
 |
 |
 |
 |
 |
Revisão
|
|
| Compostos orgânicos e inorgânicos contendo selênio: revisão de métodos analíticos e perspectivas para análises químicas Organic and inorganic compounds containing selenium: analytical methods review and chemical analysis perspectives |
|
Fernanda C. O. L. MartinsI; Diego L. FrancoI; Rodrigo A. A. MuñozII; Djenaine De SouzaI,*
ILaboratório de Eletroanalítica Aplicada à Biotecnologia e à Engenharia de Alimentos, Universidade Federal de Uberlândia, Campus de Patos de Minas, Av. Getúlio Vargas, 230, Centro, 38700-126 Patos de Minas - MG, Brasil Recebido em 13/03/2017 *e-mail: djenaine@ufu.br This article critically reviews the spectrometric, chromatographic and electroanalytical methods applied for the identification and quantification of organic and inorganic compounds containing Se and their applications in food, biological and natural water samples. The main characteristics, advantages and disadvantages of each analytical method are presented, and special attention is paid to sample preparation strategies prior to identification and quantification. Perspectives of research on new analytical procedures with improved speed, sensitivity, selectivity, accuracy, and mainly environmentally friendly, are also reported. CONSIDERAÇÕES GERAIS A magnífica máquina humana necessita de diversos nutrientes, os quais são o combustível para a manutenção do seu perfeito funcionamento. Dependendo das quantidades necessárias, estes nutrientes são classificados como macronutrientes (carboidratos, proteínas, lipídios e fibras) e micronutrientes (vitaminas e minerais). Estes compostos apresentam funções específicas e, consequentemente, diferentes concentrações ideais para o desempenho de suas funções metabólicas. A ingestão diária de nutrientes dependerá da composição química da dieta alimentar.1 Dentre os principais micronutrientes destacam-se ferro, zinco, cobre, selênio, cromo, iodo, molibdênio, manganês, silício, níquel, boro, vanádio, flúor, arsênio, alumínio, lítio e estanho, seja em suas formas orgânicas ou inorgânicas. Estes compostos desempenham suas respectivas atividades metabólicas quando presentes em concentrações abaixo de microgramas por grama (µg g-1). Entretanto, acima destas concentrações podem apresentar toxicidade ao organismo humano.1 Selênio, apresentado com a sigla Se, é um micronutriente essencial para a dieta humana, em função das suas propriedades antioxidantes, auxiliando no retardamento do envelhecimento, no tratamento de doenças cardiovasculares e neoplasias, distrofia muscular, esclerose múltipla, osteoporose, reprodutividade, e desordem muscular.2,3 Alguns estudos indicam sua importância no metabolismo e funcionamento da tireóide e sua atuação no metabolismo dos ácidos graxos, juntamente com a vitamina E (α-tocoferol).4,5 Assim, o Conselho Americano de Alimentação e Nutrição (Food and Nutrition Board, EUA),6 recomenda a ingestão diária de 15 a 20 µg por dia, de 55 µg por dia e 70 µg por dia, para crianças, mulheres lactantes e outros adultos, respectivamente. Entretanto, o limite máximo de ingestão diário para uma pessoa adulta não deve ultrapassar 400 µg. A diferença entre a concentração essencial de Se, a deficiência ou a toxicidade é muito pequena. As concentrações ingeridas são fortemente dependentes do nível de concentração em alimentos e na quantidade de alimento ingerido.7 O consumo diário abaixo de 0,1 µg de Se por grama de alimento (0,1 µg g-1) resulta em problemas de saúde, tais como necrose e degenerção do fígado, dos rins, do coração e do pâncreas, além de aumentar o risco de alguns tipos de câncer. O consumo acima de 1 µg g-1 resultará na toxicidade para o organismo, promovendo queda de cabelo, unhas quebradiças, irritação na pele e nos olhos.8,9 Em função da sua importância nutricional, adubação agrícola com compostos ricos em Se é uma maneira eficaz de reduzir as deficiências alimentares e melhorar a qualidade da saúde.10 As diferenças geográficas combinadas com as condições ambientais e práticas de cultivo influenciam na biodisponibilidade das diferentes formas de Se nos solos, alterando, assim, suas concentrações em alimentos e águas naturais e, consequentemente, influenciando na quantidade ingerida na dieta alimentar.11,12 A origem natural do Se é a liberação de gases vulcânicos, aerossóis marinhos e a combustão do carvão.13 Algumas práticas como mineração, processamento de metais e carvão, adubação agrícola e uso de lodo ativo na adubação agrícola podem promover um aumento na biodisponibilidade de Se no meio ambiente. Além disto, o emprego de Se na área farmacêutica, higiene pessoal, produção de vidros, cerâmicas, tintas, bem como em produção de produtos eletrônicos e fotocópias têm promovido um aumento na quantidade de Se no meio ambiente.14 Desta maneira, a Organização Mundial da Saúde e o Conselho Nacional de Meio Ambiente definem limites máximos para a presença de Se em águas para consumo público inferior a 10 µg L-1.15,16 Quimicamente, o Se é encontrado na sua forma orgânica como selenoaminoácidos, (selenometionina ou selenocisteína, estruturas mostradas na Figura 1), e também como derivados metilados.17 Em sua forma inorgânica, ele é encontrado em diversos estados de oxidação (-2, 0, +4 e +6), mas apenas selenatos [Se(VI)] e selenitos [Se(IV)] apresentam solubidade em água e fazem parte dos processos metabólicos em humanos. Os compostos de Se (IV) apresentam toxicidade aproximadamente 10 vezes maior que os compostos de Se (VI), indicando a importância da especiação química.18
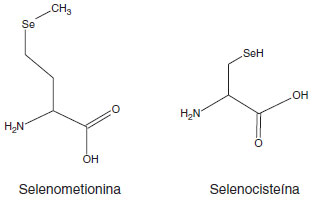 Figura 1. Estruturas moleculares das principais formas de Se
METODOLOGIAS EMPREGADAS NA DETERMINAÇÃO DE SELÊNIO As diferenças observadas entre os níveis nutricionais e níveis de toxicidade de Se mostram a necessidade do desenvolvimento de metodologias eficientes para identificação e quantificação de Se, em amostras de alimentos, amostras naturais, suplementos alimentares e amostras biológicas.14,19 Neste contexto, têm sido empregadas técnicas espectroscópicas,20 técnicas cromatográficas21 e técnicas eletroanalíticas.22 Um detalhado levantamento bibliográfico mostrou que até 1970 houve a predominância no uso de técnicas eletroanalíticas, principalmente em função da simplicidade analítica e da possibilidade de especiação química. Até este período as técnicas eletroanalíticas empregavam eletrodos de mercúrio como sensores nas análises químicas. Entretanto, com o advento de uso de uma química limpa, ou química verde, iniciou-se uma intensa política de substituição de mercúrio por outros materiais, promovendo a busca por materiais alternativos, e assim a aplicação de diferentes materiais como dispositivos analíticos. Aliados à elevada toxicidade do mercúrio, ao intenso desenvolvimento da instrumentação analítica e dos métodos de preparação de amostras, as técnicas espectroscópicas e as técnicas cromatográficas começaram a intensificar a sua aplicabilidade. A Figura 2 apresenta a relação aproximada do número de publicações científicas abordando a determinação analítica de compostos contendo Se na última década, considerando-se as três classes de métodos analíticos citados anteriormente.
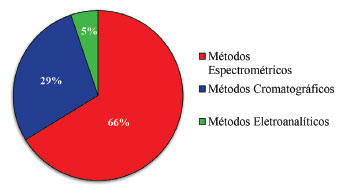 Figura 2. Determinação de Se empregando-se diferentes técnicas analíticas, considerando-se a última década (2006-2016)
Métodos espectrométricos A espectrometria atômica e a espectrometria molecular são intensamente empregadas para a identificação e quantificação de Se. De maneira geral, estas técnicas envolvem a medida da radiação eletromagnética absorvida (técnicas de absorção) ou emitida (técnicas de emissão) por espécies químicas após a incidência de energia (radiante, elétrica, calor ou química). A intensidade de radiação eletromagnética é empregada na quantificação das espécies químicas de interesse, enquanto a identificação pode ser realizada pelo valor do comprimento de onda no qual ocorre o máximo de absorção ou de emissão da radiação eletromagnética.23,24 Cada uma das técnicas espectroscópicas apresentam características específicas, proporcionando diferenças significativas na precisão, exatidão, sensibilidade, seletividade e também aplicabilidade em diferentes tipos de amostras, tais como águas naturais, solos e alimentos. Desta maneira, pôde-se observar a aplicação de diferentes técnicas espectroscópicas para identificação e quantificação das diferentes formas de Se, como apresentado na Figura 3.
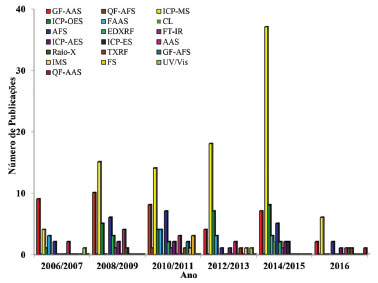 Figura 3. Evolução nas publicações científicas com quantificação de Se empregando-se técnicas espectroscópicas, dos anos 2006 a 2016
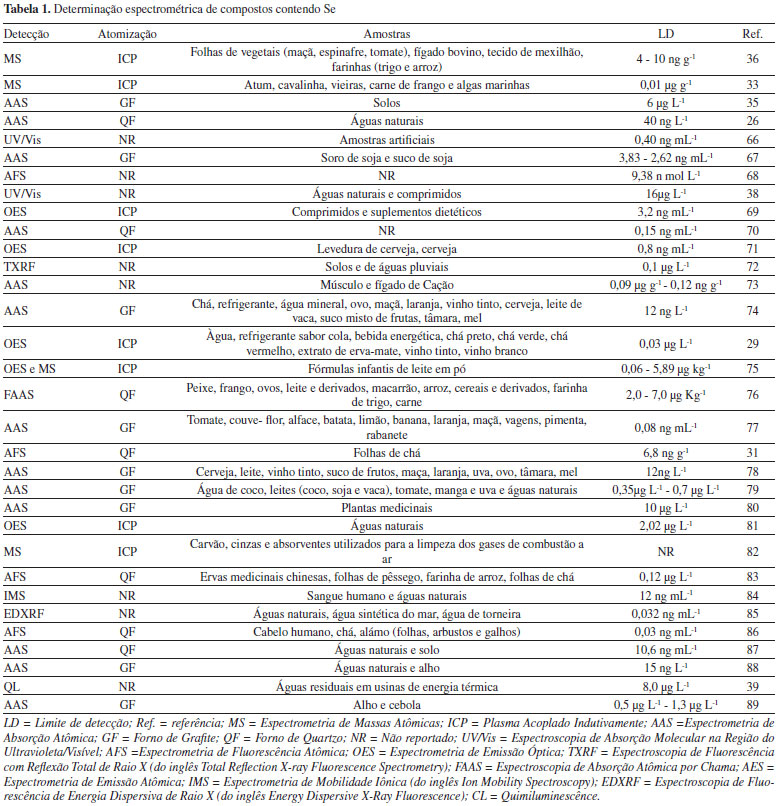
A Espectroscopia Molecular com Absorção de Radiação Ultravioleta/Visível (UV/Vis) é utilizada para a detecção de diferentes tipos de compostos orgânicos e inorgânicos. Entretanto, para compostos que possuam Se na sua estrutura química, a absorção de radiação é pouco sensível, necessitando-se, assim, de procedimentos químicos que promovam eficiência analítica e seletividade. Estes procedimentos envolvem reações com formação de complexos, ou reações de oxidação/redução entre reagentes químicos específicos e compostos contendo Se, como realizado por Wen et. al.25 e Shabani, Dadfarnia, Nozohor,26 respectivamente. Os produtos das reações químicas específicas apresentam absorção da radiação eletromagnética, promovendo melhoria na sensibilidade e seletividade analítica em compostos químicos contendo Se. A Quimiluminescência (CL, do inglês Chemiluminescence) consiste na radiação eletromagnética liberada a partir de uma reação química específica, a qual pode apresentar duração variada e diferentes comprimentos de onda para a emissão de luz. Assim, íons metálicos, ânions e algumas biomoléculas, em diversas amostras podem emitir luz e desta maneira serem quantificados. Ezoe et. al.27 empregaram a CL, originada a partir da reação química entre o Se vaporizado (H2Se) com ozônio, para a quantificação do Se presente em amostras de resíduos industriais. Na Espectrometria de Absorção Atômica (AAS) ou na Espectrometria de Emissão Atômica (AES), nas quais a intensidade de radiação eletromagnética absorvida ou emitida é relacionada à concentração, a determinação de Se e de outras espécies químicas necessita de uma prévia atomização das amostras para geração de átomos e íons gasosos. A escolha do atomizador (eletrotérmico, plasma ou chama) dependerá do limite de detecção desejável e do volume de amostra e é fator fundamental para o sucesso da análise espectroscópica.28 Os atomizadores eletrotérmicos são fornos tubulares e aquecidos em temperaturas de 300 ºC a 3000 ºC, que permitem a completa conversão atômica. O Forno de Grafite (GF, do inglês Graphite Furnace) e o Tubo de Quartzo (QF, do inglês Quartz Furnace), têm sido utilizados para a análise de elementos que possuem baixa volatilidade em sua forma atômica, já que o hidreto produzido é mais volátil. Com o uso destes atomizadores, é necessário o uso de uma fonte adicional de energia (lâmpada de cátodo oco) para promover a absorção da radiação. Etapas prévias de digestão das amostras são necessárias para a completa remoção dos componentes orgânicos e esta etapa é fundamental na sensibilidade e seletividade da análise química. Desta maneira, Rybínová et. al.29 utilizaram o QF para a produção de hidretos, os quais aceleraram os átomos livres e isolados de Se, para a detecção em amostras de águas naturais. Assim, o Se(IV) foi convertido em uma substância volátil, e posteriormente quantificado por AAS, com uma melhoria significativa na sensibilidade. Os atomizadores de chama utilizam uma mistura de combustíveis e oxidantes para a atomização das amostras, permitindo variações de temperaturas de 1700 °C até 3200 °C, com baixo custo de manutenção e simplicidade de operação. Eles são empregados na Espectroscopia de Absorção Atômica em Chama (FAAS, do inglês Flame Atomic Absorption Spectrometry),30 entretanto, são necessárias etapas prévias de extração com solvente, removendo interferentes que podem alterar os resultados da análise química, as quais nem sempre são eficientes para análises de traços. Assim, é fundamental a utilização de etapas de digestão de amostras, que são procedimentos químicos para conversão completa de substâncias nos seus constituintes mais simples (CO2, água e sais inorgânicos). Os métodos clássicos de digestão empregam o aquecimento das amostras utilizando-se ácidos fortes ou agentes oxidantes, em recipientes abertos.25 Os métodos modernos de digestão combinam a digestão ácida com a radiação de micro-ondas, no procedimento de digestão assistida por micro-ondas (MWAD, do inglês Microwave-Assisted Digestion), realizada em recipientes fechados apropriados.31 A MWAD promove melhoria na sensibilidade analítica, entretanto, são necessárias condições adequadas na liberação de gases produzidos, proporcionando segurança operacional e qualidade analítica. O Plasma Indutivamente Acoplado (ICP, do inglês Inductively Coupled Plasma) é empregado como uma fonte de átomos e de íons, com excelente sensibilidade analítica e mínimo efeito de interferentes. O ICP é um ambiente altamente energético que promove a completa atomização da amostra, ionização, excitação e também emissão da radiação eletromagnética. A quantificação dos íons formados no plasma é avaliada por Espectrometria de Massa Atômica (ICP-MS, do inglês Mass Spectrocopy), enquanto a quantificação da intensidade de radiação emitida por átomos ou íons excitados é avaliada em Espectrometria de Emissão Óptica (OES, do inglês Optic Emisson Spectroscopy). Como a temperatura do ICP é muito alta (8.000 ºC a 10.000 ºC), é necessário um isolamento térmico eficiente e um sistema de nebulização de amostras, que irá promover a conversão da amostra em gotículas finas denominadas aerossol. A manutenção do ICP apresenta custo elevado em função da quantidade de energia necessária para a permanência de altas temperaturas e o funcionamento ininterrupto do equipamento.32 A MS é um sistema de detecção empregado juntamente com o ICP, o qual permite a identificação e quantificação de quase todos os metais da tabela periódica, com excelente seletividade e sensibilidade. Os íons produzidos no ICP separados empregando-se os sistemas de analisadores os quais podem ser: quadrupolar, tempo de vôo, ou armadilha de íons. Desta maneira, ocorre a separação dos íons em função da razão de carga/massa (m/z) que são quantificadas em função de suas respectivas abundâncias relativas.33 A energia do plasma é suficiente para também promover a excitação dos átomos e íons e, consequentemente, a emissão de luz, a qual pode ser quantificada por OES. Escudero et. al.34 utilizaram a ICP-OES para a especiação das formas inorgânicas de Se e quantificação de suas respectivas formas em amostras de vinhos, águas naturais, refrigerantes e chás. Neste contexto, Zhang et. al.35 determinaram simultaneamente Se, As, Bi e Te em amostras de folhas de chás submetidas à digestão com HCl concentrado e 30% de H2O2, a 125 ºC por 5 horas. Após a digestão, foi empregado um GF na atomização do Se e demais metais, e a análise química foi realizada por Espectroscopia de Fluorescência Atômica (AFS, do inglês Atomic Fluorescence Spectroscopy). Análises de rotina e a detecção simultânea destes metais foram possíveis em função da eficiência da digestão ácida e o emprego de lâmpadas multielementares como fonte de energia para a emissão da radiação eletromagnética.36 Já Ohki et al.37 utilizaram a mistura de HNO3 e H2O2 na digestão ácida de selenoproteínas em alimentos. Inicialmente, a proporção entre os reagentes, o efeito da temperatura e da pressão no procedimento de digestão ácida foram avaliados para a melhoria da eficiência de digestão e a posterior quantificação por ICP-MS. Schneider et al.38 empregaram a MWAD para tratar as amostras de solo em meio ácido visando quantificar o teor de Se total, Se (IV) após extração em meio alcalino assistida por ultrassom e Se (VI) por diferença entre o teor total e o obtido para Se (IV). A quantificação das espécies de Se foi feita por geração de hidreto e posterior análise por GF AAS. Guo et. al.39 utilizaram MWAD com a mistura de HNO3 e HClO4 para a preparação de amostras de atum, cavalinha, vieiras, frango, carne e algas marinhas. A detecção foi realizada empregando-se ICP-MS. Neste trabalho a MWAD e o ICP-MS promoveram melhoria considerável na sensibilidade analítica. A Espectrometria de Fluorescência de Energia Dispersiva de Raio X (EDXRF, do inglês Energy Dispersive X-Ray Fluorescence) e a Espectrometria de Fluorescência com Reflexão Total de Raio X (TXRF, do inglês Total Reflection X-ray Fluorescence Spectrometry) são técnicas que envolvem a detecção de Raio-X emitidos pelo elemento químico de interesse, após a sua excitação por radiação do tipo gama, por um feixe de Raio-X ou por partículas carregadas, os quais ao serem incididos interagem com a amostra, provocando absorção, emissão e espalhamento de radiação eletromagnética. A vantagem no uso destas técnicas é a possibilidade de análises diretamente nas amostras, sem a necessidade de etapas de digestão ácida.28,32 A Tabela 1 apresenta uma breve revisão de trabalhos que apresentam a quantificação de Se empregando métodos espectrométricos e variados processos de atomização para diferentes tipos de amostras.
Métodos cromatográficos Na última década as técnicas cromatográficas têm sido amplamente empregadas na separação de compostos químicos contendo Se, como apresentado na Figura 4. De maneira geral, os trabalhos publicados apresentam o uso de técnicas cromatográficas com o objetivo de promover a separação de compostos orgânicos e inorgânicos contendo Se, para posteriormente realizar a quantificação empregando-se uma técnica espectrométrica (descritas no item anterior) como método de detecção. Estes sistemas são denominados técnicas hifenadas e promovem adequada separação, identificação e quantificação de espécies químicas, incluindo compostos contendo Se.
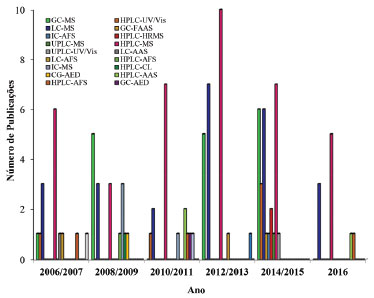 Figura 4. Evolução nas publicações científicas com quantificação de Se empregando-se técnicas cromatográficas, dos anos 2006 a 2016
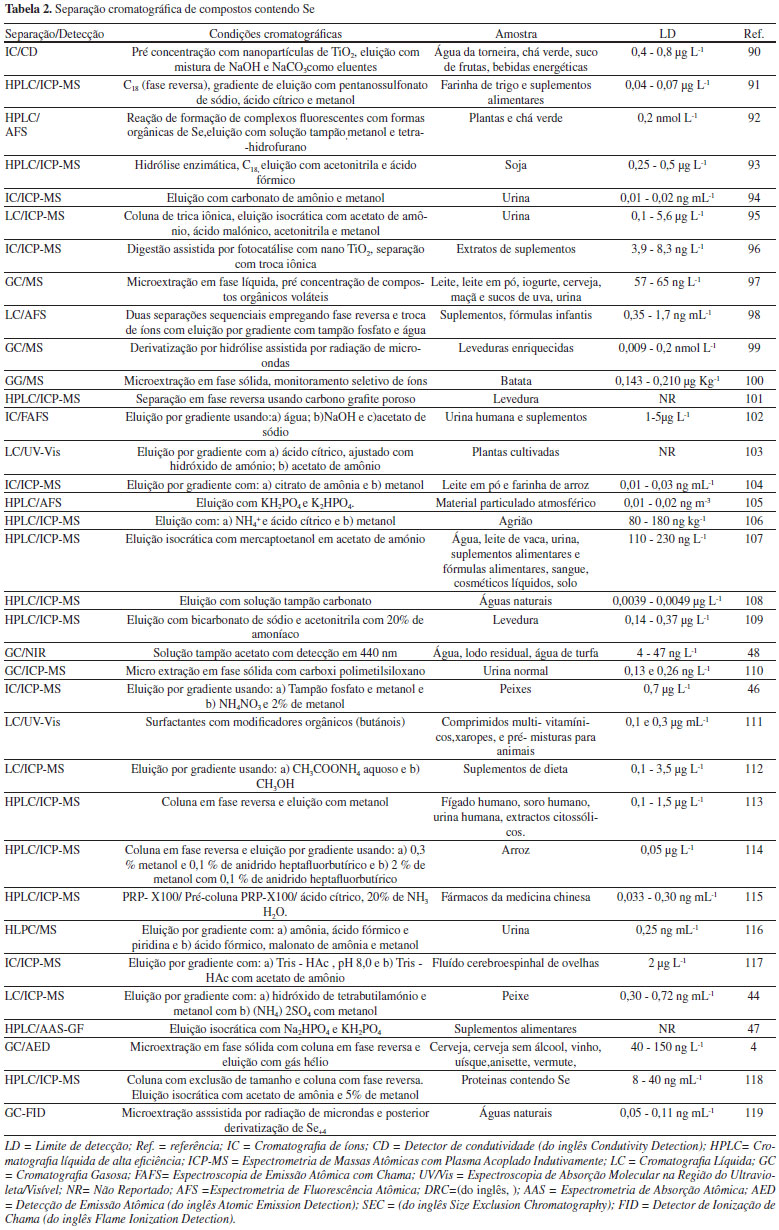
As técnicas cromatográficas envolvem a separação de componentes químicos ou remoção de interferentes numa análise química, os quais podem atenuar ou aumentar o sinal analítico. Elas estão relacionadas a mecanismos físico-químicos de interação de espécies químicas entre uma fase móvel (líquido, gás ou fluído supercrítico) e uma fase estacionária (sólida ou líquida).40 Estas interações envolvem processos de adsorção, partição, exclusão, troca iônica ou afinidade, as quais estão diretamente relacionadas às diferenças de polaridade, pois algumas espécies possuem maior interação com a fase móvel, e outras com a fase estacionária, promovendo diferenças de mobilidade e consequentemente, a separação analítica. A escolha adequada da fase móvel e da fase estacionária é responsável por curtos tempos de retenção e boa resolução entre os picos cromatográficos. Após a coluna cromatográfica, o emprego de detectores permite a identificação e quantificação das espécies químicas. A Cromatografia Líquida (LC, do inglês Liquid Chromatography), a Cromatografia Líquida de Alta Eficiência (HPLC, do inglês High Performance Liquid Chromatography), a Cromatografia Gasosa (GC, do inglês Gas Chromatography) e a Cromatografia de Íons (IC, do inglês Ion Chromatography) são empregadas na separação, identificação e quantificação de espécies químicas contendo Se em diferentes tipos de amostras.41-43 Na LC são empregados solventes orgânicos de diferentes polaridades, soluções tampão, ou misturas de soluções aquosas ou orgânicas, com eluição por gravidade. A escolha adequada do eluente e da fase estacionária proporcionam melhor separação. Assim, Bryszewska e Mage44 utilizaram a mistura de metanol, solução tampão e surfactantes, com uma coluna cromatográfica C18 para a separação de selenocisteína, selenometionina, metil-selenocisteína, selenato, selenito e óxido de selênio em amostras de peixes, mariscos e planctos. Inicialmente, as amostras foram submetidas ao processo de MWAD e extração com hidrólise enzimática. Com o emprego da LC foi possível a diferenciação das diferentes formas de Se, e o ICP-MS possibilitou a quantificação de Se total. A IC é um tipo de LC empregada para separação e detecção de compostos iônicos ou ionizáveis, de baixo peso molecular, que emprega fase móvel e fase estacionária composta por substâncias trocadoras de íons. A separação é dependente da afinidade eletrostática entre o analito e substâncias trocadoras de íons, podendo ser controladas variando-se o pH e a força iônica do meio. A detecção é realizada por medidas da condutividade dos íons, e por isto a rapidez e sensibilidade proporcionadas por este tipo de sepração45 possibilita o acoplamento a um ICP-MS (LC-ICP-MS). Desta maneira, Reyes et. al.46 empregaram a IC na separação de compostos contendo As e Se em peixes, os quais foram inicialmente submetidos à Extração Enzimática Assistida por Micro-ondas (MAEE, do inglês Microwave-assisted Enzymatic Extraction), que possibilitou a extração simultânea destes compostos empregando-se a combinação de enzimas específicas em recipiente fechado e com a aplicação de radiação de micro-ondas. Após a separação, as espécies de As e Se foram quantificadas por ICP-MS. A substituição do efeito da gravidade por um sistema de bombeamento de solventes permite o emprego de diversas velocidades de vazão da fase móvel e também a eluição por gradiente, melhorando a separação, que é o fator fundamental para o emprego da HPLC e da Cromatografia Líquida de Ultra Alta Eficiência (UPLC, do inglês Ultra Performance Liquid Chromatography). Além disto, o emprego de detectores específicos permite uma excelente quantificação de substâncias, em concentrações na ordem de microgramas por litro (partes por bilhão) ou nanogramas por litro (partes por trilhão), minimizando o tempo da separação analítica e principalmente o consumo de solventes orgânicos.40 Para análise de Se e outros metalóides, os detectores mais empregados são os espectrométricos ICP-MS e AAS acoplados a GF ou QF. Assim, Niedzielski et. al.47 realizaram a separação de todas as formas de Se presente em amostras de suplementos alimentares empregando HPLC. As amostras inicialmente foram submetidas ao processo de digestão ácida e, posterior à separação por HPLC, ocorreu a atomização em GF e a detecção de Se por AAS. A GC é utilizada na separação de compostos voláteis e termicamente estáveis, em intervalos de temperaturas que variam de ambiente até cerca de 350 ºC. Apresenta excelente resolução e permite a determinação simultânea de várias substâncias químicas. Dependendo do detector utilizado é possível a quantificação na faixa de partes por trilhão empregando-se pequenas quantidades de amostras. Compostos não voláteis podem ser separados e identificados após reações químicas específicas que promovem a conversão para compostos voláteis e termicamente estáveis, as reações de derivatização. Para compostos contendo Se, estas reações envolvem a metilação dos compostos e posterior aplicação da GC, como reportado por Vriens et. al.,48 em que a derivatização produziu dimetilseleneto (H3C-Se-CH3) e dimetilsulfureto (H3C-S-CH3), os quais foram separados e identificados por MS. Cada tipo de composto químico contendo Se requer uma metodologia específica no processo de separação, proporcionando assim diferentes metodologias de separação e posterior detecção. Assim, a Tabela 2 apresenta algumas metodologias desenvolvidas para separação e identificação de Se em diferentes tipos de amostras.
Métodos eletroanalíticos Desde a década de 1950, as técnicas eletroanalíticas têm se tornado uma excelente alternativa para a identificação e quantificação de compostos químicos em amostras de interesse ambiental, indústrial e bioquímico.49,50 Estas técnicas envolvem medidas de propriedades elétricas, as quais são relacionadas à identidade e quantidade da substância de interesse. Para a detecção de Se as técnicas mais utilizadas envolvem a aplicação adequada de uma diferença de potencial elétrico e medidas de correntes elétricas originadas a partir de reações de transferência de elétrons (reações redox), as técnicas voltamétricas. Estas técnicas apresentam baixo custo, simplicidade operacional, sensibilidade, e ainda permitem a diferenciação dos vários estados de oxidação de Se.51 De maneira geral, nestas técnicas a sensibilidade analítica está diretamente relacionada ao modo de aplicação do potencial elétrico. A Voltametria de Pulso Normal (NPV, do inglês Normal Pulse Voltammetry), Voltametria de Pulso Diferencial (DPV, do inglês Differential Pulse Voltammetry) e Voltametria de Onda Quadrada (SWV, do inglês Square Wave Voltammetry) são as técnicas que envolvem a aplicação de pulsos de potencial, e as informações, qualitativas e quantitativas, são relacionadas às reações de oxidação/redução que ocorrem na interface entre um condutor elétrico (eletrodo de trabalho) e o condutor iônico (solução contendo a espécie de interesse).23,50,52 Como os sinais obtidos dependem de reações na interface eletrodo/solução, as mudanças na interface alteram significativamente os sinais analíticos, promovendo alterações na sensibilidade e seletividade analítica. A escolha do eletrodo de trabalho depende de dois fatores fundamentais: o comportamento redox do analito de interesse e os sinais de corrente obtidos no intervalo de potencial para a análise química.22 Na escolha do material para construção do eletrodo de trabalho é importante observar o intervalo de potencial de cada eletrodo, o qual é diretamente relacionado ao sobrepotencial de evolução de hidrogênio, onde se inicia a decomposição da água, que é dependente do solvente e do pH do meio. Também deve ser considerada a geometria do eletrodo, a condutividade elétrica, a reprodutibilidade da superfície, as propriedades mecânicas, o custo de fabricação, a disponibilidade e a toxicidade. Assim, uma grande variedade de materiais pode ser empregada tais como: mercúrio, metais nobres (platina e ouro), diferentes formas de carbono, e eletrodos quimicamente modificados.22 Para compostos contendo Se, até o ano 2000 havia a predominância no uso do eletrodo gotejante de mercúrio (HMDE, do inglês Hanging Mercury Dropping Electrode), isto por que o Se (IV) e Se (VI) podem ser acumulados sobre mercúrio produzindo HgSe, que é posteriormente redissolvido, produzindo uma corrente elétrica diretamente proporcional à concentração de Se. O uso do HMDE produz resultados analíticos com excelente sensibilidade e reprodutibilidade em função da constante renovação da gota de mercúrio em cada uma das análises efetuadas. Piech e Kubiak53 utilizaram um HMDE modificado com cobre para a determinação de Se em amostras de águas naturais, cogumelos e fígado bovino, as quais foram previamente tratadas empregando-se MWAD. A detecção foi realizada empregando-se a CSV com DPV. A utilização de cobre possibilitou a formação específica de compostos intermetálicos contendo Se, Cu e Hg, promovendo, assim, excelente sensibilidade analítica e limite de detecção na faixa de partes por bilhão (ppb). Apesar da excelente sensibilidade e estabilidade no sinal obtido com o uso do HMDE, as análises químicas resultam na geração de uma grande quantidade de resíduos tóxicos de mercúrio. Desta maneira, pode-se observar que na última década o HMDE começou a ser substituído por outros materiais menos tóxicos, tais como eletrodos metálicos construídos a partir de metais nobres (platina, ouro, prata e diamante dopado com boro) e eletrodos quimicamente modificados54-57 (com polímeros, aminoácidos, enzimas, nanopartículas metálicas, filmes de mercúrio, filmes de bismuto, dentre outros), como pode ser observado na Figura 5 e na Tabela 3.
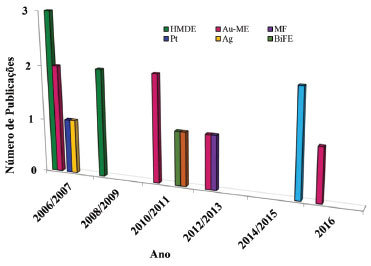 Figura 5. Evolução nas publicações científicas com quantificação de Se empregando-se técnicas eletroanalíticas com diferentes superfícies eletródicas, dos anos 2006 a 2016
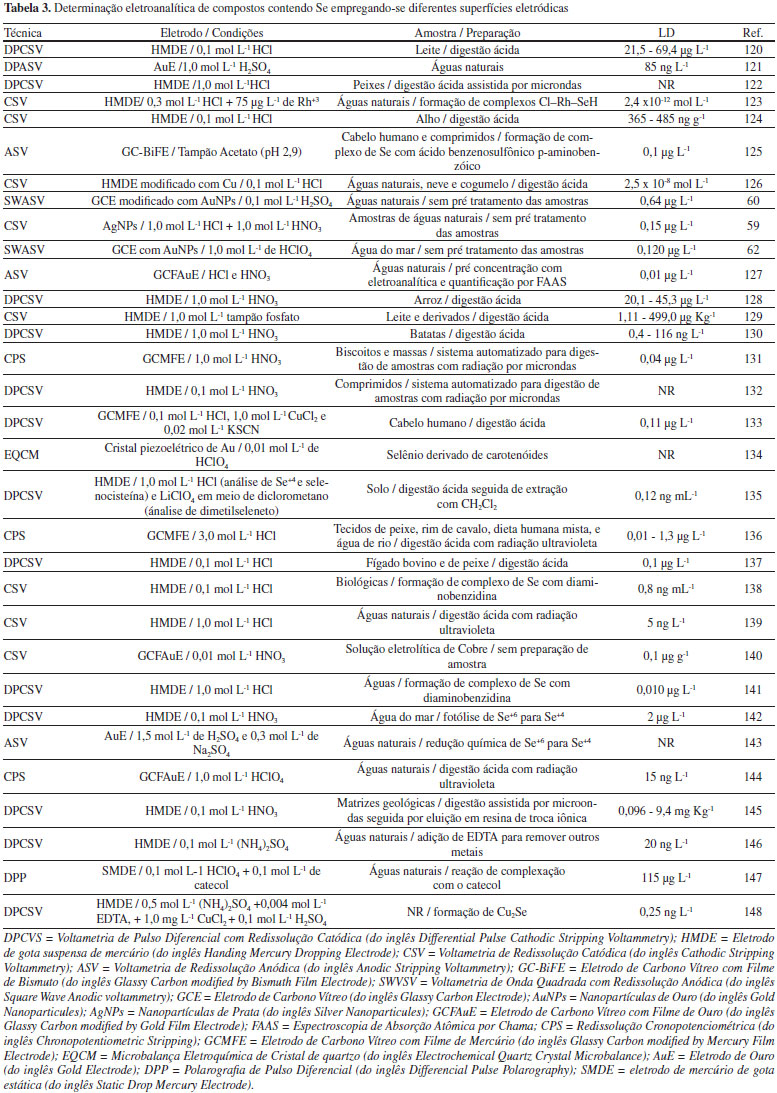
Beni, Collins e Arrigan58 utilizaram eletrodo de ouro e platina para a quantificação de Se (IV), com a comparação analítica em ambas as superfícies metálicas. O uso de um eletrodo anelar de Ag para a quantificação Se (IV) foi demonstrado por Bás, Jedlińska e Węgiel59 para análise de águas naturais com limite de detecção de 0,15 µg L-1. Eletrodos quimicamente modificados proporcionam melhoria no sinal analítico e/ou diminuição nos valores de potencial de redução e ou oxidação dos compostos de interesse. Neste contexto, Idris, Mabuba e Arotiba60 utilizaram o eletrodo de carbono vítreo modificado com nanopartículas de ouro aliado à técnica de SWV para a quantificação de Se em águas naturais. Segura et. al.61 empregou nanopartículas de prata, possibilitando a determinação de Se em águas naturais com limite de detecção inferior a 0,64 µg L-1, empregando a Voltametria de Redissolução Anódica (ASV) ou Catódica (CSV), mostrando que a modificação com nanopartículas promove melhoria significativa no sinal analítico e nos valores de limite de detecção. A determinação de seleno-cistina em eletrodos de ouro modificados com filme de selênio foi demonstrada por Walker et. al.62 mostrando que é possível investigar o par redox selenocistina/selenocisteína em condições fisiológicas. Além da alta detectabilidade oferecida pelos métodos eletroquímicos de redissolução que possuem uma etapa prévia de pré-concentração in situ (na solução eletrolítica em que é feita a medida eletroquímica), a detecção seletiva de diferentes espécies de Se, sem a necessidade de qualquer separação prévia, é uma clara vantagem dos métodos eletroanalíticos de análise.
CONCLUSÕES E PERSPECTIVAS O desenvolvimento de métodos analíticos para identificação e quantificação de compostos químicos contendo Se apresenta grande importância científica, em função das propriedades nutricionais, funções metabólicas e dosagens essenciais. De maneira geral, a utilização de técnicas espectrométricas e cromatográficas necessita de etapas prévias de processamento, digestão e purificação das amostras, proporcionando elevado custo, aumento no tempo necessário para aquisição dos dados analíticos e principalmente, erros associados à preparação e manipulação das amostras. Assim, torna-se fundamental o desenvolvimento de metodologias que minimizem as etapas de preparação de amostras. O desenvolvimento de instrumentação que automatize a manipulação de amostras, diminuindo os erros experimentais é uma alternativa para a aplicação de métodos espectroscópicos e cromatográficos para a determinação de Se em amostras complexas, tais como solos, alimentos e fluidos biológicos. O desenvolvimento de fontes de emissão de radiação eletromagnética (lâmpadas de cátodo oco) com melhor reprodutibilidade na intensidade de emissão é uma necessidade para a ampliação da aplicabilidade da AFS. Além disto, a aplicação de laser na excitação de Se, o qual apresenta excelente sensibilidade, ainda apresenta alto custo da instrumentação e dificuldade na obtenção de lasers específicos de alta potência. Já a utilização das técnicas eletroanalíticas permite a minimização na manipulação e tratamento das amostras, reduzindo o tempo, o custo e os erros associados à manipulação das amostras. Entretanto, na maioria dos casos, a aplicação destas técnicas envolve o uso de HMDE, que produz resíduos de mercúrio. O emprego de eletrodos sólidos, como prata, ouro e platina, além de eletrodos quimicamente modificados, vem sendo investigado em substituição aos eletrodos de mercúrio. Aliar as vantagens associadas ao uso de técnicas eletroanalíticas (rapidez, sensibilidade e robustez) com as vantagens do emprego de HMDE (reprodutibilidade da superfície eletródica) pode permitir a identificação e quantificação de compostos orgânicos contendo Se e outros metalóides em amostras de interesse ambiental, industrial e biológico. Neste contexto, o uso de Eletrodos Sólidos de Amálgama (SAE, do inglês solid amalgam electrode) são promissores no desenvolvimento de procedimentos para detecção e quantificação de compostos orgânicos e inorgânicos.63,64 Estes eletrodos podem ser construídos empregando-se diferentes proporções entre mercúrio e outros metais tais como cobre, prata, platina, ouro, bismuto, irídio ou cádmio e apresentam alta estabilidade mecânica, longa durabilidade e podem ter a superfície tratada eletroquimicamente, promovendo assim uma excelente reprodutibilidade nos sinais analíticos.65 O emprego de SAE juntamente com DPV ou SWV se mostra como uma excelente alternativa para o desenvolvimento de metodologias para identificação e quantificação de Se.
AGRADECIMENTOS Os autores agradecem o apoio financeiro da FAPEMIG (processos APQ 01938-11, APQ 02052-12, APQ 02528-14, APQ 00711-14), do CNPq (308174/2013-5 e 460799/2014-2), da PROPP-UFU e da Rede Mineira de Química (RQ-MG).
REFERÊNCIAS 1. Neto, F. A.; Nutrição clínica, 1a ed., Guanabara Koogan: Rio de Janeiro, 2012. 2. Panigati, M.; Falciola, L.; Mussini, P.; Beretta, G.; Facino, R. M.; Food Chem. 2007, 105, 1091. 3. Himoto, T.; Yoneyama, H.; Kurokohchi, K.; Inukai, M.; Masugata, H.; Goda, F.; Haba R.; Watababe, S.; Kubota, S.; Senda, S.; Masaki, T.; Nutr. Res. 2011, 31, 829. 4. Campillo, N.; Peñalver, R.; López-García, I.; Hernández-Córdoba, M.; J. Chromatogr. A. 2009, 1216, 6735. 5. Organização Mundial da Saúde; Elementos traço na nutrição e saúde humanas, 1a ed., São Paulo, Brasil, 1998. 6. Food and Nutrition Board, Institute of Medicine, Dietary reference intakes from vitamin C, vitamin E, selenium and carcinoids, National Academic Press: New York, 2000. 7. National Research Council, Recommended Dietary Allowances, 10th ed., National Academy Press: Washington, 2000. 8. Zingaro, A. R.; Cooper, C. W.; Selenium. In The toxicology of selenium and its compounds, 1st ed., Van Nostrand Reinhold Company/Litton Educational Publishing: New York, 1974. 9. Nascimento, P. C.; Jost, C. L.; De Carvalho, L. M.; Bohrer, D.; Koschinsky, A.; Anal. Chim. Acta 2009, 648, 162. 10. Mattila, P.; Konko, K.; Eurola, M.; Pihlava, J. M.; Astola, J.; Vahteristo, L.; Hietaniemi, V.; Kumpulainen, J.; Valtonen, M.; Piironen, V.; Food Chem. 2001, 49, 2343. 11. Kelly, S.; Baxter, M.; Chapman, S.; Rhodes, C.; Dennis, J.; Brereton, P.; Eur. Food Res. Technol. 2002, 214, 72. 12. Gupta, U. G.; Gupta, S. C.; Gupta, M. D.; Commun. Soil Sci. Plan. 2000, 31, 11. 13. Latorre, C. H.; García, J. B.; Martín, S. G.; Crecente, R. M. P.; Anal. Chim. Acta 2013, 804, 37. 14. Tan, L. C.; Nancharaiah, Y. V.; van Hullebusch, E. D.; Lens, P. N. L.; Biotechnol. Adv. 2016, 34, 886. 15. Awual, M. R.; Yaita, T.; Suzuki, S.; Shiwaku, H.; J. Hazard .Mater, 2015, 291, 111. 16. Resolução CONAMA n°357, Classificação de águas, doces, salobras e salinas do Território Nacional, Brasil, 2005. 17. Suzuki, K. T.; J. Health Sci. 2005, 51, 107. 18. Akramipour, R.; Hemati, M.; Fattahi, N.; Pirsaheb, M.; Ahmadi-Jouibari, T.; J. Pharm. Biomed. Anal. 2017, 140, 155. 19. Lemy, D.; Finger, S. E.; Nelson, M. K.; Environ. Toxicol. Chem. 1993, 12, 2265. 20. Falandysz, J.; Food Chem. 2013, 138, 242. 21. Jagtap, R.; Maher, W.; Microchem. J. 2016, 124, 422. 22. Wang, J.; Analytical Eletrochemistry, 2nd ed., John Wiley & Sons: New York, 2001. 23. Harris, D. C.; Explorando a Química Analítica, 4a ed., LTC: Rio de Janeiro, 2011. 24. Silverstein, R. M.; Bassler, G. C.; Kiemle, D. J.; Spectrometric identification of organic compounds, 7th ed., Jonh Wiley & Sons: New York, 2005. 25. Wen, X.; Zhang, Y.; Li, C.; Fang, X.; Zhang, X.; Spectrochim. Acta, Part A 2014, 123, 200. 26. Shabani, A. M. H.; Dadfarnia, S.; Nozohor, M.; Spectrochim. Acta, Part A 2013, 116, 1. 27. Ezoe, K.; Ohyama, S.; Hashem, A.; Ohira, S.; Toda, K.; Talanta 2016, 148, 609. 28. Pavia, D. L.; Lampman, G. M.; Kriz, G. S.; Introduction to Spectroscopy, 3rd ed., Thomson Learning: Washintgon, 2001. 29. Rybínová, M.; Červený, V.; Hraníček, J.; Rychlovský, P.; Microchem. J. 2016, 124, 584. 30. Broekaert, J. A. C.; Analytical Spectrometry with Flames and Plasmas, Wiley-VCH: New York, 2002. 31. Wang, N.; Wang, P.; Chem. Eng. J. 2016, 283, 193. 32. Ebdon, L.; Fisher, A.; Hill, S. J. In An introduction to analytical atomic Spectrometry, Evans, E. H., eds.; John Wiley & Sons: New York, 1998. 33. de Souza, J. R.; da Silva, L.; da Rocha, M. S.; Saint'Pierre, T. D.; Food Anal. Met. 2017, doi: 10.1007/s12161-017-0861-y. DOI: http://dx.doi.org/10.1007/s12161-017-0861-y 34. Escudero, L. A.; Pacheco, P. H.; Gasquez, J. A.; Salonia, J. A.; Food Chem. 2015, 169, 73. 35. Zhang, N.; Fu, N.; Fang, Z.; Feng, Y.; Ke, L.; Food Chem. 2011, 124, 1185. 36. Bruttel, P. A.; Schäfer, J.; Sample Preparation Techniques in Voltammetric Trace Analysis, 1st ed.; Metrohm AG, Herisau: Switzerland, 1992. 37. Ohki, A.; Nakajima, T.; Hirakawa, S.; Hayashi, K.; Takanashi, H.; Microchem. J. 2016, 124, 693. 38. Schneider, M.; Pereira, E. R.; Castilho, I. N. B.; Carasek, E.; Welz, B.; Martens, I. B. G.; Microchem. J. 2016, 125, 50. 39. Guo, W.; Hu, S.; Wang, Y.; Zhang, L.; Hu, Z.; Zhang, J.; Microchem. J. 2013, 108, 106. 40. Colins, C. H.; Braga, G. L.; Bonato, P. S.; Fundamentos de Cromatografia, 1a ed., Unicamp: Brasil, 2006. 41. Braithwaite, A.; Smith, F. J.; Chromatographic Methods, 5th ed., Blackie: Londres, 1996. 42. Sewll, P.; Clarke, C.; Chromatographic Separations, Wiley: Nova York, 1988. 43. Jonsson, J. A.; Chromatographic Theory and Basic Principles, Marcel Dekker: Nova York, 1987. 44. Bryszewska, M. A.; Mage, A.; J. Trace Elem. Med. Biol. 2015, 29, 91. 45. Eith, C.; Kolb, M.; Seubert, A.; Práticas em Cromatografia de íons - uma introdução, 2 th ed., Metrohm: São Paulo, 2006. 46. Reyes, L. H.; Mar, J. L. G.; Rahman, G. M. M.; Seybert, B.; Fahrenholz, T.; Kingston, H.M. S.; Talanta 2009, 78, 983. 47. Niedzielski, P.; Rudnicka, M.; Wachelka, M.; Kozak, L.; Rzany, M.; Wozniak, M.; Kaskow, Z.; Food Chem. 2016, 190, 454. 48. Vriens, B.; Mathis, M.; Lenny H. E. Winkel, L. H. E.; Berg, M.; J. Chromatogr. A 2015, 1407, 11. 49. Bond, A. M.; Compton, R. G.; Fiedler, D. A.; Inzelt, G.; Kahlert, H.; Komorsky-Lovric´ Š.; Lohse, H.; Lovric´, M.; Maeken, F.; Neudeck, A.; Retter, U.; Scholz, F.; Stojet, Z. In Eletroanalytical Methods, Scholz, F., ed.; 2th ed., Springer: New York, 2009. 50. De Souza, D.; Machado, S. A. S.; Avaca, L. A.; Quim. Nova 2003, 26, 81. 51. Dahmen, E. A. M. F.; Electroanalysis: Theory and applications in aqueous and non-aqueous media and in automatec chemical control, Elsevier: Amsterdam, 1986. 52. Mirčeski, V.; Komorsky-Lovrić, Š.; Lovrić, M. In Square-Wave Voltammetry, Theory and Application, Scholz, F. eds., Springer: New York, 2007. 53. Piech, R.; Kubiak, W. W.; Electrochem. Acta 2007, 53, 584. 54. Sajid, M.; Nazal, M. K.; Mansha, M.; Alsharaa, A.; Jillani, S. M. S.; Basheer, C.; Trends Anal. Chem. 2016, 76, 15. 55. Šebková, S.; Navrátil, T.; Kopanica, M.; Anal. Lett. 2005, 38, 1747. 56. Fagan-Murphy, A.; Allen, M. C.; Patel, B. A.; Electrochim. Acta 2015, 152, 249. 57. Zima, J.; Švancara, I.; Bareka, J.; Vytřas, K.; Crit. Rev. Anal. Chem. 2009, 39, 204. 58. Beni, V.; Collins, G.; Arrigan, D. W. M.; Anal. Chim. Acta 2011, 699, 127. 59. Baś, B.; Jedlińska, K.; Węgiel, K.; Electrochem. Commun. 2014, 49, 79. 60. Idris, A. O.; Mabuba, N.; Arotiba, O. A.; J. Electroanal. Chem. 2015, 758, 7. 61. Segura, R.; Pizarro, J.; Díaz, K.; Placencio, A.; Godoy, F.; Pino, E.; Recio F.; Sens. Actuators, B 2015, 220, 263. 62. Walker, L. M.; Karnaukh, E. A.; Dewan, F., Buzzeo, M. C.; Electrochem. Commun. 2016, 69, 28. 63. Yosypchuk, B.; Novotný, L.; Crit. Rev. Anal. Chem. 2002, 32, 141. 64. Yosypchuk, B.; Barek, J.; Crit. Rev. Anal. Chem. 2009, 39, 189. 65. De Souza, D.; Melo, L. C.; Correia, A. N.; Lima-Neto, P.; Fatibello-Filho, O.; Mascaro, L. H.; Quim. Nova 2011, 34, 3. 66. Khajehsharifi, H.; Mousavi, M. F.; Ghasemi, J.; Shamsipur, M.; Anal. Chim. Acta 2004, 512, 369. 67. Yan, L.; Deng, B.; Shen, C.; Long, C.; Deng, Q.; Tao, C.; J. Chromatogr. A 2015, 1395, 173. 68. Song, C.; Fei, Q.; Shan, H.; Feng, G.; Cui, M.; Liu, Y.; Huan, Y.; Spectrochim. Acta, Part A 2013, 116, 497. 69. Tyburska, A.; Jankowski, K.; J. Pharm. Biomed. Anal. 2013, 74, 268. 70. Duben, O.; Boušek, J.; Dědina, J.; Kratzer, J.; Spectrochim. Acta, Part B 2015, 111, 57. 71. Tyburska, A.; Jankowski, K.; Rodzik, A.; Spectrochim. Acta, Part B 2011, 66, 517. 72. Marguí, E.; Floor, G. H.; Hidalgo, M.; Kregsamer, P.; Roman-Ross, G.; Streli, C.; Queralt, I.; Spectrochim. Acta, Part B 2010, 65, 1002. 73. Nunes, D. L.; dos Santos, E. P.; Barin, J. S.; Mortari, S. R.; Dressler, V. L.; Flores, E. M. de M.; Spectrochim. Acta, Part B 2005, 60, 731. 74. Tuzen, M.; Pekiner, O. Z.; Food Chem. 2015, 188, 619. 75. Khan, N.; Jeong, I. S.; Hwang, I. M.; Kim, J. S.; Choi, S. H.; Nho, E. Y.; Choi, J. Y.; Kwak, B.; Ahn, J.; Yoon, T.; Kim, K. S.; Food Chem. 2013, 141, 3566. 76. Sigrist, M.; Brusa, L.; Campagnoli, D.; Beldoménico, H.; Food Chem. 2012, 134, 1932. 77. Shrivas, K.; Patel, D. K.; Food Chem. 2011, 124, 1673. 78. Tuzen, M.; Pekiner, O. Z.; Food Chem. 2015, 188, 619. 79. Oliveira, A. P.; Neto, J. A. G.; Nóbrega, J. A.; Correia, P. R. M.; Oliveira, P. V.; Food Chem. 2005, 93, 355. 80. Kolachi, N. F.; Kazi, T. G.; Afridi, H. I.; Khan, S.; Wadhwa, S. K.; Shah, A. Q.; Shah, F.; Baig, J. A.; Sirajuddin; Food Chem. Toxicol. 2010, 48, 3327. 81. Awual, R.; Hasan, M.; Khaleque, A.; Sens. Actuators, B 2015, 209, 194. 82. Díaz-Somoano, M.; López-Antón, M. A.; Martínez-Tarazona, M. R.; Fuel 2004, 83, 231. 83. Liu, Z.; Sun, H.; Shen, S.; Li, L.; Shi, H.; Anal. Chim. Acta 2005, 550, 151. 84. Shahdousti, P.; Alizadeh, N.; Anal. Chim. Acta 2011, 684, 67. 85. Kocot, K.; Leardi, R.; Walczak, B.; Sitko, R.; Talanta 2015, 134, 360. 86. Li, Z.; Guo, Y.; Talanta 2005, 65, 1318. 87. Maleki, N.; Safavi, A.; Doroodmand M. M.; Talanta 2005, 66, 858. 88. Martinis, E. M.; Escudero, L. B.; Berton, P.; Monasterio, R. P.; Filippini, M. F.; Wuilloud, R. G.; Talanta 2011, 85, 2182. 89. Izgi, B.; Gucer, S.; Jaćimović, R.; Food Chem. 2006, 99, 630. 90. Xu, S.; Zheng, M.; Zhang, X.; Zhang, J.; Lee, Y.; Microchem. J. 2012, 101, 70. 91. Hsieh, Y.; Jiang, S.; Microchem. J. 2013, 110, 1. 92. Zhang, H.; Gao, P.; Guo, X.; Wang, H.; Microchem. J. 2013, 110, 192. 93. Tie, M.; Li, B.; Zhuang, X.; Han, J.; Liu, L.; Hu, Y.; Li, Y.; Microchem. J. 2015, 123, 70. 94. Wang, R.; Hsu, Y.; Chang, L.; Jiang, S.; Anal. Chim. Acta 2007, 590, 239. 95. Lu, Y.; Rumpler, A.; Francesconi, K. A.; Pergantis, S. A.; Anal. Chim. Acta 2012, 731, 49. 96. Tsai, Y.; Lin, C.; Hsu, I.; Sun, Y. Anal. Chim. Acta 2014, 806, 165. 97. Ghasemi, E.; Sillanpää, M.; Najafi, N. M.; J. Chromatogr. A 2011, 1218, 380. 98. Viñas, P.; López-García, I.; Merino-Meroño, B.; Campillo, N.; Hernández-Córdoba, M.; Anal. Chim. Acta 2005, 535, 49. 99. Zhang, X.; Yang, L.; Mester, Z.; Anal. Chim. Acta 2012, 744, 54. 100. Gionfriddo, E.; Naccarato, A.; Sindona, G.; Tagarelli, A.; Anal. Chim. Acta 2012, 747, 58. 101. Arnaudguilhem, C.; Bierla, K.; Ouerdane, L.; Preud'homme, H.; Yiannikouris, A.; Lobinski, R.; Anal. Chim. Acta 2012, 757, 26. 102. Liang, L.; Mo, S.; Zhang, P.; Cai, Y.; Mou, S.; Jiang, G.; Wen, M.; J. Chromatogr. A 2006, 1118, 139. 103. Pedrero, Z.; Encinar, J. R.; Madrid, Y.; Cámara, C.; J. Chromatogr. A 2007, 1139, 247. 104. Kuo, C.; Jiang, S.; J. Chromatogr. A 2008, 1181, 60. 105. Moscoso-Pérez, C.; Moreda-Piñeiro, J.; López-Mahía, P.; Muniategui-Lorenzo, S.; Fernández-Fernández, E.; Prada-Rodríguez, D.; J. Chromatogr. A 2008, 1215, 15. 106. Peachey, E.; Cook, K.; Castles, A.; Hopley, C.; Goenaga-Infante, H.; J. Chromatogr. A 2009, 1216, 7001. 107. Moreno, F.; García-Barrera, T.; Gómez-Ariza, J. L.; J. Chromatogr. A 2013, 1300, 43. 108. Shih, T.; Hsu, I.; Wu, J.; Lin, C.; Sun, Y.; J. Chromatogr. A 2013, 1304, 101. 109. Zhang, Q.; Yang. G.; J. Chromatogr. A 2014, 1325, 83. 110. Bueno, M.; Pannier, F.; Talanta 2009, 78, 759. 111. Kulikov, A. U.; J. Pharm. Biomed. Anal. 2007, 43, 1283. 112. Zembrzuska, J.; Matusiewicz, H.; Polkowska-Motrenko, H.; Chajduk, E.; Food Chem. 2014, 142, 178. 113. Yu, H.; Chen, C.; Gao, Y.; Li, B.; Chai, Z.; Chin. J. Anal. Chem. 2006, 34, 749. 114. Tao, Z.; Yu-Xi, G.; Bai, L.; Yu-Feng, L.; Chun-Ying, C.; Gang, W.; Chin. J. Anal. Chem. 2008, 36, 206. 115. Yu-Pin, C.; Li-Zhen, Y.; Hong-Li, H.; Bi-Yang, D.; Chin. J. Anal. Chem. 2015, 43, 1329. 116. Kokarnig, S.; Tsirigotaki, A.; Wiesenhofer, T.; Lackner, V.; Francesconi, K. A.; Pergantis, S. A.; Kuehnelt, D.; J. Trace Elem. Med. Biol. 2015, 29, 83. 117. Humann-Ziehank, E.; Ganter, M.; Michalke, B.; J. Trace Elem. Med. Biol. 2016, 33, 14. 118. Chitta, K. R.; Landero-Figueroa, J. A.; Kodali, P.; Caruso, J.; A. ; Merino E. J.; Talanta 2013, 114, 25. 119. Najafi, N. M.; Tavakoli, H.; Abdollahzadeh, Y.; Alizadeh, R.; Anal. Chim. Acta 2012, 714, 82. 120. İnam, R.; Somer, G.; Food Chem. 2000, 69, 345. 121. Ochab, M.; Gęca, I.; Korolczuk, M.; Talanta 2017, 165, 364. 122. Lambert, D. F.; Turoczy, N. J.; Anal. Chim. Acta 2000, 408, 97. 123. Lange, B.; Van Den Berg, C. M. G.; Anal. Chim. Acta 2000, 418, 33. 124. Inam, R.; Somer, G.; Food Chem. 1999, 66, 381. 125. Zhang, Q.; Li, X.; Shi, H.; Hongzhou, Y. Z.; Electrochim. Acta 2010, 55, 4717. 126. Piech, R.; Kubiak, W. W.; Electrochem. Acta 2007, 53, 584. 127. Cacho, F.; Jankuv, L.; Lauko, L.; Kroliak, M.; Manova, A.; Beinrohr, E.; Talanta 2013, 116, 195. 128. Panigati, M.; Falciola, L.; Mussini, P.; Beretta, G.; Facino, R. M.; Food Chem. 2007, 105, 1091. 129. Shahbazi, Y.; Ahmadi, F.; Fakhari, F.; Food Chem. 2016, 192, 1060. 130. Ferri, T.; Favero, G.; Frasconi, M.; Microchem. J. 2007, 85, 222. 131. Švarc-Gajić, J.; Stojanović, Z.; Talanta 2013, 115, 474. 132. Stoica, A.; Babaua, G.; Iorgulescu, E.; Marinescu, D.; Baiulescu, G.; J. Pharm. Biomed. Anal. 2002, 30, 1425. 133. Wang, Y.; Liu, Z.; Yao, G.; Zhu, P.; Hu, X.; Yang, C.; Xu, Q.; Anal. Chim. Acta 2009, 649, 75. 134. Foss, B.; Ion, A.; Partali, V.; Sliwka, H.; Banica, F.; J. Electroanal. Chem. 2006, 593, 15. 135. Ochsenkühn-Petropoulou, M.; Tsopelas, F.; Anal. Chim. Acta 2002, 467, 167. 136. Adeloju, S. B.; Young T. M.; Anal. Chim. Acta 1994, 296, 69. 137. Adeloju, S. B.; Bond, A. M.; Hughes, H. C.; Anal. Chim. Acta 1983, 148, 59. 138. Stará, V.; Kopanica, M.; Anal. Chim. Acta 1988, 208, 231. 139. Elleouet, C.; Quentel, F.; Madec, C.; Water Res. 1996, 30, 909. 140. Hamilton, T. W.; Ellis, J.; Florence, T. M.; Anal. Chim. Acta 1979, 110, 87. 141. Breyer, P.; Gilbert, B. P.; Anal. Chim. Acta 1987, 201, 33. 142. Batley, G. E.; Anal. Chim. Acta 1986, 187, 109. 143. Bryce, D. W.; Izquierdo, A.; De Castro, M. D. L.; Anal. Chim. Acta 1995, 308, 96. 144. Gil, E. P.; Ostapczuk, P.; Anal. Chim. Acta 1994, 293, 55. 145. Ferri, T.; Rossi, S.; Sangiorgio, P.; Anal. Chim. Acta 1998, 361, 113. 146. Baltensperger, U.; Hertz, J.; Anal. Chim. Acta 1985, 172, 49. 147. McCrory-Joy, C.; Rosamilia, J. M.; Anal. Chim. Acta 1982, 142, 231. 148. Esteban, M.; Ariño, C.; Ruisánchez, I.; Larrechi, M. S.; Rius, F. X.; Anal. Chim. Acta 1993, 284, 435. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access