
 |
 |
 |
 |
 |
Artigo
| More active and sulfur resistant bimetallic Pd-Ni catalysts |
|
Carolina BettiI; Nicolás CarraraI; Juan BadanoI; Cecilia LederhosI; Carlos VeraI,II; Mónica QuirogaI,II,*
I Instituto de Investigaciones en Catálisis y Petroquímica, INCAPE (FIQ-UNL, CONICET), Colectora Ruta Nac. N° 168 Km 0, Paraje El Pozo, 3000 Santa Fe, Argentina Recebido em: 07/07/2017 *e-mail: mquiroga@fiq.unl.edu.ar The influence of the kind of metal precursor and the sequence of impregnation on the properties of Pd-Ni catalysts was evaluated during the test reaction of selective hydrogenation of styrene to ethylbenzene by means of physicochemical characterization. The focus was put on the final hydrogenating activity and the resistance to deactivation by sulfided compounds (thiophene). The used techniques of characterization were ICP, XPS, XDR, TPR, CO chemisorption and TEM. XPS results indicated the presence of different Pd species: Pdδ-, Pdº and Pdδ+. In the case of the Ni containing catalysts, Niº and NiO species were also detected. These palladium and nickel species would be responsables of the variation of activity and sulfurresistance of the catalysts. NiClPd catalysts had a higher resistance to deactivation by sulfur poisoning. This was associated to a higher concentration of Pdη+ClxOy species that would prevent the adsorption of thiophene by both steric and electronic effects. It could also be due to the lower concentration of Pdº and Niº on these catalysts, as compared to those shown by the PdNiCl catalysts. Both the Pdº and Niº species are more prone to poisoning because of their higher electronic availability. INTRODUCTION Many petrochemical processes and applications in biotechnology, pharmacy and agrochemicals are based on the catalytic heterogeneous hydrogenation of hydrocarbons. The main objectives of these processes are to get the highest possible yields in conditions of reasonably mild pressure and temperature, and at the lowest cost. Petroleum hydrocarbon mixtures contain unsaturated compounds and many aromatic compounds. For this reason it is important that the reaction may be selective to the desired products. Particularly the heterogeneous reactions of selective hydrogenation of vinylic compounds that keep the aromatic nucleus intact are of great interest and usefulness in the petrochemical industry. A clear example are the pyrolysis gasolines that are a low quality subproduct of cracking processes for olefin production.1,2 Selective hydrogenation permits obtaining processed gasoline that meets the quality and octane conditions for its addition to the gasoline pool. Besides aromatic compounds like Benzene, Toluene and Xylene (BTX) can be extracted and used for other petrochemical uses. The hydrogenation of styrene to ethylbenzene is considered as a model for the study of the purification of pyrolysis gasoline because styrene is the most refractory compound for hydrogenation. Both Pd and Ni catalysts are used for styrene hydrogenation.3,4 The main problem of the PyGas streams is that they contain high amount of sulfided compounds, usually between 300 and 2000 ppm1,3,5 that poison the catalysts. Thiophene is one of the main sulfided compounds causing deactivation of the catalysts by poisoning. In a previous work with monomethalic catalysts the effect of chlorine for the sulfurresistance was studied. In Badano et al.,6 it was found that chloride complex species (Pdxδ+OyClz and the Ptxδ+OyClz, with δ < 2) present in the surface could hinder the adsorption of poisons via a steric factor (big size of chlorine ligands) or an electronic one (high electronegativity). Besides, bimetallic catalysts are potentially thioresistant systems. In a previous work our group studied Pt-Ni and Pt-W catalysts, and reported electronic and steric effects that improved the activity or the sulfur resistance.7,8 The objectives of this work were three: (i) the synthesis of bimetallic Pd-Ni catalysts varying the order of impregnation of the metals and the kind of precursor; chlorided and nitrogenated precursors were used; (ii) the catalytic evaluation of these catalysts; (iii) the assessment of the resistance of the catalysts to deactivation by sulfur poisoning. Four bimetallic catalysts were synthesized: PdNiN, NiNPd, PdNiCl and NiClPd. Results of activity and thioresistance were compared to the properties of a monometallic Pd catalyst.
EXPERIMENTAL Catalysts preparation Bimetallic catalysts were prepared by successive impregnation of the metal precursors in solution. The used support was γ-Al2O3 Ketjen CK 300. This was previously calcined in order to stabilize its surface area (SBET: 224 m2 g -1). For the incorporation of the metals to the support the technique of incipient wetness impregnation was used. Impregnating solutions with dissolved PdCl2, NiCl2 or Ni(NO3)2.6H2O were used to obtain monometallic catalysts of Pd, NiCl and NiN. Monometallic catalysts were dried with 24 h in a stove at 373 K, then they were calcined in dried air for 3 h at 823 K and finally they were reduced for 1 h at 673 K in a hydrogen flow of 110 mL min -1. During the impregnation of the second metal, an acidified solution of PdCl2 was impregnated over the monometallic catalysts of NiN and NiCl, for obtaining the bimetallic catalysts NiNPd and NiClPd respectively. Similarly, acidified solutions of NiCl2 and Ni(NO3)2.6H2O were impregnated over Pd monometallic catalysts to obtain the PdNiCl and PdNiN catalysts, respectively. The resulting bimetallic catalysts were dried for 24 h in a stove at 373 K, calcined for 3 h at 823 K and reduce for 1 h at 673 in a hydrogen stream of 110 mL min -1 before the reaction test. Catalysts characterization The mass concentration of Pd and Ni in the final catalysts was determined by plasma induction atomic emission spectroscopy (ICP-OES). The equipment used was a Perkin Elmer 2100 and the samples had to be previously digested in diluted sulfuric acid for the analysis. The electronic state of the Pd and Ni surface species and their atomic ratios were determined by means of X-ray photoelectron spectroscopy (XPS). XPS spectra were recorded in a Multitecnica UniSpecs equipment. It had an XR50 model X-ray dual Mg/Al source and a hemispheric Phoibos 150 analyzer working in fixed analyzer transmission mode (FAT). The spectra were obtained with an energy pass of 30 eV and a Mg anode operated at 200 W. The pressure during the measurements was lower than 2 10-8 mBar. The samples were previously reduced in situ in the reaction chamber of the instrument, with a stream of H2:Ar at 673 K for 10 min. The reference binding energy used as a reference was Al 2p at 74.1 eV. X-ray diffraction (XRD) spectra of the powdered samples were obtained in a Shimadzu XD-1 instrument using CuKα radiation (λ = 1.5405 Å) filtered with Ni, in the 15°< 2q < 85° range and at a scan speed of 1° min -1. The samples were powdered and reduced ex situ under a hydrogen flow. Then they were cooled down to room temperature in nitrogen flow and put into the chamber of the equipment to record the spectrum. The study of the reducibility of the surface species was performed by temperature programmed reduction (TPR) in a Micromeritics Auto Chem II apparatus equipped with a thermal conductivity detector. A cold water trap was placed before the thermal detector to condense water. Before the TPR tests the samples were pretreated in situ in an air stream at 773 K for 30 min. After that the samples were cooled up to 308 K in an Ar stream (AGA purity 99.99%). Then the temperature was increased up to 1173 K at 10 K min -1 in a gas flow (5% (v/v) hydrogen in argon) at a total flow rate of 40 mL min -1. CO chemisorption experiments were performed in a chemisorption equipment designed ad-hoc. The catalyst was placed in a quartz reactor and first over reduced in situ in a hydrogen stream (673 K, 1 h, 60 cm3 min-1). Then the carrier was switched to N2 and the adsorbed hydrogen was desorbed (673 K, 60 cm3 min-1) for 1h; then the cell was cooled down up to room temperature. Then 0.42 cm3 pulses of diluted CO (3.04% CO in N2) were fed to the reactor until the outlet peaks were constant. Non-chemisorbed CO was quantitatively transformed into CH4 over a Ni/Kieselgur catalyst and detected in a flame ionization detector connected on-line. The metal dispersion was calculated on the assumption of adsorption stoichiometry of CO to either Pd or Ni equal to 1 molecule of CO per atom of exposed metal.9 Transmission electron microscopy (TEM) experiments were carried out in a JEOL 100 CX II operated at 100 kV. Samples were dispersed in ethanol by sonication and dropped on a copper grid coated with carbon film. Catalytic evaluation of catalysts The hydrogenation reaction of styrene to ethylbenzene was performed in batch mode using a PTFE coated, stainless steel, stirred tank reactor. The reaction of styrene hydrogenation was performed at 333 K, 20 bar hydrogen pressure, 1200 rpm stirring rate, 0.3 g catalyst mass and 200 mL solution of 5% (v/v) of styrene in toluene, n-decane was used as an internal standard for chromatography analysis. Some tests were performed with the feed of the standard test additivated with 600 ppm of thiophene, in order to assess the catalysts resistance to sulfur poisoning. Reactants and products were analyzed in a Shimadzu gas chromatograph equipped with a flame ionization detector and a 30 m, J&W InnoWax capillary column (cat. number 19091N-213).
RESULTS AND DISCUSSION Catalysts characterization Table 1 contains the catalyst notation used and chemical composition of the catalysts. Mass concentration values were determined by ICP. The results indicate that total Pd and Ni load were established at 0.8 and 3.7 mass percent for all the prepared catalysts, the error of the technique was ±13%, with an atomic ratio of Ni/Pd ≅ 7.7. The bulk atomic ratio of the bimetallic catalysts with both chlorine precursor were the highest ones.
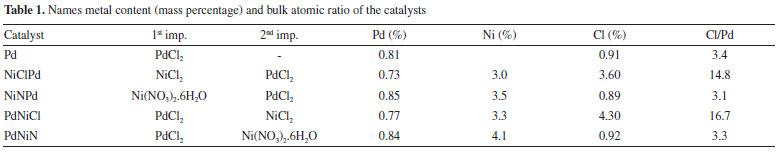
Figures 1 and 2 show the XPS spectra of the samples in the Pd 3d5/2 and Ni 2p3/2 region. The binding energies (BE) were determined by curve fitting of the spectrum line and they are tabulated in Table 2.
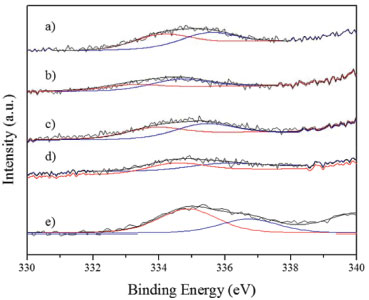 Figure 1. XPS spectra. Pd 3d5/2region. a) NiClPd. b) PdNiN. c) NiNPd. d) PdNiCl. e) Pd
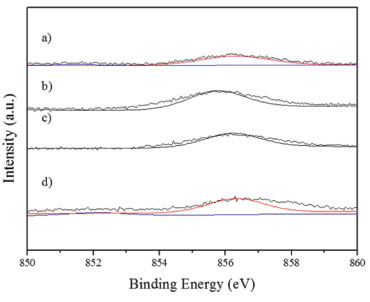 Figure 2. XPS spectra. Ni 2p3/2region. a) NiClPd. b) PdNiN. c) NiNPd. d) PdNiCl
As it can be seen in Figure 1 and Table 2 the catalysts had different surface Pd species. The peak located at about 334.1 - 334.8 eV was assigned to Pdº.10 On the monometallic catalyst and on some bimetallic ones also a peak at about 335.4 - 336.7 eV can be seen that was attributed to the presence of electrodeficient chlorinated species of Pdη+ stabilized by the presence of remaining chlorine ions. These species would have the formula, Pdη+ClxOy with 0 < η < 2. 6 The bimetallic PdNiN and NiNPd catalysts had a peak at 333.5 ± 0.3 eV that according to some reports would have a higher availability of electrons, i.e. they would be species Pdδ- (with δ < 0). These species could be due to the formation of metal bonds or alloys.11,12 When the region of Ni binding energies of the bimetallic PdNiCl and NiClPd catalysts is analyzed (Figure 2 and Table 2) a peak at about 851.7 ± 0.4 eV can be seen that would correspond to Niº.10 The peak at 856.0 ± 0.4 eV in all bimetallic catalysts would correspond to the presence of nickel oxide (Ni +2) that according with literature reports would have a strong interaction with the alumina support.10,13,14 Some authors suggested the presence of NiO.x(Al2O3) (0 ≤ x ≤ 1) species that would be formed during the thermal treatments.15 With respect to the atomic ratios obtained by XPS (Table 2) similar values were obtained in the case of the Pd/Al ratio. All XPS spectra had a peak at 198.5 eV corresponding to Cl 2p3/2 that could be due to surface chlorine species that were not eliminated during the thermal pretreatment stages.10 PdNiCl and NiClPd catalysts had high Cl/Pd ratios because these catalysts were impregnated with chlorine-containing metal precursors. The Ni/Pd ratios are higher for the PdNiN and PdNiCl catalysts. This would be related to the preparation procedure because Ni is impregnated in second place, thus staying in an outer position.7 The metal dispersion of the catalysts depends strongly on the chemical nature of the precursors and on the thermal treatments the catalysts underwent. As seen in Table 2 the monometallic catalyst was the one with higher metal dispersion. The lower dispersion and hence the higher particle size of the bimetallic catalysts could be related to the double thermal treatment they were exposed to. This long thermal treatment would cause more sintering of Pd and Ni particles.16 Bimetallic catalysts with Ni as a precursor chlorine had higher dispersion than the bimetallic ones with Ni nitrate as precursor. This could be related to the presence of complex species of the Pdη+ClxOy kind, formed during the calcination treatment. The smaller particle size is related to a high dispersion and a higher value of binding energy (BE).17,18 The monometallic Pd catalyst would have the smallest particles and the bimetallic catalysts prepared from Ni nitrate (NiNPd and PdNiN) would have the bigger particles. The size of the palladium cristallites was affected by the addition of Ni to the bimetallic catalysts. The growth pattern of palladium cristallites in the presence of Ni has been previously explained by the Pd-Ni competition for the support sites during catalyst preparation.19 The X-ray diffractograms of all catalysts had only three peaks at 2θ = 37.7°, 46.0° (400) and 67.0° (440), corresponding to the structure of g-Al2O3.20 For this reason the difractograms are not presented. No peak at 39.7°, related to the (111) reflections of Pd021 could be found, neither the peaks corresponding to the PdO or the peaks due to bulk NiO. This was attributed to the small particle size and the relatively high limit of detection of the XRD technique.22 Heracleous et al.23 have reported that in catalysts of Ni supported over alumina, a minimum Ni mass content of 15% is needed in order to detect the NiO (bulk) diffraction lines at 2θ = 43.3°, 63.0°, 75.5° and 79.5°. Salagre et al.24 have reported that the low intensity diffraction lines of NiO particles of Ni/Al2O3 catalysts would only be detected at Ni contents higher than 26.6%. In our case, due to this low sensitivity of the XRD method the Pd and NiO phases cannot be detected. Figure 3 shows the TPR traces of the catalysts. These tests were performed in order to determine the reducibility of the Pd and Ni species on the surface of alumina.
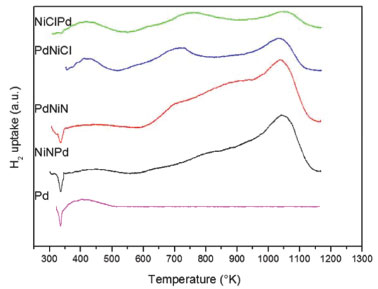 Figure 3. TPR-H2traces
The Pd monometallic catalyst (Pd) and the bimetallic PdNi of nitrate precursor (PdNiN and NiNPd) had a negative peak at low temperature, about 343 K, which is attributed to the decomposition of the Pd β-hydride phase.25 The β-PdH phase is not seen in the bimetallic catalysts with chlorinated precursor (PdNiCl and NiClPd) due to the excessive presence of chlorine that would prevent the formation of this phase. What is seen in the two latter ones is a broad peak between 373 and 473 K that could be assigned according to some authors, to the reduction of PdO species strongly interacting with the support. Other authors suggest that the consumption of hydrogen near these temperatures is due to the reduction of Pdη+ClxOy (0 < η < 2) species or to the reduction of Pd +2 ions stabilized by neighbouring Cl -, that were not eliminated during calcination and remain on the alumina surface.26-28 Figure 3 shows that above 503 K multiple hydrogen consumption peaks appear due to the reduction of NiO species that interact with the support. The first peaks at 520-620 can be assigned to the reduction of NiOx with weak or null interaction with the support (bulk NiO). 19,29-32 Reduction peaks at higher temperatures in the 620-770 K and 773-1073 K range, are attributed to the reduction of well dispersed NiO, interacting strongly with the support, and in the range 1000-1273 K to the reduction of the NiAl2O4 spinel.29,31,32 In the case of the bimetallic catalysts prepared from nickel chloride salts (PdNiCl and NiClPd) a well defined TPR peak corresponding to bulk NiO in the temperature range 623-773 K can be seen. In the case of the bimetallic catalysts prepared from nitrate salt (PdNiN and NiNPd) a bigger presence of NiO interacting strongly with the support is seen. The higher chlorine content would prevent a strong interaction of Ni with alumina. For all prepared catalysts the peak at temperatures higher than 1000 K would correspond to the presence of nickel refractory species, NiAl2O4. According to the reduction temperature employed during the synthesis of the catalysts (1 h at 673 in a hydrogen stream), bimetallic catalysts prepared with chloride salt (PdNiCl and NiClPd) would be at least in part reduced as Pdº and Niº. This would coincide with the XPS information because both bimetallic catalysts had Pdº and Niº. While bimetallic prepared from nitrate salt (PdNiN and NiNPd) will present β-PdH phase and NiOx surface species interacting with support. These results were corroborated by XPS. TEM micrographs of the catalysts are shown in Figure 4. Small spherical Pd particles with diameters in the range of 2-3.5 nm can be seen in the case of the monometallic catalyst. For all bimetallic catalysts an increase of the size was detected (2.5-4 nm) while the size distribution was less homogeneous. This would point to a higher dispersion of the monometallic catalyst.
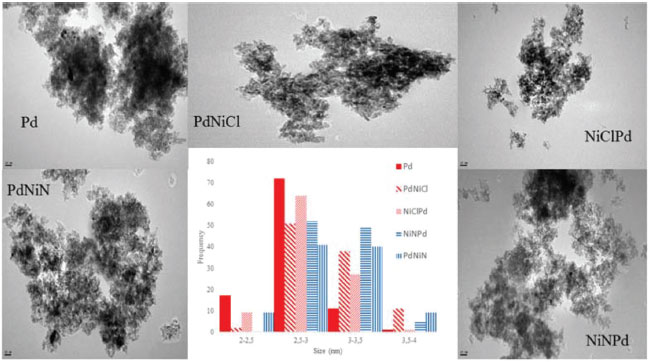 Figure 4. TEM results. Micrographs and size distribution plot
Catalytic Test In all the performed catalytic tests the selectivity to ethylbenzene was higher than 98%. Figure 5 shows values of total conversion as a function of time for the reaction with thiophene-free feed.
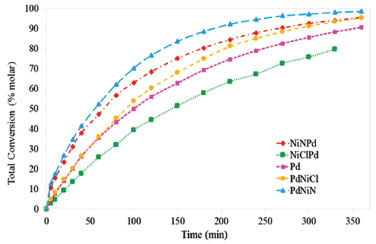 Figure 5. Total conversion of styrene as a function of time. Tests with thiophene-free feed
It can be seen that PdNiN and NiNPd were the most active materials, with conversion values higher than 90%, for reaction time values of 300 min. The ordering of the catalysts according to their activity was: PdNiN > NiNPd > PdNiCl > Pd > NiClPd. During the reaction of hydrogenation the dissociative adsorption of the H-H bond is favored by the interaction of the d orbitals of the metals rich in electrons with the antibonding molecular orbitals of hydrogen.33 The high conversion values of PdNiN and NiNPd could be due to the presence of Pdδ- with a higher availability of electrons (lower BE). The most active catalyst is PdNiN that has both Pdδ- and Pdº species, both with d10 orbitals that favor the dissociative adsorption of hydrogen. The most active of the bimetallic catalysts with chlorinated nickel precursor is PdNiCl, that has a high concentration of metallic palladium and has also metallic nickel (d10 and d8). Both favor the dissociative adsorption of hydrogen. The results of the tests with a sulfur doped feed can be seen in Figure 6. All catalysts show a decrease of the total conversion as a consequence of the loss of active sites. However no big differences between the different catalysts can be seen.
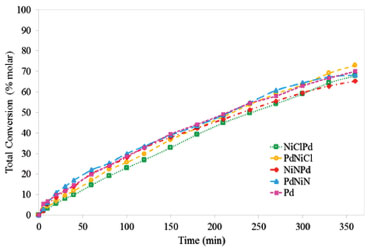 Figure 6. Total conversion of styrene as a function of time. Feed poisoned with thiophene
The resistance to sulfur poisoing was assessed by calculating the initial reaction rates for the reactions sulfur free (rºsf) and sulfur poisoned (rºsp). From the values of initial reaction rate and considering a simple model of linear deactivation (zero order deactivation), the fraction of poisoned sites (α) can be calculated as α = 1-rºsp / rºsf. Table 3 shows values of initial values of hydrogenation and fraction of poisoned sites (α). The found order of activity of the tests without poison was: PdNiN > NiNPd >> PdNiCl ≥ Pd >>NiClPd. In the presence of thiophene the found activity order was: PdNiN > NiNPd ≥ Pd > PdNiCl >> NiClPd. The order of the catalysts with respect to poisoning resistance was: PdNiN ≅ NiNPd << PdNiCl < Pd <NiClPd.
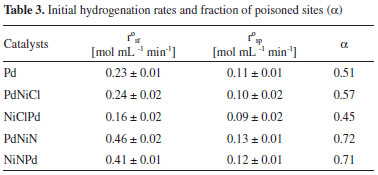
According to the most accepted model for the sulfur poisoning of Group VIII metals, poisoning occurs by a donation of electrons from the metal (Lewis basic site) to the sulfur atom (Lewis acid site).6 As seen in Table 3 the NiClPd catalyst has the lowest fraction of poisoned sites. Its high thioresistance would be due to the presence of Pdη+OxCly and NiO.xAl2O3 species that prevent the adsorption of thiophene either by steric hindrance and/or electronic effects (on Pdη+ and Ni2+, Lewis acid sites). Besides the lower thioresistance of PdNiN and NiNPd would be related to the amount of exposed Pdδ- species (Lewis basic sites) that promote a strong adsorption of the poison and thus an increase of the blocking of active sties. This would be due to the mentioned poisoning mechanism of Group VIII metals, i.e. electron donation from the metal to sulfur. Particularly thiophene would interact with the metal surface in a planar way through the electrons of the aromatic ring (weak h5 bond).6,7,34 In the case of the test reactions with thiophene some of the d electrons of Pd would be shared with sulfur atoms, thus decreasing the activity of the catalysts. Taking into account the chlorine present on the catalysts as detailed in Table 1 some other conclusions can be reached. Of the 4 bimetallic catalysts the two catalysts prepared with nitrate salt, PdNiN and NiNPd, were the least sulfur resistant. Both had a lower Cl/Pd surface ratio in comparison to the bimetallic catalysts prepared with a Ni chloride salt, NiClPd and PdNiCl. The presence of remaining chlorine would be a determining factor on the resistant to sulfided compounds and other poisons. In the case of the bimetallic catalysts prepared with Ni chloride, NiClPd and PdNiCl, it can be seen that the most thioresistant is NiClPd. This has a lower percentage of surface Pdº and Niº in comparison to the PdNiCl catalyst. Both species, Pdº and Niº, are prone to be poisoned because of their higher availability of electrons.
CONCLUSIONS The effect of the sequence of impregnation and of the kind of palladium and nickel precursor used was evaluated for a series of bimetallic Pd-Ni catalysts. The properties assessed were the catalytic activity and the sulfur resistance, during the selective hydrogenation of styrene. Excepting NiClPd, all the bimetallic catalysts were found to be more active than the Pd monometallic catalyst. The XPS results point to the presence of different Pdδ-, Pdº and Pdη+OxCly species. The presence of Pdδ- species would indicate the formation of a metallic bond or a Pd-Ni alloy. Species of Pd with high availability of electrons were found on the bimetallic catalysts suggesting that an electronic effect would be partly responsible for their higher conversion of styrene, either in the presence or the absence of thiophene. The order of hydrogenation rate of styrene in the absence of sulfur was: PdNiN > NiNPd >> PdNiCl ≥ Pd >>NiClPd. After the poisoning with 600 ppm thiophene the order changed to: PdNiN > NiNPd ≥ Pd > PdNiCl >>NiClPd. The bimetallic catalysts prepared by successive impregnation of NiCl2 and PdCl2 had a high surface content of chlorine. These catalysts were the most sulfur resistant because of steric and electronic effects. The great poisoning of PdNiN and NiNPd bimetallic catalysts would be due to the easy adsorption of the thiophene sulfur atoms over the relatively abundant Pdδ- species with high availability of electrons. Bimetallic PdNiN and NiNPd catalysts were the most active during hydrogenation of styrene in the absence of thiophene. A higher concentration of surface Pdδ- species on these catalysts would be responsible for the higher activity. PdNiCl and NiClPd had the highest Cl/Pd ratios and were also the most thioresistant. Oxychlorinated Pd species (Pdη+) probably prevented the adsorption of thiophene by means of steric hindrance (big size of Pd oxychloride species) and electronic effects (high electronegativity of Cl). NiClPd was the more sulfur resistant than PdNiCl. This could be due to a lower percentage of Pdº and Niº in comparison to PdNiCl, since both species (Pdº and Niº) are more prone to be poisoned due to their higher electronic availability.
ACKNOWLEDGEMENTS We thank CONICET, ANPCyT and UNL for the financial assistance of this work (Grants PIP 112-201101-00410 and 112-201301-00457, PICT and CAI + D).
REFERENCES 1. Hatch, L. F.; Matar, S.; From Hydrocarbon to Petrochemicals, Gulf Publishing Company: Houston, 1981, ch. 3 and 7. 2. Zhou, Z.; Zeng, T.; Cheng, Z.; Yuan, W.; Chem. Eng. Sci. 2010, 65, 1832. 3. Nijhuis, T. A.: Dautzenberg, F. M.; Moulijn, J. A.; Chem. Eng. Sci. 2003, 58, 1113. 4. Gaspar, A. B.; dos Santos, G. R.; de Souza Costa, R.; da Silva, M. A. P.; Catal. Today 2008, 133-135, 400. 5. Choi, J. S.; Mauge, F.; Pichon, C.; Fourcade, J. O.; Jumas, J. C.; Clair, C. P.; Uzio, D.; Appl. Catal., A 2004, 267, 203. 6. Badano, J.; Quiroga, M.; Betti, C.; Vera, C.; Canavese, S.; Coloma-Pascual, F.; Catal. Lett. 2010, 137, 35. 7. Betti, C.; Badano, J. M.; Maccarrone, M. J.; Mazzieri, V.; Vera, C.; Quiroga, M.; App. Catal., A 2012, 181-186, 435. 8. Betti, C.; Badano; J.; Rivas, I.; Mazzieri, V.; Maccarrone, M. J.; Vera, C.; Quiroga, M.; J. Chem. 2012, 8, 1. 9. Chen, H.; Li, T.; Jiang, F.; Wang, Z.; J. Mol. Catal. A: Chem. 2016, 421, 167. 10. NIST, X-ray Photoelectron Spectroscopy Database NIST Standard Reference Database 20, Version 3.5 (Web Version), National Institute of Standards and Technology, USA, 2007. 11. Park, K. W.; Choi, J. H.; Sung, Y. E.; J. Phys. Chem. B 2003, 107, 5851. 12. Abu Bakar, N. H. H.; Bettahar, M. M.; Abu Bakar, M.; Monteverdi, S.; Ismail, J.; Alnot, M.; J. Catal. 2009, 265, 63. 13. Wagner, C. D.; Riggs, W. M.; Davis, R. D.; Moulder, J. F.; Muilenberg, G. E.; Handbook of X-ray Photoelectron Spectroscopy Perkin-Elmer Corp., Physical Electronics Division, Eden Prairie, Minnesota, USA, 1979. 14. Castaño, P.; Pawelec, B.; Fierro, J. L. G.; Arandes, J. M.; Bilbao, J.; Fuel 2007, 86, 2262. 15. Salagre, P.; Fierro, J. L. G.; Medina, F.; Sueiras, J. E.; J. Mol. Catal. A: Chem. 1996, 106, 125. 16. Ivanova, A. S.; Slavinskaya, E. M.; Gulyaev, R. V.; Zaikovskii, V. I.; Stonkus, O. A.; Danilova, L. G.; Plyasova, L. M.; Polukhina, I. A.; Boronin, A. I.; Appl. Catal., B 2010, 97, 57. 17. Voogt, E. H.; Mens, A. J. M.; Gijzeman, O. L. J.; Geus, J. W.; Surf. Sci. 1996, 350, 21. 18. Beketov, G.; Heinrichs, B.; Pirard, J. P.; Chenakin, S.; Kruse, N.; Appl. Surf. Sci. 2013, 287, 293. 19. Hilli, Y.; Kinnunen, N. M.; Suvanto, M.; Savomaki, A.; Kallinen, K.; Pakkanen, T. A.; Appl. Catal., A 2015, 497, 85. 20. Huang, S.; Zhang, C.; He, H.; Catal. Today 2008. 139, 15. 21. Cobo, M.; Quintero, A.; Montes de Correa, C.; Catal. Today 2008, 133-135, 509. 22. Telkar, M. M.; Nadgeri, J. M.; Rode, C. V.; Chaudhari, R. V.; Appl. Catal., A 2005, 295, 23. 23. Heracleous, E.; Lee, A. F.; Wilson, K.; Lemonidou, A. A.; J. Catal. 2005, 231, 159. 24. Salagre, P.; Fierro, J. L. G.; Medina, F.; Sueiras, J. E.; J. Mol. Catal. A: Chem. 1996, 106, 125. 25. Sá, J.; Arteaga, G. D.; Daley, R. A.; Bernardi, J.; Anderson, J. A.; J. Phys. Chem. B 2006, 110, 17090. 26. Sales, E. A.; Jove, J.; De Jesús Mendez, M.; Bozon-Verduraz, F.; J. Catal. 2000, 195, 88. 27. Noronha, F. B.; Aranda, D. A. G.; Ordine, A. P.; Smachl, M.; Catal. Today 2000, 57, 275. 28. Pinna, F.; Menegazzo, F.; Signoretto, M.; Canton, P.; Fagherazzi, G.; Pernicone, N.; Appl. Catal., A 2001, 219, 195. 29. Kim, P.; Kim, H.; Joo, J. B.; Kim, W.; Song, I: K.; Yi, J.; J. Mol. Catal. A: Chem. 2006, 256, 178. 30. Hoffer, B. W.; Dick van Langeveld, A.; Janssens, J. P.; Bonne, R. L. C.; Lok, C. M.; Moulijn, J. A.; J. Catal. 2000, 192, 432. 31. Juan-Juan, J.; Roman-Martinez, M. C.; Illan-Gomez, M. J.; Appl. Catal., A 2006, 301, 9. 32. Hou, Z.; Yokota, O.; Tanaka, T.; Yashima, T.; Appl. Catal., A 2003, 253, 381. 33. Shriver, D. F.; Atkins, P. W.; Langford, C. H.; Inorganic Chemistry, 3rd ed., WH Freeman and Co: New York, 1994, p. 258. 34. Arcoya, A.; Seoane, X. L.; Gomez-Sainero, L. M.; Appl. Surf. Sci. 2003, 211, 341. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access