovskita sobre nanofolhas de TiO2, por spin-coating, seguido de evaporação do eletrodo de ouro, obtendo-se a célula solar apresentada na Figura 9. As células solares obtidas apresentaram PCE de 5,5% (Jsc = 16,1 mA cm-2, Voc = 0,63 V e FF = 0,57), a qual foi aumentada para 8,0% por Etgar et al.106 em outro trabalho expandindo a espessura da perovskita. Atualmente, o recorde de eficiência de células solares sem condutor de buracos é de 11,2%, tratando a perovskita com um antisolvente, reportado por Cohen et al.107 O sucesso no funcionamento desses dispositivos indica que MAPbI3 pode agir também como um transportador de buracos eficiente. Porém, o Voc desses dispositivos são mais baixos do que aqueles produzidos com spiro-MeOTAD, indicando que a falta da camada transportadora de buracos resulta em um aumento da recombinação. Assim, com dispositivos comprovando que as perovskitas podem transportar tanto elétrons como buracos, essa família de perovskitas é considerada com uma natureza de transporte ambipolar de cargas.36,43,59,107
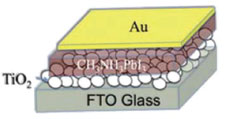 Figura 9. Esquema da célula solar sensibilizada por perovskita sem a camada HTL (Adaptado com permissão da referência 59)
O efeito mais significativo da camada mesoporosa é como sua espessura afeta a eficiência de conversão de energia da célula solar. Nas duas configurações, a de junção líquida e em sua equivalente de estado sólido, demonstrou-se que quanto menor for a espessura da camada mesoporosa, melhor é o desempenho do dispositivo.77,101 Além disso, a dependência exata da espessura da camada mesoporosa na PCE é determinada pela natureza da distribuição da perovskita dentro da camada de TiO2, bem como da espessura da camada superior de perovskita, o que por sua vez depende da concentração da solução de perovskita.36,78,108,109 Uma estrutura com a camada mesoporosa de TiO2 completamente preenchida pela perovskita pode resultar em uma alta performance do dispositivo, pois haverá uma alta densidade de elétrons no TiO2, promovendo uma melhora nas taxas de transporte de carga e eficiência de coleta, além de que a perovskita sobre toda a superfície da camada mesoporosa reduz a possibilidade de recombinação entre o TiO2 e o HTL.74 Diversos estudos têm sido realizados no sentido da substituição de materiais ETLs e HTLs, com o intuito de se obter melhor eficiência para as diversas arquiteturas das células solares de perovskitas.16,74 Óxido de zinco (ZnO),110,111 óxido de estanho (SnO2),103 óxido de nióbio (Nb2O5)112 e PCBM113-116 são alguns materiais que têm sido utilizado como condutores de elétrons no lugar do TiO2. A utilização de ETLs orgânicos evita a utilização de altas temperaturas, como é necessário para preparar o TiO2, permitindo, assim, a fabricação de dispositivos em baixas temperaturas, como a 100º C.1,29 Até o momento, o spiro-MeOTAD tem sido o HTL mais utilizado em células solares, pois apresenta boas propriedades de formação de filme e alta mobilidade de buracos. Entretanto, este polímero apresenta uma rápida recombinação de carga na interface TiO2/spiro-MeOTAD quando existem buracos na perovskita. Além disso, possui alto custo, dificultando sua utilização comercial.116,117 Desse modo, materiais como poli-triarilamina (PTAA),78,114,118 poli(3-hexiltiofeno)(P3HT),119-122 poli[N-900-heptadecanil-2,7-carbazol-alt-5,5-(40,70-di-2-tienil-20,10,30-benzotidiazol)] (PCDTBT),78 NiO,123-125 CuSCN,126-129 CuI,130,131 entre outras diversas moléculas orgânicas, têm sido estudados para a substituição do spiro-MeOTAD.36,74,104,132 A Figura 10 apresenta alguns dos HTLs e ETLs recentemente utilizados em células solares de perovskitas e os seus respectivos níveis energéticos.
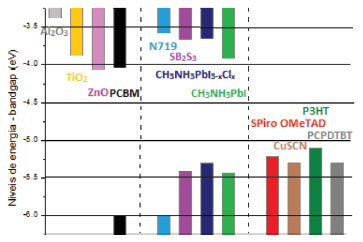 Figura 10. Níveis de energia para vários materiais usados em Células Solares de Perovskitas: transportadores de elétrons (esquerda) e buracos (direita) (Adaptado de 76)
Os pioneiros na produção das células solares de perovskitas de heterojunção planar de filmes finos foram Lee, Snaith e colaboradores, que apresentaram a estrutura FTO/TiO2 compacto/CH3NH3PbI2Cl/spiro-MeOTAD/Au com uma PCE relativamente baixa de 1,8% e demais parâmetros de Jsc = 7,13 mA cm-2, Voc = 0,64 V e FF = 0,4.42 A fabricação desses dispositivos apresenta dificuldades devido aos desafios da deposição de uma camada de perovskita livre de buracos e homogênea a partir de processos em solução, de modo que uma cobertura pobre resulta em uma pobre absorção de luz, bem como caminhos de desvios sobre a camada absorvente, reduzindo, assim, a eficiência dos dispositivos. Eperon, Snaith e colaboradores estudaram cuidadosamente diversos fatores envolvidos na fabricação das células solares de filmes finos, utilizando CH3NH3PbI3-xClx, e alcançaram uma eficiência de 11,4% (Jsc = 20,3 mA cm-2, Voc = 0,89 V e FF = 0,64) .36,126 Outras eficiências significativas também foram alcançadas com estudos realizados com diferentes materiais ETL e HTL, por exemplo como 15,4% (Jsc = 21,5 mA cm-2, Voc = 1,07 V e FF = 0,67) por Snaith, Liu e Johnston,66 que utilizaram HTL de spiro-OMeTAD contendo aditivos como sal de lítio bis(trifluorometilsifonil)imida e terc-butilpiridina, até 19,3% (Jsc = 22,75 mA cm-2, Voc = 1,13 V e FF = 0,75) pelo grupo de Yang,65 que utilizou ETL de TiO2 dopado com ítrio (Y-TiO2). Além das arquiteturas mesoporosas e planares, as PSCs também podem ser fabricadas de forma invertida, semelhante a uma célula solar orgânica regular, como mostrado na Figura 2 (c). Nesse caso, o HTL, geralmente orgânico, é depositado sobre o eletrodo condutor transparente (ITO), e sobre este, a perovskita. Na sequência, o ETL é depositado sobre a perovskita, e sobre este, o eletrodo metálico. Com a iluminação a partir do lado do semicondutor tipo p, os elétrons são transportados até o eletrodo de metal e os buracos até o condutor transparente.1,36,74,104 Atualmente, o HTL mais utilizado é o PEDOT:PPS, e os ETLs são geralmente derivados de fulerenos. O primeiro trabalho nessa direção foi realizado por Jeng et al.,52 que empregaram uma arquitetura de dispositivo consistida em poli (3,4-etileno dioxi tiofeno):poli-(estireno sulfonado) (PEDOT:PSS) como HTL e derivados de C60, como (6,6)-fenil C61- ácido butírico metil éster (PC61BM) e bisaduto indeno-C60 (ICBA) como ETLs. Em relação à deposição da camada perovskita, alguns desafios foram enfrentados para a formação de um revestimento uniforme de MAPbI3 sobre a camada de PEDOT:PPS, restringindo, assim, a espessura total da perovskita e, com isso, limitando a eficiência para 3,9% (Jsc = 10,32 mA cm-2, Voc = 0,60 V e FF = 0,63).36,52 Posteriormente, Peng, Liu e colaboradores alcançaram uma eficiência de 11,7% (Jsc = 16,1 mA cm-2, Voc = 0,99 V e FF = 74) e 11,8% (Jsc = 16,2 mA cm-2, Voc = 1,03 V e FF = 70,7) em uma estrutura composta por ITO/CH3NH3PbI3/PC61BM/Al e ITO/poli tiofeno (PT)/CH3NH3PbI3/C60/BCP/Ag.133,134 Atualmente, a eficiência de células solares de perovskita invertidas é de aproximadamente 18%, utilizando fulereno e PEDOT:PSS como ETL e HTL, respectivamente.59,135-137 Até o que se conhece atualmente, somente três cátions A são capazes de estabilizar a estrutura das perovskitas de haleto e chumbo, Cs+, CH3NH3+ (MA), e HC(NH2)+,138 sendo que atualmente a perovskita mais estudada é a MAPbI3 porém, por meio de cálculos teóricos e experimentos, tem-se demonstrado que esta composição é instável termodinamicamente, independente da umidade, iluminação ou atmosfera.139,140 Esses estudos têm demonstrado ainda que a substituição de I- por Br- e Cl-, quanto a substituição de MA por FA, levam a uma maior estabilidade das estruturas. A primeira evidência experimental dessa estabilização foi dada por Noh et al.40 com o sistema MAPb(I1-xBrx)3, em que acima de x=0,2 a estrutura passa de tetragonal para pseudocúbica e o material se torna mais resistente à umidade. O aumento dos teores de bromo leva à estabilização da estrutura e ao aumento do bandgap, entretanto, este aumento desloca o bandgap para uma região de menor eficiência no gráfico do máximo de eficiência de Shockley-Queisser. Outra composição de perovskita de grande interesse devido ao menor bandgap (1,47 eV) é a FAPbI3. Todavia, esta perovskita se transforma espontaneamente em um polimorfo sem propriedades fotovoltaicas.141 Esse problema foi solucionado pela adição de MA e Br- à estrutura (sistema (MA1-yFAy)Pb(I1-xBrx)3), ocasionando a estabilização e a alta eficiência.141 Com esse sistema são frequentemente obtidos PCE acima de 20%, atualmente.68,85,142,143 Recentemente, Saliba et al. adicionaram o íon césio (Cs+1) a esta composição e observaram melhor eficiência, estabilidade e reprodutibilidade.144 Ainda mais recente, em outro estudo, Saliba et al. adicionaram a essa composição o íon rubídio (Rb1+), obtendo uma perovskita com mistura de quatro cátions (Rb1+, Cs1+, MA e FA) com alta eficiência (21,6%) e alta estabilidade.88 Para a estabilização da estrutura da perovskita é necessário um cátion A suficientemente grande; porém, quando o cátion é muito pequeno, a estrutura preferida é do tipo NH4CdCl3, que pode ser descrito como cadeias duplas de [MI5]3-, e se temos o cátion muito grande, a estrutura preferida é de tipo CsNiBr3, que consiste em cadeias simples de octaedros, podendo o cátion A interferir na largura do bandgap, influenciando as propriedades óticas, devido a variações na estrutura.138 Dessa forma, diversos parâmetros foram modificados e estudados desde a primeira aplicação de perovskitas em dispositivos fotovoltaicos, visando a melhoria da performance destes dispositivos, como por exemplo: configuração, HTL ou ETL aplicado, ou mesmo a estrutura da perovskita. A Tabela 2 apresenta algumas das diversas arquiteturas de células solares de perovskitas relatadas, sua eficiência e demais parâmetros fotovoltaicos obtidos.
Estabilidade Dois parâmetros são considerados importantíssimos para a aplicação comercial de células solares de perovskitas: a eficiência de conversão e a estabilidade. Hoje, altas eficiências foram alcançadas (>20%) em um período de tempo relativamente curto. Entretanto, para aplicações práticas, esses dispositivos ainda apresentam diversos problemas quanto à estabilidade a longo prazo, devido à degradação das PSCs quando expostas à umidade e oxigênio, radiação UV, processo de dissolução (solventes, solutos, aditivos) e temperatura.38,74,156,157 As perovskitas mais usadas em células solares são facilmente decompostas na presença de umidade e oxigênio, devido à presença do grupo higroscópico amina em sua estrutura molecular. Niu et al. propuseram uma sequência de reações químicas para a degradação da perovskita sob diferentes condições.157,158 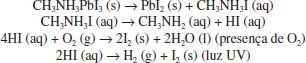 Devido a sensibilidade &ag, rave; umidade e oxigênio, PSCs geralmente são fabricadas em ambientes com atmosfera e umidade controlados (glove box) com gás inerte, para evitar sua degradação e se obter melhores eficiências de conversão; porém, a utilização de glove box eleva o custo de preparação.139-141 Em um dos primeiros trabalhos que avaliaram a estabilidade de células solares de perovskitas, Noh et al. estudaram o sistema MAPb(I1-xBrx)3, e observaram que a perovskita com x=0 (MAPbI3) apresentava valores de eficiência que decresciam lentamente em presença de 35% de umidade, mas, quando esta era aumentada para 55%, um decréscimo abrupto da eficiência acontecia.40 Avaliando as outras composições, os autores observaram que a composição com x=0,20 ou maior mantinham a eficiência mesmo com a umidade de 55%. Analisando a estrutura cristalina, as composições com x igual ou maior que 0,2 passavam da estrutura tetragonal para cúbica, o que pode estar relacionado com a maior estabilidade das células solares.40 Outro mecanismo de degradação da perovskita reportado por Ito et al.159 é causado pela extração de elétrons do I- pelo TiO2. Nesse mecanismo, pela perda de elétrons do I- na superfície do TiO2, ocorre a formação de I2, destruindo, assim, a estrutura cristalina da perovskita. O metilamônio perde, então, seu próton ácido, formando metilamina, que tem baixo ponto de ebulição e então evapora. Já o próton perdido se combina com I2 e I- formando HI, que também tem baixo ponto de ebulição e evapora. Uma representação desse mecanismo está mostrado na Figura 11 (a) e (b). Como estratégia para evitar a degradação, os autores depositaram uma camada de sulfeto de antimônio Sb2S3 sobre a TiO2 mesoporoso e observaram que os dispositivos tiveram uma estabilidade superior (Figura 11 (c)) pelo boqueio da reação entre o I- e o TiO2.159
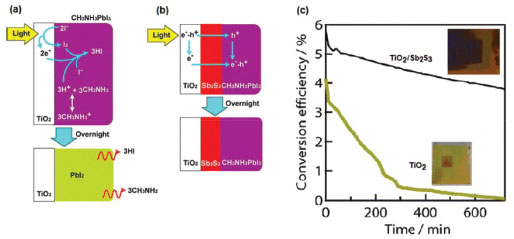 Figura 11. Esquema de degradação da perovskita MAPbI3 quando exposta à luz sobre eletrodos de (a) TiO2 mesoporoso e (b) TiO2 mesoporoso recoberto com Sb2S3. Gráfico de eficiência de conversão em função do tempo para os eletrodos de TiO2 e TiO2/Sb2S3 (Adaptado com permissão da referência 159)
Quanto aos processos de preparação da perovskita, o método de fabricação e o solvente utilizado influenciam na estabilidade do filme, bem como os materiais utilizados como HTL, ETL e aditivos. Por exemplo, o TiO2 é sensível à luz UV e o PCBM não é estável ao ar, já o spiro-MeOTAD necessita de aditivos como 4-tercbutilpiridina (TBP)160 para aumentar sua condutividade, o que causa degradação da perovskita. Assim, estudos têm sido feitos no sentido de substituir esse aditivo, bem como substituintes, por PEDOT:PSS, que apresenta natureza ácida e influencia na estabilidade das células solares.38,157 Estudos de estabilidade térmica mostraram que a exposição do material a temperaturas elevadas também pode degradar o filme de perovskita, constatando que este requer uma etapa de recozimento. Os estudos também indicaram que a estabilidade térmica pode ser afetada por variações sutis de rotas de síntese e de precursores utilizados.156,161 Atualmente, as células solares de perovskita com maior eficiência são baseadas em perovskitas com chumbo. Entretanto, o Pb é um metal com natureza tóxica, o que dificulta sua aplicação nesses dispositivos e faz com que pesquisadores tenham mais um grande desafio: substituir o chumbo por substâncias não-tóxicas, que não prejudicam o meio ambiente, tais como o estanho (Sn).162 Os substitutos mais viáveis para o Pb, são o Sn e o Ge, porém, o maior problema desses metais é que eles já apresentam uma instabilidade química no estado de oxidação necessário (2+), resultando em uma perovskita com menor estabilidade. O Sn é o metal que tem recebido maior atenção para substituir o Pb, porém, estudos têm revelado uma fácil oxidação do Sn2+, com um comportamento de semicondutor tipo n, para Sn4+, cujo comportamento passa a ser de semicondutor tipo p, o qual atua como dopante no interior do material, num processo chamado "auto dopagem".163-165 Enquanto isso, apesar da sua toxicidade, o Pb apresenta uma maior proteção contra a oxidação.10 O primeiro trabalho utilizando estanho substituindo o chumbo em perovskita foi reportado por Hao et al.,165 no início de 2014. Os autores alcançaram a eficiência de 5,73% com a perovskita MASnI3. Ainda no mesmo ano, Hao et al.,166 utilizando uma perovskita de metilamônio com uma mistura de chumbo e estanho (CH3NH3Sn(1-x)Pb(x)I3), e Noel et al.,161 com um estudo de diferentes solventes na cristalização da peroviskita, conseguiram eficiências semelhantes - 5,44 e 6,4%, respectivamente. Desse modo, demais estudos vêm sendo realizados para que se possa melhorar a estabilidade das células solares de perovskitas. Porém, são estudos recentes e não completamente entendidos, precisando de intensificação nos estudos para possíveis aplicações práticas comerciais dos dispositivos.38,156
CONCLUSÕES E PERSPECTIVAS As perovskitas representam uma classe de materiais que vem chamando cada vez mais a atenção de pesquisadores da área fotovoltaica. Suas variadas propriedades e sua diversificação quanto aos inúmeros elementos que podem fazer parte da sua estrutura permitem com que sejam utilizadas em diversos dispositivos. As células solares de perovskitas apresentaram um rápido avanço e aumento da PCE em um curto espaço de tempo, comparadas com qualquer outro tipo de tecnologia solar, e hoje têm eficiência semelhante às células solares de silício. Sua primeira aplicação em células solares foi em 2009, apresentando uma eficiência de conversão de energia de 3,8%. Hoje, de acordo com o Laboratório Nacional de Energia Renovável (NREL) dos Estados Unidos, já apresenta uma eficiência de 22,1%, mostrando um grande avanço em pouco tempo. Diversos métodos de deposição do filme de perovskita têm sido desenvolvidos, bem como a utilização de diferentes materiais nos dispositivo, como ETL, HTL e aditivos, demonstrando grande influência no controle da morfologia do filme, estabilidade e eficiência do dispositivo, tanto para células solares mesoporosas, planares e invertidas. Recentemente, perovskitas orgânica-inorgânicas têm se tornado objeto de estudo na aplicação em células solares, devido à sua forte propriedade de absorção de luz. Novos métodos de fabricação, estrutura de dispositivo e materiais continuam a surgir. Entretanto, esses dispositivos ainda apresentam vários aspectos-chaves que precisam ser melhorados antes de sua aplicação comercial.11,48,58 Os maiores empecilhos enfrentados pelas células solares de perovskitas estão relacionados à estabilidade e toxicidade do material. As PSCs são vulneráveis à umidade e à presença de oxigênio, exposição à luz UV, processo de produção em solução e temperatura, o que resulta em dificuldades de fabricação, prejudicando, assim, as propriedades de transporte de carga e, consequentemente, a eficiência do dispositivo.59 Alguns estudos já estão sendo realizados para melhorar a estabilidade das perovskitas, tornando-as mais estáveis. Perovskitas baseadas em haletos de chumbos são as mais utilizadas na formação do filme das células solares, porém têm resultado em um impacto negativo sobre o meio ambiente, devido à toxicidade do Pb. Dessa forma, seu uso em equipamentos eletrônicos pode ser restrito, dependendo do país e de suas leis nacionais, prejudicando, assim, sua aplicação comercial.17 A substituição do Pb pelo Sn tem sido estudada, porém, estas perovskitas ainda apresentam instabilidade, sendo um desafio a ser contornado nos próximos anos.17,59 Dentre outras perspectivas esperadas, estão: reduzir a temperatura de processamento, permitindo, assim, o uso de diferentes substratos e materiais para formação do filme; além de reduzir o custo do dispositivo,17 bem como aperfeiçoar o preparo e aplicação dos pontos quânticos de perovskitas inorgânicas.81,91 Vale ressaltar, também, que além de serem utilizadas em células solares, as perovskitas podem ser utilizadas em tecnologias de células em paralelo de alta performance, nas quais utilizam tanto a perovskita, quanto tecnologias já existentes, permitindo a introdução de um produto novo e de alta tecnologia no mercado.16 Para as células solares de perovskitas, é esperado um futuro promissor, com significativos progressos e altas eficiências de conversão de energia. Entretanto, para isso, são necessários mais estudos para melhorar a estabilidade dos dispositivos e outros problemas ainda enfrentados, para então poder alcançar uma comercialização nos próximos anos.
AGRADECIMENTOS Os autores agradecem à CAPES, CNPq, FAPEMIG e FAPESP pelo apoio recebido.
REFERÊNCIAS 1. Wang, B.; Xiao, X.; Chen, T.; Nanoscale 2014, 6, 12287. 2. Armaroli, N.; Balzani, V.; Angew. Chem. Int. Ed. 2006, 45, 2. 3. Longo, C.; Paoli, Marco-A. D.; J. Braz. Chem. Soc. 2003, 14, 889. 4. Rhodes, C. J.; Sci Prog. 2010, 93, 37. 5. Rühle, S.; Shalom, M.; Zaban, A.; ChemPhysChem 2010, 11, 2290. 6. Carvalho, E. F. A.; Calvete, M. J. F.; Rev. Virtual Quim. 2010, 2, 192. 7. Machado, C. T.; Miranda, F. S.; Rev. Virtual Quim. 2015, 7, 126. 8. Jean, J.; Brown, P. R.; Jaff, R. L.; Buonassisid, T.; Bulović, V.; Energy Environ. Sci. 2015, 8, 1200. 9. Green, M. A.; Progress in Photovoltaics: Research and Applications 2005, 13, 447. 10. Polman, A., Knight, M., Garnett, E. C., Ehrler, B., Sinke, W. C.; Science 2016, 352, 307. 11. Park, N.-G.; J. Phys. Chem. Lett. 2013, 4, 2423. 12. Snaith, H. J.; J. Phys. Chem. Lett. 2013, 4, 3623. 13. Kim, H.-S.; Im, S. H., Park, N.-G.; J. Phys. Chem. C 2014, 118, 5615. 14. Hodes, G.; Cahen, D.; Nature Photon 2014, 8, 87. 15. Service, R. F.; Science 2014, 344, 458. 16. Green, M. A.; Ho-Baillie, A.; Snaith, H. J.; Nat. Photonics 2014, 8, 506. 17. Petrović, M.; Chellappan, V.; Ramakrishna, S.; Solar Energy 2015, 122, 678. 18. Green, M. A.; Emery, K.; Hishikawa, Y.; Warta, W.; Dunlop, E. D.; Progress in Photovoltaics: Research and Applications 2014, 22, 701. 19. www.nrel.gov, acessada em setembro de 2017. 20. Fraas, L.; Partain, L.; Solar Cells: A Brief History and Introduction in Solar Cells and their applications; Fraas, L., Partain, L., eds.; John Wiley & Sons, Inc.: Singapore, 2010, cap. 1. 21. Ohnishi, M.; Takeoka, A.; Nakano, S.; Kuwano, Y.; Renewable Energy 1995, 6, 275. 22. O'regan, B.; Gratzel, M.; Nature 1991, 353, 737. 23. Yu, M.; Long, Yun-Ze.; Sun, B.; Fan, Z.; Nanoscale 2012, 4, 2783. 24. Agnaldo, J. S.; Bastos, J. B. V.; Cressoni, J. C.; Viswanathan, G. M.; Rev. Bras. Ens. Fís. 2006, 28, 77. 25. Kakiage, K.; Aoyama, Y.; Yano, T.; Oya, K.; Fujisawa, J.; Hanaya, M.; Chem. Commun. 2015, 51, 15894. 26. Kuang, D.; Brillet, J.; Chen, P.; Takata, M.; Uchida, S.; Miura, H.; Sumioka, K.; Zakeeruddin, S. M.; Grätzel, M.; ACS Nano 2008, 2, 1113. 27. Lee, J.-K.; Yang, M.; Mater Sci Eng B 2011, 176, 1142. 28. Halme, J.; Vahermaa, P.; Miettunen, K.; Lund, P.; Adv. Mater. 2010, 22, E210. 29. Sugathan, V.; John, E.; Sudhakar, K.; Renewable Sustainable Energy Rev. 2015, 52, 54. 30. Shalini, S.; Balasundaraprabhu, R.; Prasanna, S.; Mallick, T. K.; Senthilarasu, S.; Renewable Sustainable Energy Rev. 2015, 51, 1306. 31. Tian, J.; Cao, G.; Nano Reviews 2013, 4, 22578. 32. Kouhnavard, M.; Ikeda, S.; Ludin, N. A.; Ahmad Khairudin, N. B.; Ghaffari, B. V.; Mat-Terid, M. A.; Ibrahim, M. A.; Sepeai, S.; Sopian, K.; Renewable Sustainable Energy Rev. 2014, 37, 397. 33. Lee, J.-W.; Son, D.-Y.; Ahn, T. K.; Shin, H.-W.; Kim, I. Y.; Hwang, S.-J.; Ko, M. J.; Sul, S.; Han, H.; Park, N.-Gyu.; Scientific Reports, 2013, 3, 1050. 34. Hod, I.; Zaban, A.; Langmuir 2014, 30, 7264. 35. Kazim, S.; Nazeeruddin, M. K.; Gratzel, M.; Ahmad, S.; Angew. Chem., Int. Ed. 2014, 53, 2812. 36. Sum, T. C.; Mathews, N.; Energy Environ. Sci. 2014, 7, 2518. 37. Park, N.-G.; Materials Today 2015, 18, 65. 38. Song, T.-B.; Chen, Q.; Zhou, H.; Jiang, C.; Wang, H.-H.; Yang, Y. M.; Liu, Y.; You, J.; Yang, Y.; J. Mater. Chem. A 2015, 3, 9032. 39. De Wolf, S.; Holovsky, J.; Moon, S. J.; Löper, P.; Niesen, B.; Ledinsky, M.; Haug, F. J.; Yum, J. H.; Ballif, C.; J. Phys. Chem. Lett. 2014, 5, 1035. 40. Noh, J. H.; Im, S. H.; Heo, J. H.; Mandal, T. N. Seok, S. I.; Nano Lett. 2013, 13, 1764. 41. Ishihara, T.; Optical Properties of Pb-based inorganic-organic perovskites, World Scientific: Singapore, 1995, pp. xix, 414 p. 42. Lee, M. M.; Teuscher, J.; Miyasaka, T.; Murakami, T. N.; Snaith, H. J.; Science 2012, 338, 643. 43. Etgar, L.; Gao, P.; Xue, Z.; Peng, Q.; Chandiran, A. K.; Liu, B.; J. Am. Chem. Soc., 2012, 134, 17396. 44. Ponseca, C. S.; Savenije, T. J.; Abdellah, M.; Zheng, K.; Yartsev, A.; Pascher, T.; Harlang, T.; Chabera, P.; Pullerits, T.; Stepanov, A.; Wolf, J. P.; Sundström, V.; J. Am. Chem. Soc. 2014, 136, 5189. 45. Stranks, S. D.; Eperon, G. E.; Grancini, G.; Menelaou, C.; Alcocer, M. J. P.; Leijtens, T.; Herz Petrozza, L. M. A.; Snaith, H. J.; Science 2013, 342, 341. 46. Xing, G.; Mathews, N.; Sun, S.; Lim, S. S.; Lam, Y. M.; Gratzel, M.; Mhaisalkar, S.; Sum, T. C.; Science 2013, 342, 344. 47. Shi, D.; Adinolfi, V.; Comin, R.; Yuan, M.; Alarousu, E.; Buin, A.; Chen, Y.; Hoogland, S.; Rothenberger, A.; Katsiev, K.; Losovyj, Y.; Zhang, X.; Dowben, P. A.; Mohammed, O. F.; Sargent, E. H.; Bakr, O. M.; Science 2015, 347, 519. 48. Jiang, M.; Wu, J.; Lan, F.; Tao, Q.; Gao, D.; Li, G.; J. Mater. Chem. A 2015, 3, 963. 49. Lv, S.; Han, L.; Xiao, J.; Zhu, L.; Shi, J.; Wei, H.; Xu, Y.; Dong, J.; Xu, X.; Li, D.; Wang, S.; Luo, Y.; Meng, Q.; Li, X.; Chem. Commun. 2014, 50, 6931. 50. Ball, J. M.; Lee, M. M.; Hey, A.; Snaith, H. J.; Energy Environ.Sci. 2013, 6, 1739. 51. Marchioro, A.; Teuscher, J.; Friedrich, D.; Kunst, M.; van de Krol, R.; Moehl, T.; Gratzel, M.; Moser, J.-E.; Nat. Photonics 2014, 8, 250. 52. Jeng, J.- Y.; Chiang, Y.-F.; Lee, M.-H.; Peng, S.-R.; Guo, T.-F.; Chen, P.; Wen, T.-C.; Adv. Mater. 2013, 25, 3727. 53. Giorgi, G.; Yamashita, K.; J. Mater. Chem. A 2015, 3, 8981. 54. King, G., Woodward, P. M.; J. Mater. Chem. 2010, 20, 578 55. Schlom, D. G.; Chen, L.-Q.; Pan, X.; Schmehl, A.; Zurbuchen, M. A.; J. Am. Ceram. Soc. 2008, 91, 2429. 56. Müller, K. A.; Kool, T. W.; Properties of perovskites and other oxides, World Scientific: Singapore, 2010. 57. Shriver, D. F.; Atkins, P. W.; Overton, T. L.; Rourke, J. P.; Weller, M. T.; Armstrong, F. A.; Química Inorgânica, 4. ed., Bookman: Porto Alegre, 2008. 58. Giorgi, G.; Fujisawa, J.-I.; Segawa, H.; Yamashita, K.; J. Phys. Chem. Lett. 2013, 4, 4213. 59. Liu, X.; Zhao, W.; Cui, H.; Xie, Y.; Wang, Y.; Xu, T.; Huang, F.; Inorg. Chem. Front. 2015, 2, 315. 60. Poglitsch, A.; Weber, D.; J. Chem. Phys. 1987, 87, 6373. 61. Shao, U.; Yuan, Y.; Huang, J.; Nat. Energy 2016, 1, 15001 62. Wu, C.-G.; Chiang, C.-H.; Tseng, Z.-L.; Nazeeruddin, Md. K.; Hagfeldt, A.; Grätzel, M.; Energy Environ. Sci. 2015, 8, 2725. 63. Son, D.-Y.; Lee, J.-W.; Choi, Y. J.; Jang, I.-H.; Lee, S.; Yoo, P. J.; Shin, H.; Ahn, N.; Choi, M.; Kim, D.; Park, N.-G.; Nat. Energy 2016, 1, 16081. 64. Noh, J. H.; Im, S. H.; Heo, J. H.; Mandal, T. N.; Seo k, S. I. ; Nano Lett., 2013, 13, 1764. 65. Zhou, H.; Chen, Q.; Li, G.; Luo, S.; Song, T.-b.; Duan, H. -S.; Hong, Z.; You, J.; Liu, Y.; Yang, Y.; Science 2014, 345, 542. 66. Liu, M.; Johnston, M. B.; Snaith, H. J.; Nature 2013, 501, 395. 67. Yang, W. S.; Noh, J. H.; Jeon, N. J.; Kim, Y. C.; Ryu, S.; Seo, J.; Seok, S. I.; Science 2015, 348, 1234. 68. Bi, D.; Tress, W.; Dar, M. I.; Gao, P.; Luo, J.; Renevier, C.; Schenk, K.; Abate, A.; Giordano, F.; Correa-Baena, J. P.; Decoppet, J. D.; Zakeeruddin, S. M.; Nazeeruddin, M. K.; Grätzel, M.; Hagfeldt, A.; Sci Adv. 2016, 2, e1501170. 69. Perumallapelli, G. R.; Vasa, S. R.; Jang, J.; Org. Electron. 2016, 31, 142. 70. Dharani, S.; Mulmudi, H. K.; Yantara, N.; Thu Trang, P. T.; Park, N. G.; Graetzel, M.; Mhaisalkar, S.; Mathews, N.; Boix, P. P.; Nanoscale 2014, 6, 1675. 71. Liang, K.; Mitzi, D. B.; Prikas, M. T.; Chem Mater 1998, 10, 403. 72. Burschka, J.; Pellet, N.; Moon, S. J.; Humphry-Baker, R.; Gao, P.; Nazeeruddin, M. K.; Grätzel, M.; Nature 2013, 499, 316. 73. Jung, H. S.; Park, N.-G.; Small 2015, 11, 10. 74. Salim, T.; Sun, S.; Abe, Y.; Krishna, A.; Grimsdale, A. C.; Lam, Y. M.; J. Mater. Chem. A 2015, 3, 8943. 75. Stranks, S. D.; Nayak, P. K.; Zhang, W.; Stergiopoulos, T.; Snaith, H. J.; Angew. Chem., Int. Ed. 2015, 54, 3240. 76. Cui, J.; Yuan, H.; Li, J.; Xu, X.; Shen, Y.; Lin, H.; Wang, M.; Sci. Technol. Adv. Mater. 2015, 16, 036004. 77. Kim, H.-S.; Lee, C.-R.; Im, J.-H.; Lee, K.-B.; Moehl, T.; Marchioro, A.; Moon, S.-J.; Humphry-Baker, R.; Yum, J.H.; Moser, J. E.; Grätzel, M.; Park, N.-G.; Sci. Rep. 2012, 2, 591. 78. Heo, J. H.; Im, S. H.; Noh, J. H.; Mandal, T. N.; Lim, C.-S.; Chang, J. A.; Lee, Y. H.; Kim, H.-j. Sarkar, A.; Nazeeruddin, M. K.; Gratzel, M.; Seok, S. I.; Nat. Photonics 2013, 7,486. 79. Sun, S.; Salim, T.; Mathews, N.; Duchamp, M.; Boothroyd, C.; Xing, G.; Sum, T. C.; Lam, Y. M.; Energy Environ. Sci. 2014, 7, 399. 80. Eperon, G. E.; Burlakov, V. M.; Docampo, P.; Goriely, A.; Snaith, H. J.; Adv. Funct. Mater. 2014, 24, 151. 81. Protesescu, L.; Yakunin, S.; Bodnarchuk, M. I.; Krieg, F.; Caputo, R.; Hendon, C. H.; Yang, R. X.; Walsh, A.; Kovalenko, M. V.; Nano Lett. 2015, 15, 3692. 82. Malinkiewicz, O.; Yella, A.; Lee, Y. H.; Espallargas, G. M.; Graetzel, M.; Nazeeruddin, M. K.; Bolink, H. J.; Nat Photonics 2014, 8, 128. 83. Chen, Q.; Zhou, H.; Hong, Z.; Luo, S.; Duan, H.-S.; Wang, H.-H.; Liu, Y.; Li, G.; Yang, Y.; J. Am. Chem. Soc. 2014, 136, 622. 84. Pang, S.; Hu, H.; Zhang, J.; Lv, S.; Yu, Y.; Wei, F.; Qin, T.; Xu, H.; Liu, Z.; Cui, G.; Chem. Mater 2014 26, 1485. 85. Im, J.-H.; Kim, H.-S.; Park, N.-G.; APL Mater. 2014, 2, 081510. 86. Correa-Baena, J. P.; Anaya, M.; Lozano, G.; Tress, W.; Domanski, K.; Saliba, M.; Matsui, T.; Jacobsson, T.J.; Calvo, M.E.; Abate, A.; Grätzel, M.; Míguez, H.; Hagfeldt, A.; Adv. Mater. 2016, 28, 5031. 87. Jacobsson, T. J.; Correa-Baena, J. P.; Pazoki, M.; Saliba, M.; Schenk, K.; Grätzel, M.; Hagfeldt, A.; Energy Environ. Sci. 2016, 9, 1706. 88. Saliba, M.; Matsui, T.; Domanski, K.; Seo, J. Y.; Ummadisingu, A.; Zakeeruddin, S. M.; Correa-Baena, J. P.; Tress, W. R.; Abate, A.; Hagfeldt, A.; Grätzel, M.; Science 2016, 354, 206. 89. Maity, P.; Dana, J.; Ghosh, H. N.; J. Phys. Chem. C 2016, 120, 18348. 90. Wang, Y., Li, X., Song, J., Xiao, L., Zeng, H., Sun, H.; Adv. Mater. 2015, 27, 7101. 91. Kulbak, M.; Gupta, S.; Kedem, N.; Levine, I.; Bendikov, T.; Hodes, G.; Cahen, D.; J. Phys. Chem. Lett. 2016, 7, 167. 92. Nedelcu, G.; Protesescu, L.; Yakunin, S.; Bodnarchuk, M. I.; Grotevent, M. J.; Kovalenko, M. V.; Nano Lett. 2015, 15, 5635. 93. Wu, K.; Liang, G.; Shang, Q.; Ren, Y.; Kong, D.; Lian, T.; J. Am. Chem. Soc. 2015, 137, 12792. 94. Akkerman, Q. A.; D'Innocenzo, V.; Accornero, S.; Scarpellini, A.; Petrozza, A.; Prato, M.; Manna, L.; J. Am. Chem. Soc. 2015, 137, 10276. 95. Kim, Y.; Yassitepe, E.; Voznyy, O.; Comin, R.; Walters, G.; Gong, X.; Kanjanaboos, P.; Nogueira, A. F.; Sargent, E. H.; ACS Appl. Mater. Interfaces 2015, 7, 25007. 96. Mitzi, D. B.; Wang, S.; Feild, C. A.; Chess, C. A.; Guloy, A. M.; Science 1995, 267, 1473. 97. Mitzi, D. B.; Prog. Inorg. Chem. 2007, 48, 1. 98. Mitzi, D. B.; Chondroudis, K.; Kagan, C. R.; J. Res. Dev. 2001, 45, 29. 99. Yin, W.-J.; Yang, J.-H.; Kang, J.; Yan, Y.; Wei, S.-H.; J. Mater. Chem. A 2015, 3, 8926. 100. Kojima, A.; Teshima, K.; Shirai, Y.; Miyasaka, T.; J. Am. Chem. Soc. 2009, 131, 6050. 101. Im, J.-H.; Lee, C.-R.; Lee, J.-W.; Park, S.-W.; Park, N.-G.; Nanoscale 2011, 3, 4088. 102. Yang, W. S.; Noh, J. H.; Jeon, N. J.; Kim, Y. C.; Ryu, S.; Seo, J.; Seok, S. I.; Science 2015, 348, 1234. 103. Anaraki, E. H.; Kermanpur, A.; Steier, L.; Domanski, K.; Matsui, T.; Tress, W.; Saliba, M.; Abate, A.; Grätzel, M.; Hagfeldt, A.; Correa-Baena, J. P.; Energy Environ. Sci. 2016, 9, 3128. 104. Li, M.-H.; Shen, P.-S.; Wang, K.-C.; Guo, T.-F.; Chen, P.; Mater. Chem. A 2015, 3, 9011. 105. Snaith, H. J.; J. Phys. Chem. Lett. 2013, 4, 3623. 106. Laban, W. A.; Etgar, L.; Energy Environ. Sci. 2013, 6, 3249. 107. Cohen, B.-E.; Aharon, S.; Dymshits, A.; Etgar, L. J.; Phys. Chem. C 2016, 120, 142. 108. Kim, H.-S.; Lee, J.-W.; Yantara, N.; Boix, P. P.; Kulkarni, S. A.; Mhaisalkar, S.; Grätzel, M.; Park, N.-G.; Nano Lett. 2013, 13, 2412. 109. Choi, J. J.; Yang, X.; Norman, Z. M.; Billinge, S. J. L.; Owen, S. J.; Nano Lett. 2013, 14, 127. 110. Zhao, X.; Shen, H.; Zhang, Y.; Li, X.; Zhao, X.; Tai, M.; Li, J.; Li, J.; Li, X.; Lin, H.; ACS Appl. Mater. Interfaces, 2016, 8, 7826. 111. Liu, D.; Kelly, T. L.; Nat. Photonics 2014, 8, 133. 112. Fernandes, S. L.; Véron, A. C.; Neto, N. F. A.; Nüesch, F. A.; da Silva, J. H. D.; Zaghete, M. A.; Graeff, C. F. de O.; Mater. Lett. 2016, 181, 103. 113. Shao, Y.; Yuan, Y.; Huang, J.; Nature Energy 2016, 1, 15001. 114. Ryu, S.; Seo, J.; Shin, S. S.; Kim, Y. C.; Jeon, N. J.; Noh, J. H.; Seok, S. I.; Mater. Chem. A 2015, 3, 3271. 115. Seo, J.; Park, S.; Kim, Y. C. ; Jeon, N. J.; Noh, J. H.; Yoon, S. C.; Seok, S. I.; Energy Environ. Sci. 2014, 7, 2642. 116. Tao, C.; Neutzner, S.; Colella, L.; Marras, S.; Kandada, A. R. S.; Gandini, M.; De Bastiani, M.; Pace, G.; Manna, L.; Caironi, M.; Bertarelli, C.; Petrozza, A.; Energy Environ. Sci. 2015, 8, 2365. 117. Ma, S.; Zhang, H.; Zhao, N.; Cheng, Y.; Wang, M.; Shen, Y.; Tu, G.; J. Mater. Chem. A 2015, 3, 12139. 118. Ryu, S.; Noh, J. H.; Jeon, N. J.; Kim, Y. C.; Yang, W. S.; Seo, J.; Seok, S. I.; Energy Environ. Sci. 2014, 7, 2614. 119. Guo, Y.; Liu, C.; Inoue, K.; Harano, K.; Tanaka, H.; Nakamura, E.; J. Mater. Chem. A 2014, 2, 13827. 120. Giacomo, F. D.; Razza, S.; Matteocci, F.; D'Epifanio, A.; Licoccia, S.; Brown, T. M.; Carlo, A. D.; J. Power Sources 2014, 251, 152. 121. Zhang, M.; Lyu, M.; Yu, H.; Yun, J. H.; Wang, Q.; Wang, L.; Chemistry 2015, 21, 434. 122. Matteocci, F.; Razza, S.; Di Giacomo, F.; Casaluci, S.; Mincuzzi, G.; Brown, T. M.; D'Epifanio, A.; Licoccia, S.; Di Carlo, A.; Phys. Chem. Chem. Phys. 2014, 16, 3918. 123. Hu, L.; Peng, J.; Wang, W.; Xia, Z.; Yuan, J.; Lu, J.; Huang, X.; Ma, W.; Song, H.; Chen, W. Cheng, Y.-B.; Tang, J.; ACS Photonics 2014, 1, 547. 124. Wang, H.; Zeng, X.; Huang, Z.; Zhang, W.; Qiao, X.; Hu, B.; Zou, X.; Wang, M.; Cheng, Y. B.; Chen, W.; ACS Appl Mater Interfaces 2014, 6, 12609. 125. Wang, K.-C.; Jeng, J.-Y.; Shen, P.-S.; Chang, Y.-C.; Diau, E. W.-G.; Tsai, C.-H.; Chao, T.-Y.; Hsu, H.-C.; Lin, P.-Y.; Chen, P; Guo, T.-F.; Wen, T.-C.; Sci Rep 2014, 4, 4756. 126. Subbiah, A. S.; Halder, A.; Ghosh, S.; Mahuli, N.; Hodes, G.; Sarkar, S. K.; J. Phys. Chem. Lett., 2014, 5, 1748. 127. Chavhan, S.; Miguel, O.; Grande, H.-J.; Gonzalez-Pedro, V.; Sánchez, R. S.; Barea, E. M.; Mora-Seró, I.; Tena-Zaera, R.; J. Mater. Chem. A, 2014, 2, 12754. 128. Qin, P.; Tanaka, S.; Ito, S.; Tetreault, N.; Manabe, K.; Nishino, H.; Nazeeruddin, M. K.; Grätzel, M.; Nat. Commun. 2014, 5, 3834. 129. Ito, S.; Tanaka, S.; Nishino, H.; J. Phys. Chem. Lett. 2015, 6, 881. 130. Chen, W.-Y.; Deng, L.-L.; Wang, S.-M. D. X.; Tian, C.-B.; Zhan, X.-X.; Xie, S.-Y.; Huang, R.-B.; Zheng, L.-S.; J. Mater. Chem. A 2015, 3, 19353. 131. Sepalage, G. A.; Meyer, S.; Pascoe, A.; Scully, A. D.; Huang, F.; Bach, U.; Cheng, Y.-B.; Spiccia, L.; Adv. Funct. Mater. 2015, 25, 5650. 132. Boix, P. P.; Nonomura, K.; Mathews, N.; Mhaisalkar, S. G.; Mater. Today 2014, 17, 16. 133. Zhang, Y.; Hu, X.; Chen, L.; Huang, Z.; Fu, Q.; Liu, Y.; Zhang, L.; Chen, Y.; Org. Electron. 2016, 30, 281. 134. Yan, W. Li, Y.; Sun, W.; Peng, H.; Ye, S.; Liu, Z.; Bian, Z.; Huang, C.; RSC Adv. 2014, 4, 33039. 135. Bi, C.; Wang, Q.; Shao, Y.; Yuan, Y.; Xiao, Z.; Huang, J.; Nat. Commun. 2015, 6, 7747 136. Heo, J. H.; Han, H. J.; Kim, K.; Ahn, T. K.; Im, S. H.; Energy Environ. Sci. 2015, 8, 1602. 137. Nie, W.; Tsai, H.; Asadpour, R.; Blancon, J. C.; Neukirch, A. J., Gupta, G., Crochet, J. J., Chhowalla, M., Tretiak, S., Alam, M. A., Wang, H. L., Mohite, A. D.; Science 2015, 347, 522. 138. Constantinos, C. S.; Mercouri, G. K.; Acc. Chem. Res. 2015, 48, 2791. 139. Nagabhushana, G. P.; Shivaramaiah, R.; Navrotsky, A.; Proc. Natl. Acad. Sci. U. S. A. 2016 113, 7717. 140. Buin, A.; Comin, R.; Xu, J.; Ip, A. H.; Sargent, E. H.; Chem. Mater., 2015, 27, 4405. 141. Koh, T. M.; Fu, K.; Fang, Y.; Chen, S.; Sum, T. C.; Mathews, N.; Mhaisalkar, S. G.; Boix, P. P.; Baikie, T. J.; Phys. Chem. C, 2014, 118, 16458. 142. Jeon, N. J.; Noh, J. H.; Yang, W. S.; Kim, Y. C.; Ryu, S.; Seo, J.; Seok, S. I.; Nature 2015, 517, 476. 143. Correa-Baena, J.-P.; Anaya, M., Lozano, G.; Tress, W.; Domanski, K.; Saliba, M.; Matsui, T.; Jacobsson, T. J.; Calvo, M. E.; Abate, A.; Grätzel, M.; Míguez, H.; Hagfeldt, A.; Adv. Mater. 2016, 28, 5031. 144. Saliba, M.; Matsui, T.; Seo, J.-Y.; Domanski, K.; Correa-Baena, J.-P.; Nazeeruddin, M. K.; Zakeeruddin, M. S.; Tress, W. Abate, A.; Hagfeldt, A.; Grätzel, M.; Energy Environ. Sci. 2016, 9, 1989. 145. Saliba, M.; Matsui, T.; Domanski, K.; Seo, J.-Y.; Ummadisingu, A.; Zakeeruddin, S. M.; Correa-Baena, J.-P.; Tress, W. R.; Abate, A.; Hagfeldt, A.; Grätzel, M. Science 2016, 354, 206. 146. Arora, N.; Orlandi, S.; Dar, M. I.; Aghazada, S.; Jacopin, G.; Cavazzini, M.; Mosconi, E.; Gratia, P.; De Angelis, F.; Pozzi, G.; Graetzel, M.; Nazeeruddin, M. K.; ACS Energy Lett. 2016, 1, 107. 147. Jung, M.; Kim, Y. C.; Jeon, N. J.; Yang, W. S.; Seo, J.; Noh, J. H.; Seok, S.; ChemSusChem 2016, 9, 2592. 148. Zhao, K.; Munir, R.; Yan, B.; Yang, Y.; Kim, T.-S.; Amassian, A.; J. Mater. Chem. A 2015, 3, 20554. 149. Hou, Y.; Zhang, H.; Chen, W.; Chen, S.; Quiroz, C. O. R.; Azimi, H.; Osvet, A.; Matt, G. J.; Zeira, E.; Seuring, J.; Kausch-Busies, N.; Lovenich, W., Brabec, C. J.; Adv. Energy Mater. 2015, 5, 1. 150. Habisreutinger, S. N.; Leijtens, T.; Eperon, G. E.; Stranks, S. D.; Nicholas, R. J.; Snaith, H. J.; Nano Lett. 2014, 14, 5561. 151. You, J.; Meng, L.; Song, T.-B.; Guo, T.-F.; Yang, Y.; Chang, W.-H.; Hong, Z.; Chen, H.; Zhou, H.; Chen, Q.; Liu, Y.; Marco, N. D.; Yang, Y.; Nature 2016, 11, 75. 152. Yin, X.; Yao, Z.; Luo, Q.; Dai, X.; Zhou, Y.; Zhang, Y.; Zhou, Y.; Luo, S.; Li, J.; Wang, N.; Lin, H.; ACS Appl. Mater. Interfaces 2017, 9, 2439. 153. Chen, W.-Y.; Deng, L.-L.; Dai, S.-M.; Wang, X.; Tian, C.-B.; Zhan, X.-X.; Xie, S.-Y.; Huang, R.-B.; Zheng, L.-S.; J. Mater. Chem. A, 2015, 3, 19353. 154. Zhou, H.; Chen, Q.; Li, G.; Luo, S.; Song, T.-B.; Duan, H.-S.; Hong, Z.; Liu, Y.; Yang, Y.; Science 2013, 27, 238. 155. Liu, D.; Kelly, T. L.; Nat. Photonics 2013, 8, 133. 156. Ahmed, M. I.; Habib, A.; Javaid, S. S.; Int. J. Photoenergy 2015, ID 592308. 157. Niu, G.; Guo, X.; Wang, L.; J. Mater. Chem. A 2015, 3, 8970. 158. Shahbazi, M.; Wang, H.; Solar Energy 2016, 123, 74. 159. Ito, S.; Tanaka, S.; Manabe, K.; Nishino, H.; J. Phys. Chem. C 2014, 118, 16995. 160. Li, W.; Dong, H.; Wang, L.; Li, N.; Guo, X.; Li, J.; Qiu, Y.; J. Mater. Chem. A 2014, 2, 13587. 161. Wang, D.; Wright, M.; Elumalai, N. K.; Uddin, A.; Sol. Energy Mater. Sol. Cells 2016, 147, 255. 162. Noel, N. K.; Stransk, S. D.; Abate, A. Wehrenfennig, C.; Guarnera, S.; Haghighirad, A.-A.; Sadhanala, A.; Eperon, G. E.; Pathak, S. K.; Johnston, M. B.; Petrozza, A.; Herz, L. M.; Snaith, H. J.; Energy Environ. Sci. 2014, 7, 3061. 163. Emami, S.; Andrade, L.; Mendes, A.; U.Porto Journal of Engineering 2015, 1, 52. 164. Stoumpos, C. C.; Malliakas, C. D.; Kanatzidis. M. G. Inorg. Chem. 2013, 52, 9019. 165. Hao, F.; Stoumpos, C. C; Cao, D. H,Chang, R. P. H; Kanatzidis, M. G.; Nat. Photonics 2014, 8, 489. 166. Hao, F.; Stoumpos, C. C.; Chang, R. P. H.; Kanatzidis, M. G.; J. Am. Chem. Soc. 2014, 136, 8094. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access