
 |
 |
 |
 |
 |
Revisão
|
|
| Quimioinformática: uma introdução Cheminformatics: an introduction |
|
Vinicius M. AlvesI; Rodolpho C. BragaI; Eugene N. MuratovI,II,III; Carolina Horta AndradeI,*
I Laboratório de Planejamento de Fármacos e Modelagem Molecular, Faculdade de Farmácia, Universidade Federal de Goiás, Goiânia, GO, 74605-220, Brasil Recebido em 11/06/2017 *e-mail: carolina@ufg.br Cheminformatics is an interdisciplinary field between chemistry and informatics, which has evolved considerably since its inception in the 1960s. Initially, the cheminformatics community dealt primarily with practical and technical aspects of chemical structure representation, manipulation, and processing, while modern research explores a new role: the exploration and interpretation of large chemical databases and the discovery of new compounds with desired activity and safety profiles. Despite the recent release of several hallmark reviews addressing methods and application of cheminformatics written in Portuguese, so far there are no scientific articles presenting cheminformatics research to the Brazilian scientific community yet. To address this gap, we aim to introduce the field of cheminformatics to both students and researchers in a simple and didactic way by narrating important historical facts and contextualizing information within the scope of various applications. INTRODUÇAO A quimioinformática é uma ciência interdisciplinar que utiliza recursos das ciências da computaçao e informaçao para resolver problemas da química.1 O termo quimioinformática foi cunhado por Frank Brown em 1998, definindo-a como “mistura de recursos de informaçao para transformar dados em informaçao e informaçao em conhecimento, no intuito de tomar decisoes melhores e mais rápidas na área de identificaçao e otimizaçao de compostos líderes”.2 Em uma definiçao mais abrangente, em 1999, Greg Paris, entao pesquisador da companhia farmacêutica Novartis, definiu a quimioinformática como “um termo genérico que engloba a concepçao, criaçao, organizaçao, gestao, recuperaçao, análise, disseminaçao, visualizaçao e utilizaçao de informaçao química”.3 Apesar do nome recente, a quimioinformática nao foi estabelecida ou fundada. Trata-se uma ciência que evoluiu e se consolidou ao longo de décadas.1,4 A revista científica mais importante da área, Journal of Chemical Information and Modeling, da American Chemical Society (ACS), existe desde 1961, chamada, na época, de Journal of Chemical Documentation (1961-1974) e, posteriormente, Journal of Chemical Information and Computer Sciences (1975-2005), chegando no nome atual em 2005 (http://pubs.acs.org/toc/jcisd8/current). Nesse intervalo, várias áreas que hoje compoem a quimioinformática foram se consolidando como (i) representaçao, visualizaçao, manipulaçao e processamento de estruturas químicas, (ii) organizaçao de bases de dados de estruturas químicas e (iii) estudos das relaçoes quantitativas entre estrutura e atividade/propriedade (QSAR/QSPR, do inglês, quantitative structure-activity/property relationships).5 O campo da quimioinformática evoluiu bastante, passando de aspectos práticos e técnicas de representaçao, manipulaçao e processamento de estruturas químicas individuais até o seu papel primordial na atualidade: exploraçao de bases de dados químicas e descoberta de novos compostos com atividade e/ou propriedade desejadas. Ao se explorar bases de dados é possível extrair várias informaçoes que auxiliam na compreensao do comportamento de determinado grupo de compostos e é possível gerar modelos computacionais que sao utilizados para predizer a atividade de moléculas que carecem de dados experimentais, ou seja, dados de ensaios in vitro e in vivo.6,7 Essa mudança de cenário se deve, principalmente, à grande quantidade de dados biológicos e químicos produzidos graças a evoluçao de ensaios de biológicos de alta vazao (HTS, do inglês, High Throughput Screening) e da química combinatória.8 A quantidade de dados cresceu tanto que, atualmente, tornou-se inviável extrair informaçao de bases de dados e, principalmente, transformar essa informaçao em conhecimento sem o uso de um computador.6,9 Ainda na década de 1960, quando os primeiros estudos de relaçao quantitativa estrutura entre atividade foram publicados, as análises eram realizadas a poucos compostos de séries congêneres e as equaçoes deduzidas manualmente. Atualmente, modelos de QSAR/QSPR sao gerados usando milhares de compostos e descritores moleculares, aplicando-se os mais variáveis algoritmos computacionais.10 É importante ressaltar que a quimioinformática está altamente relacionada e comumente converge com outras subáreas da química que trabalham com computadores,11 como a química computacional,5 que aplica de métodos de química teórica para se calcular a estrutura e propriedades de moléculas; a modelagem molecular,12 que usa gráficos 3D e técnicas de otimizaçao para ajudar a compreender a natureza e açao de compostos químicos e proteínas; e o planejamento de fármacos auxiliado por computador,13 que diz respeito ao uso de técnicas computacionais para auxiliar na descoberta e planejamento de novas moléculas bioativas. Nas últimas duas décadas, foram publicados artigos de revisao sobre QSAR/QSPR,14,15 artigos abordando metodologias de QSAR/QSPR,16-20 e de suas aplicaçoes,21-24 além de revisoes sobre modelagem molecular25-28 em língua portuguesa. Contudo, nao existe até o presente momento nenhum artigo científico que apresente a quimioinformática ao público acadêmico e científico em português. Este artigo de revisao visa apresentar a quimioinformática de uma forma simples e didática. A intençao dos autores é que esse artigo sirva de material introdutório em aulas de modelagem molecular e química medicinal e/ou computacional, e como um primeiro contato tanto para alunos de graduaçao quanto de pós-graduaçao, assim como para pesquisadores que tenham interesse em se aventurar nessa área da ciência.
REPRESENTAÇAO DE ESTRUTURAS QUIMICAS Representaçao gráfica A representaçao gráfica de um composto químico demonstra como seus átomos estao dispostos e conectados entre si (Figura 1 A, representaçao bidimensional e Figura 1 B, representaçao tridimensional). Representaçoes gráficas sao úteis para que compostos sejam visualizados e compreendidos por seres humanos, mas sao ineficientes para um computador capturar e processar a informaçao contida naquela representaçao.29 Diversas representaçoes computacionais de estruturas químicas já foram propostas. Aqui descrevemos as mais inovadoras e importantes. Em 1949, William Wiswesser propôs um método de representaçao estrutural através de linhas, denominado de Wiswesser Line Notation (WLN). A notaçao de linhas de Wiswesser foi utilizada por anos, se mostrando inovadora na triagem de subestruturas, mas esse método nao produzia um código canônico (único) para cada estrutura e nao era fácil de ser gerado, dando espaço à outras representaçoes unidimensionais (1D).30
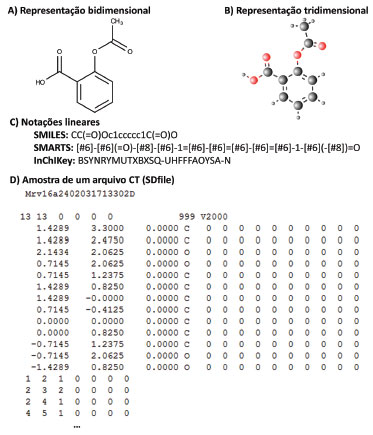 Figura 1. Métodos de representaçao de estruturas químicas, utilizando o ácido acetilsalicílico (Aspirina) como exemplo
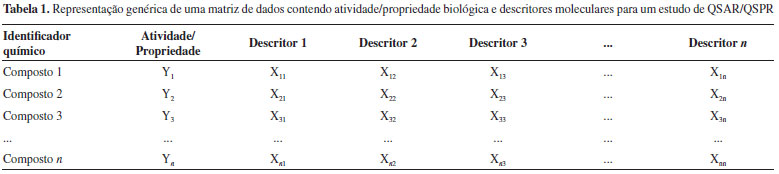
Na década de 1960, Harry Morgan desenvolveu um algoritmo capaz de representar estruturas químicas de forma única.31 Esse algoritmo representou um importante passo na representaçao de estruturas, pois foi essencial para criar um registro numérico único automatizado para cada estrutura química que fosse depositada no Sistema de Registros Químicos do Chemical Abstract Service (CAS),32 que pertence à Sociedade Americana de Química.33 Esse sistema representou um marco na pesquisa química, por ser uma forma simples de se identificar substâncias químicas, o que é essencial para a construçao de bases de dados de compostos. Foi na década de 1980 que David Weininger, fundador da Daylight Chemical Information Systems, Inc., propôs a primeira versao do SMILES (do inglês, Simplified Molecular-Input Line-Entry System) (Figura 1C). Weininger também considerou a teoria dos grafos para propor seu sistema de representaçao de estruturas. Cada átomo é representado por seu símbolo na tabela periódica, parênteses sao usados para indicar pontos de ramificaçao e os rótulos numéricos designam pontos de conexao de anéis. A gramática básica do SMILES também inclui informaçoes isotópicas, configuraçao sobre ligaçoes duplas e quiralidade, conhecido como SMILES isomérico.34,35 Desde sua criaçao, o SMILES foi modificado e expandido pela Daylight para incluir novos recursos, denominando-o de SMARTS (do inglês, SMiles ARbitrary Target Specification).36 Posteriormente, a IUPAC (do inglês, International Union of Pure and Applied Chemistry) desenvolveu sua própria notaçao linear para representar estruturas químicas, o InChIKey (do inglês, International Union of Pure and Applied ChemistryKey), baseado no seu identificador InChI.37 Nessa mesma época, diversos formatos CT (Chemical Table38 ou Connection Table39) foram desenvolvidos pela Molecular Design Limited (MDL), sendo o MDL MOL (ou molfile) e MDL SDF (ou SDfile) os mais utilizados (Figura 1D). Esses formatos representam as estruturas químicas como se fossem grafos e as informaçoes sao armazenadas em uma tabela. A teoria dos grafos descreve a relaçao de objetos em determinado conjunto através de vértices. Em arquivos CT, os átomos mais pesados que o hidrogênio correspondem aos vértices e ligaçoes químicas às arestas.38,39 Existem vários softwares que interpretam essa informaçao e representam as estruturas químicas graficamente. Softwares como o MarvinView (https://www.chemaxon.com/) e PyMOL (http://pymol.org/), interpretam essa informaçao e representam as estruturas químicas graficamente. Existem atualmente vários softwares e plataformas na web para que o usuário possa desenhar e visualizar representaçoes de estruturas químicas, denominados editores moleculares. Um dos editores moleculares mais conhecidos, o ChemDraw40 (http://www.cambridgesoft.com/software/overview.aspx), foi desenvolvido em 1985, sendo atualmente comercializado pela empresa PerkinElmer Inc. Alternativas gratuitas de livre acesso estao disponíveis e vem sendo amplamente utilizadas, como o MarvinSketch (https://www.chemaxon.com/) e Avogadro (https://avogadro.cc/). O JMSE41 (http://peter-ertl.com/jsme/) é um editor molecular escrito em JavaScript, que pode ser incorporado em outras ferramentas, como por exemplo, o OSRA (Optical Structure Recognition, https://cactus.nci.nih.gov/cgi-bin/osra/index.cgi), programa projetado para converter representaçoes gráficas de estruturas químicas, como aparecem em artigos de revistas, documentos de patentes, livros didáticos, revistas comerciais, em formatos interpretáveis por um computador, como SMILES ou SDfile. Uma lista completa de editores moleculares para diversas plataformas (Windows, macOS, Linux, Java, editores online e aplicativos para celular) pode ser encontrada na página de editores moleculares do Wikipedia (https://en.wikipedia.org/wiki/Molecule_editor). Descritores moleculares Para que uma estrutura química seja compreendida e processada por um computador, ela precisa ser descrita em uma sequência numérica única. Descritores moleculares representam estruturas químicas incorporando muito mais informaçao do que os métodos anteriores. Um descritor molecular é o resultado final de um procedimento matemático e lógico que transforma informaçao química codificada em uma representaçao simbólica de uma molécula em um número útil ou é o resultado de algum experimento padronizado.42 Os descritores sao dispostos em uma matriz (Tabela 1) ou em um vetor de bits (bit vector ou STD logic vector). Como pode-se observar na Tabela 1, os dados de atividade/propriedade (Yn) sao armazenados na coluna “Atividade/Propriedade” e cada descritor segue em uma nova coluna, de forma que todos os descritores (Xn) de um determinado composto estejam na mesma linha.43
Diferentes tipos de descritores químicos refletem diferentes níveis de representaçao estrutural. Esses descritores podem ser classificados quanto à sua “dimensionalidade” em unidimensionais (1D), baseados em propriedades físico-químicas e da fórmula molecular (ex., massa molecular, refratividade molar, logP, entre outros); bidimensionais (2D), que descrevem propriedades que podem ser calculadas de uma representaçao 2D (ex., número de átomos, número de ligaçoes, índices de conectividade, entre outros); e tridimensionais (3D), que dependem da conformaçao 3D das moléculas (ex., volume de Van der Waals, área de superfície acessível ao solvente, entre outros).44 Outros níveis de representaçao, como descritores 4D, propostos por Hopfinger et al.,45 constituem uma abordagem 3D que utiliza uma a conformaçao obtida por meio de simulaçao de dinâmica molecular. Os descritores 5D propostos por Vedani e Dobler46 foram uma extensao do 4D proposto por Hopfinger et al.,45 adicionando liberdade conformacional, permitindo assim uma representaçao múltipla da topologia dos ligantes no sítio ativo. O mesmo grupo propôs em seguida descritores 6D, que consideram vários modelos de solvataçao simultaneamente.47 Outra classificaçao diz respeito à natureza desses descritores, podendo ser: (i) constitucionais, que sao derivados da composiçao atômica do composto (ex., peso molecular, números de átomos e ligaçoes); (i) topológicos (ex., índice de conteúdo de informaçoes de ligaçoes); (iii) geométricos, que sao derivados de coordenadas 3D (ex., volume molecular, área de superfície polar, entre outros); (iv) eletrostáticos, que sao derivados das cargas parciais (ex., índices de polaridade, carga parciais, entre outros); e (v) mecânico-quânticos, que sao derivados das funçoes de onda dos elétrons (ex., energia dos orbitais moleculares).48 Vários programas de computador estao disponíveis para o cálculo de descritores moleculares, como DRAGON r (Talete SRL, Milan, Italy), CDK r (http://www.rguha.net/code/java/cdkdesc.html), CODESSA r (http://www.semichem.com/codessa/), RDKit (http://www.rdkit.org/), entre outros.
BASES DE DADOS Atualmente, existem várias bases de dados de compostos químicos que trazem diversas informaçoes relevantes para as mais diversas áreas da química. Como exemplo, as bases de dados ChemSpider (http://www.chemspider.com/) e Chemicalize (http://chemicalize.com/) trazem relevantes informaçoes relacionadas à estrutura das moléculas, como nome químico, nome comercial, identificadores (e.g., número do CAS), propriedades físicas, espectro interativo, referências na literatura e fornecedores de produtos químicos. O SciFinder (https://scifinder.cas.org/) traz informaçoes estruturais, reaçoes químicas e publicaçoes científicas e patentes relacionadas às mais de 100 milhoes de estruturas químicas depositadas no CAS (https://www.cas.org/), base de dados de compostos químicos da American Chemical Society, fundada em 1907.49 A KnowItAll U (http://www.knowitallu.com/) provê acesso a mais de 2 milhoes de espectros, incluindo infravermelho, ressonância magnética nuclear, massas, etc. Algumas bases de dados trazem informaçoes biológicas para estruturas químicas, como resultados de ensaios in vitro, in vivo e principalmente resultados de triagem de alta vazao (HCS/HTS). Como exemplo, cita-se a ChEMBL50,51 (https://www.ebi.ac.uk/chembl/) e PubChem52,53 (http://pubchem.ncbi.nlm.nih.gov/). Essas últimas tiveram um crescimento excepcional nos últimos anos, principalmente devido à expansao dos ensaios de triagem de alta vazao e da química combinatória, que aceleraram a quantidade de dados biológicos produzidos para moléculas pequenas.8 O DrugBank54,55 (https://www.drugbank.ca/) é uma base de dados de fármacos aprovados com informaçoes mais abrangentes, como estrutura química, propriedades físico-químicas calculadas e experimentais, uso terapêutico e informaçoes mais detalhadas, como propriedades farmacocinéticas, toxicológicas, farmacodinâmicas e sobre seu alvo molecular, quando disponíveis. Vale ainda mencionar bases de dados de macromoléculas, como o PDB (Protein Data Bank, https://www.rcsb.org/) e o BMRDB (Biological Magnetic Resonance Data Bank,http://www.bmrb.wisc.edu/), que sao repositórios de proteínas, ácidos nucleicos e outras biomacromoléculas complexas que contribuem em estudos relacionados às ciências da saúde, planejamento de fármacos, agricultura, etc.56,57
ANALISE DE SIMILARIDADE QUIMICA Frequentemente, moléculas semelhantes possuem propriedades semelhantes,58,59 mas isso nem sempre se aplica a todos os casos.60 A similaridade química é um dos conceitos mais explorados em quimioinformática e em outras áreas da química, como a química medicinal61 e toxicologia.62 A análise de moléculas semelhantes é extremamente importante para se estabelecer relaçoes entre estrutura e atividade ou propriedade (SAR, do inglês, Structure-Activity Relationships) e compreender o comportamento de determinado grupo de moléculas. Essa tarefa contribui para se encontrar erros experimentais e os denominados cliffs de atividade em subgrupos de moléculas. Um cliff de atividade é definido como um par de estruturas químicas semelhantes com atividade/propriedade muito diferentes.63,64 Essa análise, obviamente, pode ser feita manualmente por um bom químico, contudo, se torna inviável quando o conjunto de dados é demasiadamente grande.61 A similaridade química é calculada em um computador aplicando-se uma funçao de similaridade (também chamada de coeficiente de similaridade) a descritores moleculares.61 Dentre as funçoes de similaridade mais utilizadas, podem-se citar o coeficiente de Tanimoto65 (também conhecido como similaridade de Jaccard),66 e as distâncias Euclideana67 e de Mahalanobis.68 Qualquer tipo de descritor pode ser utilizado na análise de similaridade, mas os descritores baseados em fragmentos moleculares, principalmente os do tipo impressao digital ou fingerprints, sao os mais utilizados, por serem mais fáceis de se interpretar.69 Além do auxílio na compreensao das relaçoes entre estrutura e atividade, o uso da similaridade molecular possui várias outras aplicaçoes. Pode-se utilizar coeficientes de similaridade para se identificar o núcleo comum de um determinado número de estruturas para se fazer uma análise de pares moleculares combinados (do inglês, matched molecular pair analysis, MMPA). A MMPA é uma abordagem para se identificar e comparar pares de moléculas semelhantes de um conjunto de compostos e avaliar a mudança de propriedade associada.70 Os pares combinados sao moléculas que diferem apenas por uma subestrutura particular ou um fragmento bem definido. Portanto, a MMPA pode revelar mudanças nas propriedades biológicas entre estruturas com alta similaridade, que diferem por apenas um grupo químico. Por fim, podemos citar o uso da similaridade molecular em ferramentas de busca dentro das bases de dados químicas (ver seçao anterior), além de catálogos de compostos químicos comerciais, como a ChemBridge (http://www.chembridge.com/) e Sigma-Aldrich (http://www.sigmaaldrich.com/).
RELAÇOES QUANTITATIVAS ENTRE ESTRUTURA QUIMICA E ATIVIDADE/PROPRIEDADE (QSAR/QSPR) Breve história e evoluçao A partir da década de 1930, com base nos conhecimentos acumulados em físico-química e química orgânica, a área de relaçoes entre estrutura e atividade ou propriedade começou a avançar.71 Em 1940, o estudo publicado por Hammett representou um marco no entendimento de propriedades moleculares de compostos orgânicos e na história do QSAR.72 Ele avaliou a ionizaçao de derivados do ácido benzoico e demonstrou, pela primeira vez, uma relaçao linear entre a variaçao dos grupos substituintes e a propriedade biológica estudada. Essa relaçao ficou conhecida como equaçao de Hammett. Quase quatro décadas se passaram quando Hansch e Fujita73 consolidaram as bases dos estudos de QSAR/QSPR com um estudo inovador. Nesse estudo, os autores demonstraram que a atividade biológica poderia ser linearmente correlacionada com diferentes parâmetros físico-químicos, relacionados a efeitos hidrofóbicos, estéricos e eletrônicos. Na mesma época, Free e Wilson74 desenvolveram uma abordagem para descrever a atividade biológica de compostos através de equaçoes estabelecidas a partir da análise de séries congêneres. Na década de 1970, Kubinyi refinou o modelo de Hansch, desenvolvendo um modelo que descrevia a dependência nao linear da atividade biológica sobre o caráter hidrofóbico.75-77 Na década de 1980, com os avanços em termos de hardware e software, o estudo das propriedades de estruturas tridimensionais (3D) tornou-se possível e entao diversas abordagens de QSAR-3D foram propostas e desenvolvidas. Em 1980, Hopfinger e colaboradores desenvolveram um método baseado na forma molecular, denominado Análise da Forma Molecular (MSA, do inglês, Molecular Shape Analysis), o qual utiliza análise conformacional para obtençao de descritores 3D da forma molecular.78 Em 1988, Cramer, Patterson e Bunce desenvolveram a metodologia de Análise Comparativa de Campos Moleculares (CoMFA, do inglês, Comparative Molecular Fields Analysis).79 Essa técnica usa descritores 3D estéricos e eletrostáticos em campos moleculares, empregando o método dos mínimos quadrados parciais (PLS, do inglês, partial least squares) para estabelecer a relaçao com a atividade biológica. Em 1989, o CoMFA foi implementado na plataforma comercial SYBYL (http://www.tripos.com) e se tornou um dos métodos mais utilizados e difundidos nas últimas duas décadas.80 No entanto, o CoMFA possui um processo de modelagem lento, além de ser dependente do alinhamento e da conformaçao 3D (bioativa) das estruturas do conjunto de dados.10 Uma proposta similar ao CoMFA, mas que adicionou os efeitos de ligaçoes de hidrogênio (doador e aceptor) e hidrofóbico foi desenvolvida pelo mesmo grupo e denominada de Análise Comparativa de Indices de Similaridade Molecular (CoMSIA, do inglês, Comparative Molecular Similarity Index Analysis),81 também implementado na plataforma SYBYL (http://www.tripos.com). Outras metodologias de QSAR inovadoras foram desenvolvidas, como o QSAR-4D, proposto por Hopfinger e colaboradores.45 Nesse método, os descritores sao calculados para uma amostragem de conformaçoes obtidas por meio de simulaçao de dinâmica molecular, reduzindo a dificuldade em encontrar a conformaçao bioativa. Posteriormente, extensoes desse método foram propostas, como o Quasar QSAR-5D,46 que adiciona liberdade conformacional, permitindo uma representaçao múltipla da topologia dos ligantes no sítio ativo, e Quasar QSAR-6D, que considera vários modelos de solvataçao simultaneamente.47 Desde o final dos anos 1990, métodos usados em QSAR/QSPR apresentaram vários avanços e crescente interesse de grupos de pesquisa e indústrias farmacêuticas.82 Inúmeros descritores moleculares, métodos de aprendizado de máquina e parâmetros de validaçao foram desenvolvidos e vêm sendo aplicados. Os estudos de QSAR/QSPR consolidaram a quimioinformática como uma ciência capaz de transformar a informaçao química em conhecimento. Inicialmente, modelos de QSAR/QSPR eram utilizados principalmente para otimizaçao de compostos químicos. Atualmente, o QSAR/QSPR tem sido utilizado desde as fases de descoberta e avaliaçao de compostos químicos, até otimizaçao de compostos líderes, tendendo a ser cada vez mais empregado, devido ao aprimoramento de recursos computacionais e o crescente aumento da disponibilidade de dados químicos e biológicos de alta qualidade.10 Princípios Um modelo de QSAR é uma equaçao matemática que relaciona a estrutura química com a propriedade biológica. A abordagem de QSAR consiste na aplicaçao de vários métodos estatísticos de análise de dados para desenvolver modelos que possam predizer corretamente determinada propriedade biológica de compostos baseados em sua estrutura química. Para se estabelecer essa relaçao, é necessário o cálculo de descritores moleculares e que a atividade biológica/propriedade tenha sido definida experimentalmente (Figura 2). O modelo de QSAR é a representaçao matemática final que, genericamente, pode ser definida através da Equaçao 1:
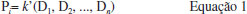
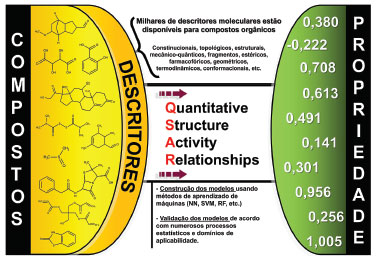 Figura 2. Esquema representando a geraçao de um modelo de QSAR/QSPR (modificado de ref. 83)
onde Pi é a atividade biológica ou propriedade da molécula, D1, D2, ..., Dn sao propriedades estruturais (descritores) calculadas (ou, em alguns casos, medidas experimentalmente) e k’ é um peso definido pelo algoritmo computacional e atribuído aos descritores para se calcular as propriedades das moléculas.43,83 Para se desenvolver um modelo de QSAR nao é necessário, obrigatoriamente, recursos computacionais. Essa equaçao pode ser derivada manualmente, contudo, dado a imensa quantidade de descritores moleculares e conjuntos de dados disponíveis, esse trabalho se tornou humanamente inviável sem o auxílio de um computador. Para contornar essa dificuldade, métodos de aprendizado de máquina têm sido amplamente empregados.84 Esses métodos estabelecem peso aos descritores, ajustando a equaçao que relaciona a estrutura química com a atividade biológica ou propriedade (Figura 2).83 O principal objetivo de um modelo de QSAR é avaliar compostos que carecem de dados experimentais (in vitro e in vivo). Essa abordagem possui aplicaçoes na (i) identificaçao racional de novos ligantes/protótipos com atividade/propriedade desejada; (ii) otimizaçao da atividade/propriedade; e na (iii) identificaçao de compostos potencialmente tóxicos.83 Alguns trabalhos que demonstram a aplicaçao de modelos de QSAR no planejamento de fármacos e prediçao de toxicidade sao comentados na seçao Aplicaçoes. Os modelos de QSAR podem ser divididos em globais e locais. Modelos globais sao gerados usando-se todo o conjunto treinamento. Nesse caso, podem existir subgrupos de moléculas que apresentam uma maior similaridade estrutural e, consequentemente, possuem características específicas. Esse subconjunto pode ser utilizado para se gerar modelos chamados de locais, ou seja, que usam um subconjunto de estruturas do conjunto de dados total baseados ou na similaridade química ou no mecanismo de açao. Apesar de modelos locais poderem contribuir com a elaboraçao de novas regras de relaçao estrutura-atividade, estudos mostram que, normalmente, nao há melhorias na precisao do modelo quando se utilizam modelos de QSAR locais em comparaçao com modelos globais.85,86 Aprendizado de máquina O aprendizado de máquina consiste em métodos de inteligência artificial que dizem respeito ao estudo e construçao de sistemas que podem aprender com dados. Comumente, o processo é desenvolvido em três etapas: (i) representaçao dos dados, (ii) otimizaçao da hipótese, e (iii) generalizaçao.87 Na geraçao de um modelo de QSAR/QSPR, os dados sao representados na forma de uma matriz com a atividade/propriedade no eixo Y e os descritores no eixo X (Tabela 1).83 Uma hipótese (equaçao) é gerada em via de se estabelecer uma relaçao dos descritores com a atividade biológica. Essa relaçao é otimizada em um determinado números de vezes, dependendo da funçao utilizada e do algoritmo selecionado. Em seguida, a capacidade de generalizaçao da hipótese é testada, avaliando-se o conjunto teste. Nesse contexto, generalizar diz respeito à habilidade da equaçao final em predizer corretamente um exemplo/tarefa (no caso, a atividade/propriedade) nao usado na construçao dos modelos (conjunto teste).87 Os métodos de aprendizado sao divididos em supervisionados e nao supervisionados. No primeiro caso, os algoritmos sao treinados com variável Y defnida na tentativa de gerar uma funçao matemática que generalize essa variável. Vários algoritmos de aprendizado supervisionados estao disponíveis atualmente, como random forest (RF),88 support vector machine (SVM),89 redes neurais (NN)90 e deep learning (DL).91 Os modelos gerados usando métodos supervisionados, uma vez validados, constituem um ponto de partida para a avaliaçao e/ou seleçao de compostos químicos que carecem de dados experimentais (ver seçao Triagem virtual à frente).10 Os métodos nao supervisionados sao usados para identificar padroes nos conjuntos de dados com base apenas nos descritores, visto que a variável Y nao é definida. Essa abordagem, quando aplicada aos conjuntos de dados químicos, identifica subgrupos homogêneos entre um conjunto de dados heterogêneo.67 Alguns dos vários usos dessa abordagem incluem (i) a verificaçao da diversidade estrutural do conjunto de dados; (ii) a avaliaçao da consistência de dados experimentais; e (iii) a exploraçao de possíveis interferências que influenciam na atividade, contribuindo para a revelaçao de novas regras de SAR.92,93 Algoritmos comumente utilizados nessa abordagem incluem análise de componentes principais (PCA, do inglês principal component analysis), análise de agrupamentos hierárquicos (HCA, do inglês hierarchical cluster analysis) e mapas auto-organizáveis (SOM, do inglês self-organizing maps).67 Boas práticas de desenvolvimento e validaçao No início dos anos 2000 surgiu uma crescente preocupaçao com a qualidade das prediçoes de modelos de QSAR gerados e publicados na literatura. Para garantir a qualidade dos modelos gerados, diretrizes e recomendaçoes de boas práticas de desenvolvimento e validaçao de modelos de QSAR foram propostas.10,83,94 Em 2004, a OECD (Organization for Economic Co-operation and Development) publicou princípios para validaçao de modelos de QSAR para o uso prático na regulamentaçao de compostos químicos. Esses princípios sao: (i) atividade biológica ou propriedade definida; (ii) algoritmo claro; (iii) domínio de aplicabilidade (DA) definido; (iv) avaliaçao apropriada da robustez e preditividade; (v) e interpretaçao mecanística, que significa encontrar relaçoes entre os descritores e a atividade biológica ou propriedade, em via de se compreender melhor o mecanismo de açao de uma estrutura química ou aprofundar o conhecimento biológico sobre a propriedade em estudo.95 Em consideraçao a esses princípios, vários elementos-chave devem ser considerados durante o desenvolvimento e validaçao de modelos de QSAR/QSPR e serao discutidos a seguir.10,83,94 Preparo químico e biológico do conjunto de dados Existe uma crescente preocupaçao sobre a irreprodutibilidade frequente de dados experimentais relatados em publicaçoes científicas.96 Modelos computacionais sao sensíveis à qualidade dos dados utilizados e, por consequência, o preparo do conjunto de dados é indispensável para tentar evitar que erros sejam propagados em outros estudos e/ou interfiram na preditividade dos modelos. Abordagens de quimioinformática auxiliam na identificaçao e remoçao de amostras que contenham erros experimentais ou de anotaçao.97 Esse processo inclui o preparo químico e biológico dos conjuntos de dados (Figura 3).
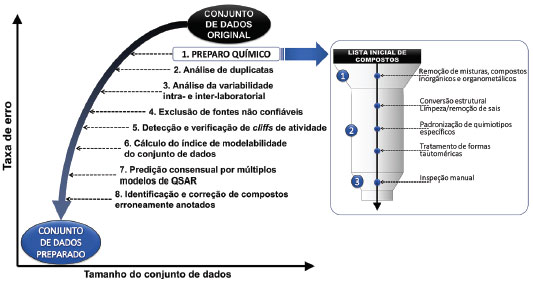 Figura 3. Fluxo geral de trabalho proposto para o preparo de conjuntos de dados (modificado de ref. 98)
Um fluxo de trabalho de preparo de conjunto de dados foi proposto por Fourches, Muratov e Tropsha (2010 e 2016).98 Esse fluxo, bastante rigoroso, se inicia com o preparo químico do conjunto de dados (Figura 3, passo 1), que segue um protocolo previamente estabelecido99 e permite a identificaçao e correçao de erros nas estruturas químicas. Nessa etapa, misturas de componentes, compostos inorgânicos e organometálicos sao removidos (quando nao é possível calcular descritores para esses compostos). É feita a padronizaçao de quimiotipos específicos, como anéis aromáticos, grupos nitro e formas tautoméricas. Contraíons sao removidos e duplicatas identificadas (compostos repetidos) sao analisadas e removidas (Figura 3, passo 2). A análise das duplicatas é importante pois permite avaliar a qualidade dos dados experimentais e remover estruturas químicas de registros duplicados com dados experimentais contraditórios, que afetam a qualidade dos modelos. Duplicatas presentes no conjunto teste superestimam a qualidade dos modelos. Uma inspeçao manual é requerida ao final do processo para garantir que todas as estruturas estejam corretas. Em conjuntos de dados grandes, essa análise pode ser feita por amostragem. Em seguida, realiza-se uma análise da variabilidade experimental intra- e interlabotarial (Figura 3, passo 3) e exclui-se de fontes de dados nao confiáveis, ou seja, dados com alta variaçao nos valores dos ensaios (passo 4), a fim de aumentar a qualidade dos dados e contribuir na tomada de decisoes sobre a combinaçao de dados de diferentes fontes. A detecçao e análise dos “cliffs” de atividade63 (passo 5) e o cálculo e ajuste do índice de modelabilidade100 do conjunto de dados (passo 6) estimam a viabilidade de obtençao de modelos preditivos para um determinado conjunto de dados e serve como indicadores adicionais de qualidade dos dados. A geraçao de modelos de QSAR de consenso (passo 7) é utilizada para tentar aumentar a confiança das prediçoes de modelos individuais. Nessa etapa, gera-se modelos de QSAR independentes, com descritores moleculares e/ou algoritmos diferentes. O modelo de consenso é aquele que considera uma média das prediçoes dos modelos independentes. Na última etapa (passo 8), dados biológicos incorretos podem ser identificados investigando-se compostos que tiveram atividade biológica predita muito diferente do valor experimental. Detecçao de amostras atípicas ou anômalas (“outliers”) Amostras atípicas ou “outliers” sao compostos que apresentam atividade/propriedade inesperada. Essas amostras podem resultar de erros experimentais, representar erros na estrutura química ou representarem “cliffs” de atividade. Um cliff de atividade representa estruturas químicas semelhantes com atividade/propriedade muito diferentes.63,64 Por esse motivo, amostras atípicas nao devem ser removidas sem explicaçao, visto que sua remoçao, frequentemente feita indiscriminadamente no passado para melhorar os parâmetros estatísticos do modelo, é considerada manipulaçao. A melhoria nos parâmetros estatísticos pode nao representar melhoria real na preditividade externa do modelo. Se um composto possui um dado de atividade/propriedade dentro do intervalo do conjunto de dados e esse composto está dentro do domínio de aplicabilidade do conjunto de dados, ele nao deve ser removido. Se o composto representar um cliff de atividade, sua má prediçao pelo modelo precisa ser interpretada e discutida.10 Validaçao dos modelos A utilidade de um modelo de QSAR/QSPR depende de sua preditividade, ou seja, da sua capacidade em predizer determinada propriedade biológica com elevada taxa de acerto. O procedimento de validaçao serve para analisar a robustez e a preditividade de um modelo de QSAR. A validaçao é dividida em interna e externa. A validaçao interna é importante para determinar a robustez de um modelo, ou seja, a capacidade de reproduzir suas prediçoes. Apesar da validaçao interna ser importante principalmente durante a geraçao dos modelos para se ajustar parâmetros do algoritmo, a real preditividade do modelo de QSAR/QSPR gerado só é avaliada usando-se um conjunto de validaçao externa, ou seja, que nao foi utilizado para gerar, derivar, ou selecionar os modelos.101,102 A validaçao interna normalmente é feita retirando-se um composto (leave-one-out) ou vários (leave-many-out) compostos do conjunto treinamento. Assim, um novo modelo é gerado e os compostos retirados sao entao preditos. Essa prática é realizada por um determinado número de vezes. O modelo final é gerado utilizando todo o conjunto treinamento. Já a validaçao externa é realizada separando-se parte dos compostos do conjunto modelagem para validaçao externa. Normalmente, separa-se entre 20% e 30% do conjunto de dados original. Um método ainda mais rigoroso de validaçao externa consiste no método de validaçao externa n-fold. Nesse caso, o conjunto de dados total é dividido em n partes. Uma das partes é utilizada como validaçao externa e as outras usadas para geraçao dos modelos. Isso é repetido n vezes, até que cada um dos n subconjuntos passe uma vez pelo conjunto de validaçao externa. Ao final, um consenso entre as prediçoes individuais é realizado. A diferença entre a validaçao interna e externa, é que, na interna, o modelo final utiliza todos os compostos que foram utilizados para validaçao, ao passo que na validaçao externa, os compostos do conjunto de validaçao externa nao sao usados para gerar os modelos.103,104 Um modelo é considerado rigorosamente validado quando ele atinge os requisitos mínimos de validaçao interna e externa. Diferentes métricas estatísticas foram propostas, sendo aquelas usadas para modelos contínuos diferentes das usadas para modelos categóricos. Um modelo contínuo é gerado quando a variável Y do conjunto de dados corresponde a uma escala numérica (p. ex., pIC50, ponto de fusao, ponto de ebuliçao, etc.). Um modelo categórico é gerado quando a variável Y é dividida em classes (p. ex., ativo/inativo, tóxico/nao-tóxico, solúvel/insolúvel, etc.). Um modelo continuo possui boa robustez quando seu coeficiente de correlaçao cruzada (R2) é maior que 0.7.104 Apesar de necessária, essa métrica é insuficiente para se avaliar a preditividade externa de um modelo,101 por isso é necessário se avaliar o coeficiente de correlaçao de validaçao cruzada externa (Q2), que é considerado válido quando Q2 ≥ 0.6.104 Os modelos categóricos sao validados usando-se a acurácia balanceada (ou taxa de classificaçao correta),105 sensibilidade e especificidade106 e os valores de preditividade positiva e negativa,107 sendo que todas essas métricas devem estar acima de 0.6. As métricas aqui citadas sao as mais comuns em quimioinformática, contudo, existem várias outras que podem ser utilizadas em via de se garantir uma alta preditividade do modelo, como curva ROC (receiver operating characteristic),108 diferentes fórmulas para calcular o Q2,104,109 entre outras que podem ser encontradas na literatura.110,111 Por fim, vale mencionar a técnica de aleatorizaçao da variável Y (Y-randomization), que é recomendada para se garantir que os resultados dos modelos de QSAR nao sejam provenientes do acaso. Nesse procedimento, a variável Y é aleatorizada e novos modelos sao gerados. Caso a preditividade dos modelos com variável Y aleatória seja melhor que os modelos de QSAR, os modelos devem ser descartados, uma vez que os descritores moleculares nao descrevem bem Y.112 Definiçao do domínio de aplicabilidade (DA) O domínio de aplicabilidade ou domínio de aplicaçao é uma característica importante de qualquer modelo de QSAR.113,114 O DA representa o espaço químico definido pelo conjunto de moléculas do conjunto de treinamento de determinado modelo de QSAR. Prediçoes sao consideradas confiáveis quando a molécula predita se insere dentro do DA do modelo em questao. É importante ressaltar que o limite do DA é totalmente definido pelo tamanho e diversidade do conjunto utilizado para desenvolver os modelos. Quando a molécula está fora do domínio de um modelo, a prediçao pode ser incorreta, visto ela pode ser muito diferente e nao ser completamente compreendida pelo modelo. Por esse motivo, a determinaçao do DA é uma etapa essencial para aceitabilidade de um modelo de QSAR.83 Recentemente, um estudo115 demonstrou que modelos de QSAR gerados para cosméticos, fármacos e pesticidas podem ser utilizados mutualmente, ou seja, um modelo desenvolvido usando principalmente fármacos e moléculas fármaco-semelhantes pode ser usado para se avaliar cosméticos e pesticidas. Contudo, é imprescindível que antes seja verificado se a molécula a ser avaliada computacionalmente está dentro do DA daquelas usadas para gerar o modelo em questao.
TRIAGEM VIRTUAL A triagem virtual (VS, do inglês Virtual Screening) compreende o processo de triagem de bibliotecas de compostos químicos através de modelos computacionais com a finalidade de avaliar e/ou selecionar compostos com propriedades desejadas.116 Essa é uma abordagem bem estabelecida no moderno processo de planejamento e desenvolvimento de fármacos,116 assim como na avaliaçao de potenciais compostos perigosos ao meio ambiente.8 A triagem virtual é uma alternativa rápida e de baixo custo para a triagem e seleçao de hits,117 o que reduz o número de compostos selecionados para avaliaçao experimental.118 Habitualmente, a taxa de sucesso da triagem de alta vazao (HTS, do inglês, High Throughput Screening) varia entre 0,01% e 0,14%, enquanto as taxas de sucesso para a VS geralmente varia entre 1% e 40%.119-121 Os métodos computacionais usados para VS sao normalmente divididos em métodos baseados no ligante (LBVS, do inglês, Ligand-based Virtual Screening) e na estrutura (SBVS, do inglês, Structure-based Virtual Screening).122 Abordagens baseadas no ligante usam estruturas químicas associadas a dados conhecidos para desenvolver modelos, tais como análise de similaridade,123 modelos de QSAR,84 e modelos farmacofóricos.124 Por outro lado, os métodos baseados na estrutura utilizam a estrutura tridimensional (3D) do alvo biológico. Neste caso, as moléculas sao acopladas no sítio de ligaçao e classificadas com base na sua afinidade de ligaçao predita ou complementaridade. Modelos farmacofóricos e métodos baseados na estrutura estao fora do escopo desse artigo. Informaçoes adicionais sobre essas técnicas podem ser encontradas na literatura.120,125-128 Um método de triagem virtual que se popularizou bastante dentro da toxicologia computacional compreende o uso de alertas estruturais. Alertas estruturais129 sao subestruturas moleculares que estao associadas com determinada propriedade da molécula.130 Comumente, alertas baseiam-se no conhecimento humano e sao destinados a entender a base química do mecanismo de toxicidade ou, pelo menos, o evento molecular iniciante no caso de propriedades mais complexas.131 Os alertas estruturais sao usados para sinalizar potenciais perigos e agrupar compostos em categorias para propiciar a comparaçao de análogos químicos por interpolaçao.132,133 Contudo, foi demonstrado que alertas estruturais sao extremamente promíscuos.134 Se usados sozinhos para predizer a toxicidade, podem ser prejudiciais tanto para a avaliaçao da segurança quanto no planejamento de fármacos. Entretanto, os alertas estruturais, quando validados estatisticamente por modelos de QSAR, podem contribuir para o planejamento de compostos mais seguros. Em resumo, alertas estruturais devem ser usados apenas como proposta de mecanismo de açao e somente quando validados um método estatístico robusto, no caso, modelos de QSAR.134
APLICAÇOES DA QUIMIOINFORMATICA As aplicaçoes da quimioinformática sao ilimitadas. Mesmo que seja uma área mais aplicada para a avaliaçao da segurança de compostos químicos, para fins ambientais e no processo de planejamento e desenvolvimento de fármacos,6 a quimioinformática pode ser utilizada em todas as áreas da química. Contribui para a reduçao do custo do desenvolvimento de novas moléculas; reduçao do número de animais utilizados em ensaios experimentais, e promoçao da química verde. Além disso, pode aumentar a eficiência do processo de pesquisa e desenvolvimento, pois diminui resíduos gerados, visto que compostos mais improváveis de terem sucesso sao descartados antes de seguirem para ensaios experimentais.43,135 Um fluxograma de aplicaçao da quimioinformática para o planejamento de moléculas de interesse encontra-se representado na Figura 4. Dados biológicos de compostos químicos provenientes de estudos experimentais (fase 0) estao sempre sendo publicados em artigos científicos e/ou depositados em repositórios de dados (fase 1). Esses dados servem de ponto de partida para os estudos de quimioinformática. Na fase 2, os dados químicos e biológicos sao compilados, preparados e integrados. Em seguida (fase 3), a análise do conjunto de dados e de similaridade é realizada e os modelos de QSAR sao gerados. Os modelos de QSAR gerados sao entao utilizados para triar virtualmente uma biblioteca com novos compostos e/ou compostos que nunca foram avaliados experimentalmente para a propriedade em estudo (fase 4). Na fase 5, os melhores hits sao selecionados para avaliaçao experimental. Compostos avaliados com propriedades indesejadas sao descartados. Os compostos selecionados podem ser sintetizados ou adquiridos de empresas especializadas, como a ChemBridge (http://www.chembridge.com/) e Sigma-Aldrich (https://www.sigmaaldrich.com/). Os compostos entram novamente na fase 0 (avaliaçao experimental), fechando o ciclo. A avaliaçao experimental é a melhor forma de validaçao de um modelo computacional. Após essa etapa, os compostos sao utilizados para enriquecer os modelos disponíveis. Essa prática aumenta o espaço químico de cobertura dos modelos e contribui para aumentar a sua preditividade.10
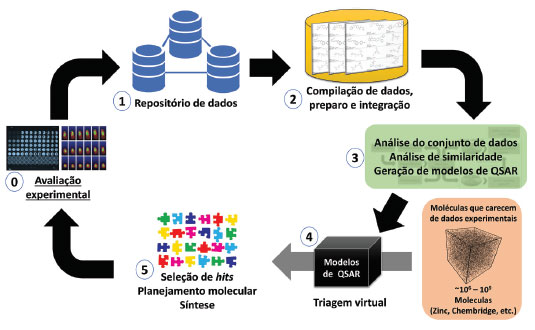 Figura 4. Aplicaçao da quimioinformática na avaliaçao, seleçao e planejamento de compostos com propriedades otimizadas (modificado de ref 10).
Modelos de QSAR têm sido empregados com sucesso na triagem virtual para a descoberta de compostos promissores. Por exemplo, Neves e colaboradores136 descobriram novos compostos com atividade contra Schistosoma mansoni integrando técnicas de QSAR e triagem de alta vazao. Em um estudo complementar, Melo-Filho e colaboradores137 identificaram novos esqueletos moleculares com atividade esquistosomicida. Braga e colaboradores138,139 desenvolveram um servidor online baseado em QSAR para a prediçao de toxicidade cardíaca (bloqueio da hERG). Zhang e colaboradores140 descobriram 25 novos compostos com atividade antimalárica e com baixa citotoxicidade, através do emprego de modelos de QSAR. Alves e colaboradores, analisando dados experimentais e de prediçoes de modelos de QSAR, demonstraram baixa correlaçao entre as propriedades de permeabilidade e sensibilizaçao cutânea,141,142 descritas como correlacionadas na literatura.143 Além disso, analisando-se dados experimentais de sensibilizaçao cutânea de pele humana e dados do modelo animal preferencial realizado em camundongos, foi demonstrado que o modelo animal nao descreve bem essa propriedade em seres humanos.144 Um recente e famoso caso da aplicaçao do QSAR para a descoberta de fármacos consistiu na descoberta de uma nova classe de inibidores potentes e seletivos do receptor androgênico com um novo mecanismo de açao, resultando no maior acordo de licenciamento acadêmico na história do Canadá, totalizando 142 milhoes de dólares.145 Nesse estudo, os autores empregaram vários métodos computacionais, dentre eles, modelos de QSAR-2D, 3D e 4D, modelos farmacofóricos e acoplamento molecular. Os autores trabalharam durante todo o projeto com experimentalistas em via de otimizar os modelos e as estruturas que eram propostas. Além dos trabalhos citados, outros estudos inovadores que compreendem a aplicaçao da quimioinformática para modelagem de peptídeos,146,147 misturas de componentes148-150 e nanopartículas151 também foram descritos. As aplicaçoes dos princípios de QSAR/QSPR sao várias, de forma que diversas derivaçoes da nomenclatura têm surgido na literatura, como QSRR (quantitative structure-(chromatographic) retention relationships),152 QNAR (quantitative nanostructure-activity relationships),153 QSTR (quantitative structure-toxicity relationships),154 entre outros.155-157 Existem atualmente softwares para elucidaçao de estruturas químicas através de seus espectros de ressonância magnética nuclear, como ACD/Structure Elucidator Suite (http://www.acdlabs.com/products/com_iden/elucidation/struc_eluc/). Além disso, vários softwares como o MarvinSketch (https://www.chemaxon.com/) permitem cálculo de propriedades como lipofilicidade, solubilidade em água, além da prediçao de espectros de massa e de ressonância magnética nuclear. Dessa maneira, observamos que métodos de quimioinformática têm sido amplamente aplicados para resolver problemas em várias áreas da química. A quimioinformática tem se consolidado em uma época em que repositórios de dados químicos e/ou biológicos com informaçao de livre acesso têm expandido rapidamente e sua utilidade e impacto na ciência tende a aumentar nas próximas décadas.6
CONSIDERAÇOES FINAIS A quimioinformática é uma ciência interdisciplinar que usa recursos computacionais e de tecnologia da informaçao para transformar informaçao química em conhecimento. Essa área se consolidou como uma área científica independente, evoluindo desde a década de 1960, quando lidava com aspectos práticos de representaçao, manipulaçao e processamento de estruturas químicas individuais até o seu papel primordial na atualidade: exploraçao de bases de dados químicos e biológicos para a descoberta de novos compostos com atividade ou propriedades desejadas. A quimioinformática representa moléculas através de grafos armazenados em tabelas, linhas ou em descritores moleculares. Medidas de similaridade química ou relaçoes hierárquicas entre estruturas químicas sao utilizadas para se estabelecer relaçoes entre estrutura-atividade e compreender o comportamento biológico de determinado grupo de compostos. Além disso, a quimioinformática utiliza métodos da inteligência artificial (aprendizado de máquina) para gerar modelos de QSAR que, quando devidamente validados, podem ser usados para predizer a atividade ou propriedade biológica de compostos que carecem de dados experimentais. Neste artigo de revisao, nosso principal objetivo foi apresentar, pela primeira vez em língua portuguesa, a quimioinformática de forma simples e didática, abordando aspectos históricos, conceitos, métodos e também aplicaçoes práticas e perspectivas do progresso dessa área específica da Química. Esperamos que este material possa servir de referência para estudantes, professores e pesquisadores que estao iniciando sua jornada ou se aventurando nessa área da ciência.
AGRADECIMENTOS Os autores agradecem ao CNPq, à CAPES e à FAPEG pelo auxílio financeiro dos projetos do grupo de pesquisa e pelas bolsas concedidas. E.M. é professor na University of North Carolina at Chapel Hill (EUA) e atualmente é pesquisador visitante especial (PVE) no LabMol-UFG (CNPq # 400760/2014-2). C.H.A. é bolsista de produtividade em pesquisa do CNPq.
REFERENCIAS 1. Gasteiger, J.; Engel, T.; Chemoinformatics: a textbook, Gasteiger, J.; Engel, T., eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, FRG, 2003. 2. Brown, F. K.; Annu. Rep. Med. Chem. 1998, 33, 375. 3. Warr, W. A.; Extract from 218th ACS National Meeting and Exposition New Orleans, Louisiana, August 22-26, 1999 http://www.warr.com/warrzone2000.html, acessada em outubro 2017. 4. Hann, M.; Green, R.; Curr. Opin. Chem. Biol. 1999, 3, 379. 5. Willett, P.; Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2011, 1, 46. 6. Fourches, D.; Em Application of Computational Techniques in Pharmacy and Medicine; Gorb, L., Kuz’min, V., Muratov, E., eds.; Springer Netherlands: Dordrecht, 2014, cap. 16. 7. Gasteiger, J.; Molecules 2016, 21, 151. 8. Zhu, H.; Zhang, J.; Kim, M. T.; Boison, A.; Sedykh, A.; Moran, K.; Chem. Res. Toxicol. 2014, 27, 1643. 9. Tetko, I. V; Engkvist, O.; Chen, H.; Future Med. Chem. 2016, 8, 1801. 10. Cherkasov, A.; Muratov, E. N.; Fourches, D.; Varnek, A.; Baskin, I. I.; Cronin, M.; Dearden, J.; Gramatica, P.; Martin, Y. C.; Todeschini, R.; Consonni, V.; Kuz’min, V. E.; Cramer, R.; Benigni, R.; Yang, C.; Rathman, J.; Terfloth, L.; Gasteiger, J.; Richard, A.; Tropsha, A.; J. Med. Chem. 2014, 57, 4977. 11. Wild, D.; Introducing cheminformatics: an intensive self-guided study, 2nd ed., McGraw-Hill Open Publishing, 2013. 12. Hinchliffe, A.; Molecular Modelling for Beginners, Wiley: West Sussex, 2003. 13. Young, D. C.; Computational drug design: A guide for computational and medicinal chemists, John Wiley & Sons, Inc.: Hoboken , 2009, vol. 14. 14. Tavares, L. C.; Quim. Nova 2004, 27, 631. 15. Gaudio, A. C.; Zandonade, E.; Quim. Nova 2001, 24, 658. 16. Ferreira, M. M. C.; Montanari, C. A.; Gaudio, A. C.; Quim. Nova 2002, 25, 439. 17. Arroio, A.; Honório, K. M.; da Silva, A. B. F.; Quim. Nova 2010, 33, 694. 18. Montanari, M. L. C.; Montanari, C. A.; Gaudio, A. C.; Quim. Nova 2002, 25, 231. 19. Cormanich, R. A.; Nunes, C. A.; Freitas, M. P.; Quim. Nova 2012, 35, 1157. 20. Martins, J. P. A.; Ferreira, M. M. C.; Quim. Nova 2013, 36, 554. 21. Freitas, H. F.; Paz, O. S.; Castilho, M. S.; Quim. Nova 2009, 32, 2114. 22. Almeida, V. L. de; Lopes, J. C. D.; Oliveira, S. R.; Donnici, C. L.; Montanari, C. A.; Quim. Nova 2010, 33, 1482. 23. Andrade, J. G.; Freitas, H. F.; Castilho, M. S.; Quim. Nova 2012, 35, 466. 24. Walter, M. E.; Almeida, V. L.; Nunes, R. J.; Quim. Nova 2013, 36, 691. 25. Carvalho, I.; Pupo, M. T.; Borges, A. D. L.; Bernardes, L. S. C.; Quim. Nova 2003, 26, 428. 26. Sant’Anna, C. M. R.; Rev. Virtual Quim. 2009, 1, 49. 27. Barreiro, E. J.; Rodrigues, C. R.; Albuquerque, M. G.; Sant’Anna, C. M. R.; Alencastro, R. B.; Quim. Nova 1997, 20, 1. 28. Andrade, C. H.; Trossini, G. H. G.; Ferreira, E. I.; Rev. Eletronica Farm. 2010, VII, 1. 29. Brecher, J.; Pure Appl. Chem. 2006, 78. 30. Wiswesser, W. J.; J. Chem. Inf. Model. 1982, 22, 88. 31. Morgan, H. L.; J. Chem. Doc. 1965, 5, 107. 32. CAS History; https://www.cas.org/about-cas/cas-history, acessada em outubro 2017. 33. Figueras, J.; J. Chem. Inf. Model. 1993, 33, 717. 34. Weininger, D.; J. Chem. Inf. Model. 1988, 28, 31. 35. Anderson, E.; Veith, G. D.; Weininger, D.; SMILES: a line notation and computerized interpreter for chemical structureshttps://cfpub.epa.gov/si/si_public_record_report.cfmdirEntryId=33186?, acessada em outubro 2017. 36. Daylight Inc.; SMARTS - A Language for Describing Molecular Patterns http://www.daylight.com/dayhtml/doc/theory/theory.smarts.html, acessada em outubro 2017. 37. Pletnev, I.; Erin, A.; McNaught, A.; Blinov, K.; Tchekhovskoi, D.; Heller, S.; J. Cheminform. 2012, 4, 39. 38. Dalby, A.; Nourse, J. G.; Hounshell, W. D.; Gushurst, A. K. I.; Grier, D. L.; Leland, B. A.; Laufer, J.; J. Chem. Inf. Model. 1992, 32, 244. 39. Elsevier MDL; CTFile Formats, Elsevier: San Leandro, 2005. 40. Mills, N.; J. Am. Chem. Soc. 2006, 128, 13649. 41. Bienfait, B.; Ertl, P.; J. Cheminform. 2013, 5, 24. 42. Consonni, V.; Todeschini, R.; Em Recent Advances in QSAR Studies; Puzyn, T., Leszczynski, J., Cronin, M. T., eds.; Springer: Dordrecht, 2010, cap. 3. 43. Cronin, M. T.; Em Recent Advances in QSAR Studies; Puzyn, T., Leszczynski, J., Cronin, M. T., eds.; Springer: Dordrecht , 2010, cap. 1. 44. Xue, L.; Bajorath, J.; Comb. Chem. High Throughput Screening 2000, 3, 363. 45. Hopfinger, A. J.; Wang, S.; Tokarski, J. S.; Jin, B.; Albuquerque, M.; Madhav, P. J.; Duraiswami, C.; J. Am. Chem. Soc. 1997, 119, 10509. 46. Vedani, A.; Dobler, M.; J. Med. Chem. 2002, 45, 2139. 47. Vedani, A.; Dobler, M.; Lill, M. A.; J. Med. Chem. 2005, 48, 3700. 48. Todeschini, R.; Consonni, V.; Handbook of Molecular Descriptors, Todeschini, R., Consonni, V., eds.; Wiley: Weinheim, 2000. 49. Chemical Abstracts Service; Introduction to CAS: a division of the American Chemical Society. 50. Willighagen, E. L.; Waagmeester, A.; Spjuth, O.; Ansell, P.; Williams, A. J.; Tkachenko, V.; Hastings, J.; Chen, B.; Wild, D. J.; J. Cheminform. 2013, 5, 23. 51. Gaulton, A.; Bellis, L. J.; Bento, A. P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.; Overington, J. P.; Nucleic Acids Res. 2012, 40, D1100. 52. Wang, Y.; Xiao, J.; Suzek, T. O.; Zhang, J.; Wang, J.; Zhou, Z.; Han, L.; Karapetyan, K.; Dracheva, S.; Shoemaker, B. A.; Bolton, E.; Gindulyte, A.; Bryant, S. H.; Nucleic Acids Res. 2012, 40, D400. 53. Canny, S. A.; Cruz, Y.; Southern, M. R.; Griffin, P. R.; Bioinformatics 2012, 28, 140. 54. Wishart, D. S.; Knox, C.; Guo, A. C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J.; Nucleic Acids Res. 2006, 34, D668. 55. Wishart, D. S.; Knox, C.; Guo, A. C.; Cheng, D.; Shrivastava, S.; Tzur, D.; Gautam, B.; Hassanali, M.; Nucleic Acids Res. 2008, 36, D901. 56. Berman, H. M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T. N.; Weissig, H.; Shindyalov, I. N.; Bourne, P. E.; Nucleic Acids Res. 2000, 28, 235. 57. Berman, H. M.; Bhat, T. N.; Bourne, P. E.; Feng, Z.; Gilliland, G.; Weissig, H.; Westbrook, J.; Nat. Struct. Biol. 2000, 7 Suppl, 957. 58. Johnson, M. A.; Maggiora, G. M.; Concepts and applications of molecular similarity, John Wiley & Sons, Inc.: New York, 1990, vol. 13. 59. Heikamp, K.; Bajorath, J.; J. Chem. Inf. Model. 2011, 51, 2254. 60. Kubinyi, H.; Pespect. Drug Discovery Des. 1998, 9-11, 225. 61. Maggiora, G.; Vogt, M.; Stumpfe, D.; Bajorath, J.; J. Med. Chem. 2014, 57, 3186. 62. Floris, M.; Manganaro, A.; Nicolotti, O.; Medda, R.; Mangiatordi, G. F.; Benfenati, E.; J. Cheminform. 2014, 6, 39. 63. Maggiora, G. M.; J. Chem. Inf. Model. 2006, 46, 1535. 64. Guha, R.; Van Drie, J. H.; J. Chem. Inf. Model. 2008, 48, 646. 65. Rogers, D. J.; Tanimoto, T. T.; Science 1960, 132, 1115. 66. Jaccard, P.; Bull. Soc. Vaudoise Sci. Nat. 1901, XXXVII, 83. 67. Ferreira, M. M. C.; Quimiometria: Conceitos, Métodos e Aplicaçoes, UNICAMP: Campinas, 2015. 68. Mahalanobis, P. C.; Proc. Natl. Inst. Sci. India 1936, 2, 49. 69. Willett, P.; Drug Discov. Today 2006, 11, 1046. 70. Kenny, P. W.; Sadowski, J.; Em Chemoinformatics in Drug Discovery.; Oprea, T. I., ed.; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, 2005, cap. 11. 71. Hammett, L. P.; J. Am. Chem. Soc. 1937, 59, 96. 72. Hammett, L. P.; Physical organic chemistry: reaction rates, equilibria, and mechanisms, 1st ed., McGraw-Hill: New York, 1940. 73. Hansch, C.; Fujita, T.; J. Am. Chem. Soc. 1964, 86, 1616. 74. Free, S. M.; Wilson, J. W.; J. Med. Chem. 1964, 7, 395. 75. Kubinyi, H.; Arzneim. Forsch. 1977, 27, 750. 76. Kubinyi, H.; Farmaco Sci. 1979, 34, 248. 77. Debnath, A. K.; Mini Rev. Med. Chem. 2001, 1, 187. 78. Hopfinger, A. J.; J. Am. Chem. Soc. 1980, 102, 7196. 79. Cramer, R. D.; Patterson, D. E.; Bunce, J. D.; J. Am. Chem. Soc. 1988, 110, 5959. 80. Zhang, L.; Tsai, K.-C.; Du, L.; Fang, H.; Li, M.; Xu, W.; Curr. Med. Chem. 2011, 18, 923. 81. Klebe, G.; Abraham, U.; Mietzner, T.; J. Med. Chem. 1994, 37, 4130. 82. Cramer, R. D.; J. Comput. Aided. Mol. Des. 2012, 26, 35. 83. Tropsha, A.; Mol. Inform. 2010, 29, 476. 84. Melville, J. L.; Burke, E. K.; Hirst, J. D.; Comb. Chem. High Throughput Screening 2009, 12, 332. 85. Sheridan, R. P.; J. Chem. Inf. Model. 2014, 54, 1083. 86. Helgee, E. A.; Carlsson, L.; Boyer, S.; Norinder, U.; J. Chem. Inf. Model. 2010, 50, 677. 87. Welling, M.; A first encounter with Machine Learning, University of California: Irvine, 2011. 88. Breiman, L. E. O.; Mach. Learn. 2001, 45, 5. 89. Cortes, C.; Vapnik, V.; Mach. Learn. 1995, 20, 273. 90. Dreiseitl, S.; Ohno-Machado, L.; J. Biomed. Inform. 2002, 35, 352. 91. LeCun, Y.; Bengio, Y.; Hinton, G.; Nature 2015, 521, 436. 92. Downs, G. M.; Barnard, J. M.; Em Reviews in Computational Chemistry; Lipkowitz, K. B., Boyd, D. B., eds.; John Wiley & Sons, Inc.: Hoboken , 2003, vol. 18, cap. 1. 93. Mercier, D.; Clustering large datasets, http://ldc.usb.ve/~mcuriel/Cursos/WC/Transfer.pdf, acessada em outubro 2017. 94. Dearden, J. C.; Cronin, M. T. D.; Kaiser, K. L. E.; SAR QSAR Environ. Res. 2009, 20, 241. 95. OECD; OECD principles for the validation, for regulatory purposes, of (Quantitative) Structure-Activity Relationship models, http://www.oecd.org/chemicalsafety/risk-assessment/37849783.pdf, acessada em outubro 2017. 96. Frye, S. V; Arkin, M. R.; Arrowsmith, C. H.; Conn, P. J.; Glicksman, M. A.; Hull-Ryde, E. A.; Slusher, B. S.; Nat. Rev. Drug Discov. 2015, 14, 733. 97. Fourches, D.; Muratov, E.; Tropsha, A.; Nat. Chem. Biol. 2015, 11, 535. 98. Fourches, D.; Muratov, E.; Tropsha, A.; J. Chem. Inf. Model. 2016, 56, 1243. 99. Fourches, D.; Muratov, E.; Tropsha, A.; J. Chem. Inf. Model. 2010, 50, 1189. 100. Golbraikh, A.; Muratov, E.; Fourches, D.; Tropsha, A.; J. Chem. Inf. Model. 2014, 54, 1. 101. Golbraikh, A.; Tropsha, A.; J. Mol. Graph. Model. 2002, 20, 269. 102. Gramatica, P.; QSAR Comb. Sci. 2007, 26, 694. 103. Tropsha, A.; Gramatica, P.; Gombar, V. K.; QSAR Comb. Sci. 2003, 22, 69. 104. Chirico, N.; Gramatica, P.; J. Chem. Inf. Model. 2011, 51, 2320. 105. Wang, X. S.; Tang, H.; Golbraikh, A.; Tropsha, A.; J. Chem. Inf. Model. 2008, 48, 997. 106. Altman, D. G.; Bland, J. M.; BMJ 1994, 308, 1552. 107. Altman, D. G.; Bland, J. M.; BMJ 1994, 309, 102. 108. McClish, D. K.; Med. Decis. Making 1989, 9, 190. 109. Chirico, N.; Gramatica, P.; J. Chem. Inf. Model. 2012, 52, 2044. 110. Roy, K.; Mitra, I.; Comb. Chem. High Throughput Screening 2011, 14, 450. 111. Roy, K.; Mitra, I.; Kar, S.; Ojha, P. K.; Das, R. N.; Kabir, H.; J. Chem. Inf. Model. 2012, 52, 396. 112. Kuz’min, V. E.; Muratov, E. N.; Artemenko, A. G.; Varlamova, E. V.; Gorb, L.; Wang, J.; Leszczynski, J.; QSAR Comb. Sci. 2009, 28, 664. 113. Gadaleta, D.; Mangiatordi, G. F.; Catto, M.; Carotti, A.; Nicolotti, O.; International Journal of Quantitative Structure-Property Relationships 2016, 1, 45. 114. Mathea, M.; Klingspohn, W.; Baumann, K.; Mol. Inform. 2016, 35, 160. 115. Alves, V. M.; Muratov, E. N.; Zakharov, A.; Muratov, N. N.; Andrade, C. H.; Tropsha, A.; Food Chem. Toxicol. 2017, no prelo. 116. Schneider, G.; Nat. Rev. Drug Discov. 2010, 9, 273. 117. Kessel, M.; Nat. Biotechnol. 2011, 29, 27. 118. Polgár, T.; Keseru, G. M.; Comb. Chem. High Throughput Screening 2011, 14, 889. 119. Truchon, J.-F.; Bayly, C. I.; J. Chem. Inf. Model. 2007, 47, 488. 120. Forli, S.; Molecules 2015, 20, 18732. 121. Clark, D. E.; Expert Opin. Drug Discovery 2008, 3, 841. 122. Ma, X. H.; Zhu, F.; Liu, X.; Shi, Z.; Zhang, J. X.; Yang, S. Y.; Wei, Y. Q.; Chen, Y. Z.; Curr. Med. Chem. 2012, 19, 5562. 123. Eckert, H.; Bajorath, J.; Drug Discovery Today 2007, 12, 225. 124. Yang, S.-Y.; Drug Discovery Today 2010, 15, 444. 125. Braga, R. C.; Alves, V. M.; Silva, A. C.; Nascimento, M. N.; Silva, F. C.; Liao, L. M.; Andrade, C. H.; Curr. Top. Med. Chem. 2014, 14, 1899. 126. Braga, R. C.; Andrade, C. H.; Curr. Top. Med. Chem. 2013, 13, 1127. 127. Kar, S.; Roy, K.; Expert Opin. Drug Discovery 2013, 8, 245. 128. Ripphausen, P.; Nisius, B.; Peltason, L.; Bajorath, J.; J. Med. Chem. 2010, 53, 8461. 129. OECD; Report of the workshop on structural alerts for the OECD (Q)SAR application toolbox, http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=env/jm/mono(2009)4&doclanguage=en, acessada em outubro 2017. 130. Blagg, J.; Em Burger’s Medicinal Chemistry and Drug Discovery, John Wiley & Sons, Inc.: Hoboken , 2010. 131. Allen, T. E. H.; Goodman, J. M.; Gutsell, S.; Russell, P. J.; Chem. Res. Toxicol. 2014, 27, 2100. 132. Enoch, S. J.; Roberts, D. W.; Em Chemical Toxicity Prediction: Category Formation and Read-Across; Cronin, M., Madden, J., Enoch, S., Roberts, D., eds.; Royal Society of Chemistry, 2013, cap. 2. 133. ECHA; Agrupamento de substâncias e métodos comparativos por interpolaçao https://echa.europa.eu/pt/support/registration/how-to-avoid-unnecessary-testing-on-animals/grouping-of-substances-and-read-across, acessada em outubro 2017. 134. Alves, V.; Muratov, E.; Capuzzi, S.; Politi, R.; Low, Y.; Braga, R.; Zakharov, A. V.; Sedykh, A.; Mokshyna, E.; Farag, S.; Andrade, C.; Kuz’min, V.; Fourches, D.; Tropsha, A.; Green Chem. 2016, 18, 4348. 135. Varnek, A.; Baskin, I. I.; Mol. Inform. 2011, 30, 20. 136. Neves, B. J.; Dantas, R. F.; Senger, M. R.; Melo-Filho, C. C.; Valente, W. C. G.; de Almeida, A. C. M.; Rezende-Neto, J. M.; Lima, E. F. C.; Paveley, R.; Furnham, N.; Muratov, E.; Kamentsky, L.; Carpenter, A. E.; Braga, R. C.; Silva-Junior, F. P.; Andrade, C. H.; J. Med. Chem. 2016, 59, 7075. 137. Melo-Filho, C. C.; Dantas, R. F.; Braga, R. C.; Neves, B. J.; Senger, M. R.; Valente, W. C. G.; Rezende-Neto, J. M.; Chaves, W. T.; Muratov, E. N.; Paveley, R. A.; Furnham, N.; Kamentsky, L.; Carpenter, A. E.; Silva-Junior, F. P.; Andrade, C. H.; J. Chem. Inf. Model. 2016, 56, 1357. 138. Braga, R. C.; Alves, V. M.; Silva, M. F. B.; Muratov, E.; Fourches, D.; Liao, L. M.; Tropsha, A.; Andrade, C. H.; Mol. Inform. 2015, 34, 698. 139. Braga, R. C.; Alves, V. M.; Silva, M. F. B.; Muratov, E.; Fourches, D.; Tropsha, A.; Andrade, C. H.; Curr. Top. Med. Chem. 2014, 14, 1399. 140. Zhang, L.; Fourches, D.; Sedykh, A.; Zhu, H.; Golbraikh, A.; Ekins, S.; Clark, J.; Connelly, M. C.; Sigal, M.; Hodges, D.; Guiguemde, A.; Guy, R. K.; Tropsha, A.; J. Chem. Inf. Model. 2013, 53, 475. 141. Alves, V. M.; Muratov, E. N.; Fourches, D.; Strickland, J.; Kleinstreuer, N.; Andrade, C. H.; Tropsha, A.; Toxicol. Appl. Pharmacol. 2015, 284, 273. 142. Alves, V. M.; Muratov, E. N.; Fourches, D.; Strickland, J.; Kleinstreuer, N.; Andrade, C. H.; Tropsha, A.; Toxicol. Appl. Pharmacol. 2015, 284, 262. 143. OECD; The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins, http://www.oecd-ilibrary.org/environment/the-adverse-outcome-pathway-for-skin-sensitisation-initiated-by-covalent-binding-to-proteins_9789264221444-en, acessada em outubro 2017. 144. Alves, V. M.; Capuzzi, S. J.; Muratov, E.; Braga, R. C.; Thornton, T.; Fourches, D.; Strickland, J.; Kleinstreuer, N.; Andrade, C. H.; Tropsha, A.; Green Chem. 2016, 18, 6501. 145. Ban, F.; Dalal, K.; Li, H.; LeBlanc, E.; Rennie, P. S.; Cherkasov, A.; J. Chem. Inf. Model. 2017, 57, 1018. 146. Lejon, T.; Strom, M. B.; Svendsen, J. S.; J. Pept. Sci. 2001, 7, 74. 147. Lejon, T.; Stiberg, T.; Strom, M. B.; Svendsen, J. S.; J. Pept. Sci. 2004, 10, 329. 148. Muratov, E. N.; Varlamova, E. V.; Artemenko, A. G.; Polishchuk, P. G.; Nikolaeva-Glomb, L.; Galabov, A. S.; Kuz’Min, V. E.; Struct. Chem. 2013, 24, 1665. 149. Oprisiu, I.; Varlamova, E.; Muratov, E.; Artemenko, A.; Marcou, G.; Polishchuk, P.; Kuz’Min, V.; Varnek, A.; Mol. Inform. 2012, 31, 491. 150. Muratov, E. N.; Varlamova, E. V.; Artemenko, A. G.; Polishchuk, P. G.; Kuz’min, V. E.; Mol. Inform. 2012, 31, 202. 151. Fourches, D.; Pu, D.; Li, L.; Zhou, H.; Mu, Q.; Su, G.; Yan, B.; Tropsha, A.; Nanotoxicology 2016, 10, 374. 152. Kaliszan, R.; Chem. Rev. 2007, 107, 3212. 153. Fourches, D.; Tropsha, A.; Em Nanotoxicology: Progress toward nanomedicine; Monteiro-Riviere, N. A., Lang Tran, C., eds.; CRC Press, 2014. 154. Roy, K.; Ghosh, G.; J. Chem. Inf. Comput. Sci. 2004, 44, 559. 155. El-Kommos, M. E.; El-Gizawy, S. M.; Atia, N. N.; Hosny, N. M.; J. Fluoresc. 2015, 25, 1695. 156. Urbano-Cuadrado, M.; Carbó, J. J.; Maldonado, A. G.; Bo, C.; J. Chem. Inf. Model. 47, 2228. 157. Garzia, A.; Villanti, A.; Tuccini, G.; J. Pharm. Sci. 1979, 68, 1081. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access