
 |
 |
 |
 |
 |
Artigo
| DFT study on the mechanistic, energetic and structural aspects of adsorption of tirapazamine onto pristine and functionalized carbon nanotubes |
|
Sadaf Avarand; Ali Morsali*,#; Mohammad M. Heravi; S. Ali Beyramabadi
Department of Chemistry, Mashhad Branch, Islamic Azad University, Mashhad, Iran & Research Center for Animal Development Applied Biology, Mashhad Branch, Islamic Azad University, Mashhad 917568, Iran Recebido em 16/08/2017 *e-mail: almorsali@yahoo.com; Using density functional theory, noncovalent interactions and two mechanisms of covalent functionalization of drug tirapazamine with pristine, COOH and COCl functionalized carbon nanotube (NT, NTCOOH and NTCOCl) have been investigated. Quantum molecular descriptors of noncovalent configurations were studied. It was specified that binding of drug tirapazamine with NTCOOH has more binding energy than NTCOCl and NT, so NTCOOH can act as a favorable system for tirapazamine drug delivery within biological and chemical systems (noncovalent). NTCOOH and NTCOCl can bond to the amino group of tirapazamine through OH (COOH mechanism) and Cl (COCl mechanism) groups, respectively. The activation parameters of two pathways were calculated and compared with each other. The activation parameters related to COOH mechanism are higher than those related to COCl mechanism and therefore COCl mechanism is suitable for covalent functionalization. These results could be generalized to other similar drugs. INTRODUCTION Carbon nanotubes (CNTs) enjoy unique electronic, mechanical, photonic and chemical properties,1-6 qualifying them to be used in biological and medical research.7-9. Much attention has focused on the functionalization of CNTs with DNA, polymers, peptides, and drugs.10,11 Conventional methods such as hydrogenation,12 cycloaddition,13 fluorination,14 ozonization15 and attachment of peptides, nucleic acids, polymers and drugs16,17 are used to functionalize CNTs. Also, the interaction of carbon nanotubes with organic and inorganic molecules has been extensively studied.18-22 Physical adsorption via π - stacking or van der Waals interaction does not cause any considerable modification in CNTs.23,24 On the other hand, covalent functionalization perturbs the physical and chemical properties of the nanotube but improves the solubility and compatibility of CNTs.25 Generally, different molecules can be functionalized onto CNTs by amidation or esterification reactions.26,27 One of the important methods used in the treatment of cancer is through chemotherapy, which is accompanied by different side effects such as vomiting, hair loss, cardio-toxicity and breathing difficulties in the patients. The higher the dose of the anti-cancer drug prescribed, the higher would be the degree of toxicity in the tissues.28,29 Due to having properties such as high drug loading capacities and good cell penetration qualities,30 CNTs are more suitable to be used for drug delivery than systems such as polymers, dendrimers, and liposomes, which are being used at present.31,32 Despite some disadvantages such as low solubility and that they will not be easily discharged from the body, there has been increasing interest during the past two decades for using CNTs in drug delivery.33-36 Using CNTs for drug delivery causes reduction in the dosage of the intended drug and consequently the reduction of its side effects.37,38 Covalent and noncovalent (hydrogen bonds and van der Waals interactions) functionalizations perform a principle role in the drug delivery systems. Quantum calculations could greatly assist in the design and analysis of drug delivery systems. The granting of Nobel Prize for chemistry in 2016 for the design and manufacturing of molecular machines, capable of being used in drug deliverance as well, confirms our statement.39-41 We have used quantum calculations for analysis of more stable structures and the mechanism of functionalization of the tirapazamine drug to CNTs. Tirapazamine (1,3-amino-1,2,4-benzotriazine 1,4-N,N-dioxide) has antitumor, antiviral and anticancer activities and is highly effective in the treatment of head, neck, Prostate and cervical cancers.42,43 Such calculations could encourage researchers in manufacturing new drug delivery systems.7,44 In spite of different theoretical studies on CNTs, so far few studies have been performed on the mechanism of functionalization.
METHODOLOGY B3LYP45-47 hybrid density functional level and 6-31G(d,p) basis sets in GAUSSIAN 09 package48 have been used for the optimization of all degrees of freedom for all geometries in solution phase. We repeated the calculations for noncovalent functionalization using M06-2X functional49,50 which implicitly accounts for "medium-range"electron correlation (systems separated by about 5Å or less),51 so M06-2X can describe the dispersion interactions within many systems.52 The solvent play a key role in chemical systems explicitly53-60 or implicitly. Polarized continuum model (PCM)61,62 was used for the consideration of implicit effects of the solvent. In the PCM method, the molecular cavity is made up of the union of interlocking atomic spheres. In this model, the molecular free energy is calculated as the sum over three components:  where Ges, Gdr and Gcav represent the electrostatic free energy, dispersion-repulsion free energy and cavitation free energy, respectively. The calculations were done on tirapazamine, COOH functionalized CNT (in water), COCl functionalized CNT (in DMF) and pristine armchair (5,5) Single-Walled CNT comprising 110 atoms (10 Å) with the ends terminated by hydrogen atoms. The transition state obtained was confirmed to have only one imaginary frequency of the Hessian. The zero-point corrections were also considered to obtain activation energy. In spite of high computational cost, approximation methods such as ONIOM63 have not been used.
RESULTS AND DISCUSSION Tirapazamine (TPZ) is a planar molecule with di-N-oxide and NH2 groups as presented in Figure 1. The optimized geometries of TPZ, COOH (NTCOOH), COCl (NTCOCl) functionalized and pristine (NT) Single-Walled CNT in solution phase have been shown in Figure 1. Use of the functionalized CNTs as well as the drugs having amino groups cause the increase in the solubility of carbon nanotubes.
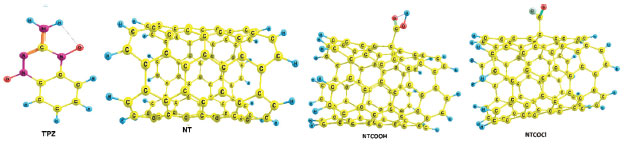 Figure 1. Optimized structures of TPZ, NT, NTCOOH and NTCOCl
Two configurations of noncovalent interactions of TPZ onto NT were studied. Figure 2 presents these modes in solution phase (water), namely, NT/TPZ1 (parallel) and NT/TPZ2 (perpendicular). The interaction between TPZ and NTCOOH or NTCOCl through amino group, forms hydrogen bonds. These reactants have been shown in Figure 3, namely, NTCOOH/TPZ and NTCOCl/TPZ (See supplementary material for Cartesian coordinates of the calculated structures).
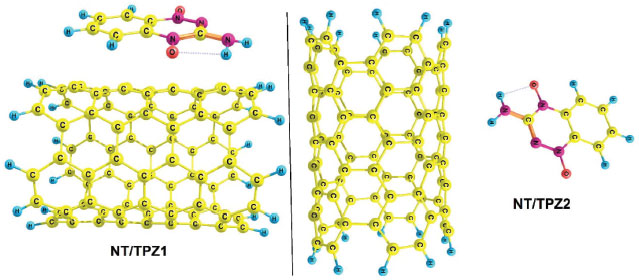 Figure 2. Optimized structures of NT/TPZ1 and NT/TPZ2
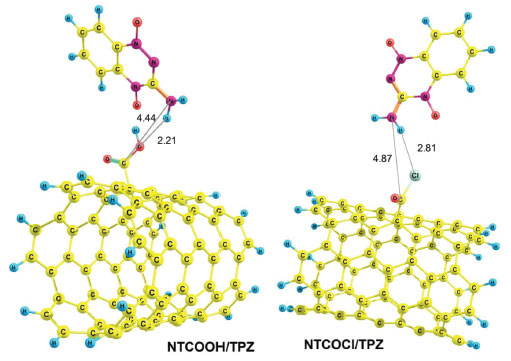 Figure 3. Optimized structures of reactants NTCOOH/TPZ and NTCOCl/TPZ
The binding energies (DE) of TPZ whit NT (in water), NTCOOH (in water) and NTCOCl (in DMF) were calculated using the following equation and presented in Tables 1 and 2: 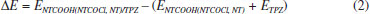
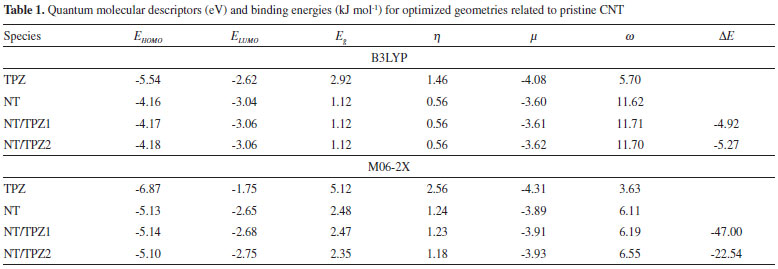
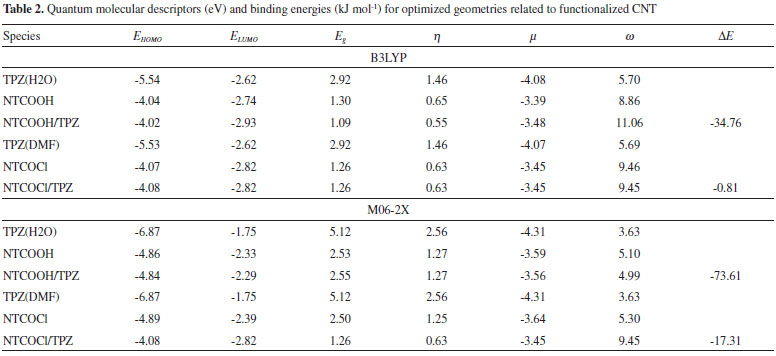
where E is electronic plus zero point energy. Using the calculated binding energies at B3LYP in Tables 1 and 2, this energy is negative for NTCOOH/TPZ (-34.76 kJ/mol) indicating TPZ is stabilized by NTCOOH surface but configurations NT/TPZ1, NT/TPZ2 and NTCOCl/TPZ have close to zero energies. We repeated the calculations using M06-2X functional to consider the contribution of dispersion interactions.51,52 The calculated binding energies related to M06-2X are more negative than those related to B3LYP. Among the two configurations of NT/TPZ1-2, the first one (parallel) has a higher stability. In parallel adsorption of TPZ onto pristine CNT, noncovalent interactions is dominated by delocalized π - π stacking between the aromatic rings of TPZ and NT in addition to interaction with di-N-oxide and NH2 groups of TPZ. Comparison between NTCOOH/TPZ and NTCOCl/TPZ shows that the first one is more stable due to the stronger hydrogen bond between TPZ and functionalized CNT. The M06-2X calculated binding energies are in good agreement with the literature.64-66 Quantum molecular descriptors such as hardness and electrophilicity index could be used to describe chemical reactivity and stability. The global hardness (η) indicates the resistance of one molecule against the change in its electronic structure (Equation 3). Decrease in η causes a decrease in the reactivity and an increase in stability.  where I = -EHOMO and A = -ELUMO are the ionization potential and the electron affinity of the molecule, respectively. Parr defined the electrophilicity index (ω) as follows:67  where chemical potential (µ) is:  Tables 1 and 2 present the quantum molecular descriptors for TPZ, NT (H2O), NT/TPZ1-2 (H2O), NTCOOH (H2O), NTCOCl (DMF), NTCOOH/TPZ (H2O) and NTCOCl/TPZ (DMF) using B3LYP and M06-2X functionals, respectively. In these tables, Eg (gap of energy between LUMO and HOMO) was also calculated. Eg notably determines a more stable system. According to the data in Tables 1 and 2, h and Eg related to the TPZ drug are higher than those of NT, NTCOOH/TPZ and NTCOCl/TPZ, showing the reactivity of TPZ increases in the presence of pristine SWCNT and COOH (COCl) functionalized SWCNT. Also, it is specified that µ of the TPZ becomes more positive in the vicinity of NT, NTCOOH and NTCOCl. w of TPZ increases in the presence of pristine SWCNT and COOH (COCl) functionalized SWCNT, showing that TPZ acts as electron acceptor. The η and Eg values in NT/TPZ and NTCOOH/TPZ are higher than NTCOCl/TPZ, suggesting NTCOCl/TPZ to be more reactive (less stable) compared to NT/TPZ and NTCOOH/TPZ. Also, in confirmation of the previous issue, the chemical potentials of the NTCOCl/TPZ are higher than those of NT/TPZ and NTCOOH/TPZ for both B3LYP and M06-2X functionals. The similar results have been obtained in other CNT/drug systems.64,68,69 Generally, for noncovalent interactions, comparison between pristine, COOH and COCl functionalized single wall carbon nanotubes shows that using the second one is more suitable due to a stronger interaction between TPZ and functionalized SWCNT and more stability. For the covalent functionalization, amino group attacks the carbon atom of COOH or COCl to transfer its proton to the OH (Cl) group. We studied these mechanisms for NTCOOH(Cl)/ TPZ. Scheme 1 shows the mechanism for the formation of covalent bond between TPZ and NTCOOH (COOH mechanism) where k1 and K1 are rate constant and equilibrium constant, respectively. In this mechanism, NTCOOH/TPZ is converted into the product NTCOTPZ/H2O. The optimized structure of product NTCOTPZ/H2O has been shown in Figure 4.
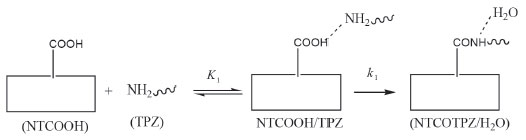 Scheme 1. COOH Mechanism of covalent functionalization
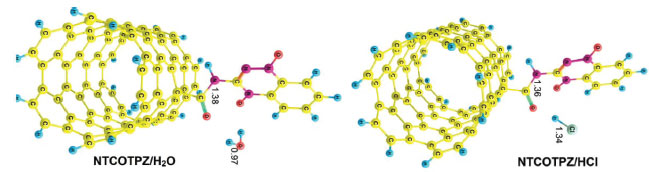 Figure 4. Optimized structures of reactants NTCOTPZ/H2O and NTCOTPZ/HCl
Using reactant NTCOOH/TPZ and product NTCOTPZ/H2O, the transition state of k1 step was optimized which we call TSk1 (Figure 5). Considering Figures 3-5, O-H and C-N bond lengths decrease (increase) from 2.21 Å and 4.44 Å (0.97 Å and 1.38 Å) for NTCOOH/ TPZ (NTCOTPZ/H2O) to 1.14 Å and 1.55Å for TSk1, respectively.
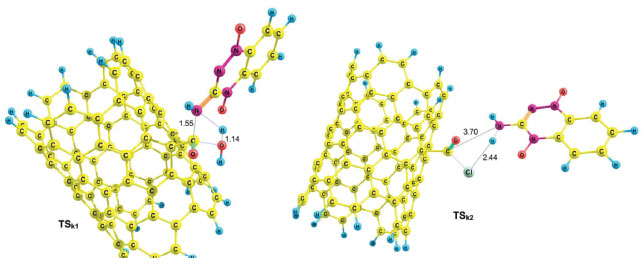 Figure 5. Optimized structures of reactants TSk1 and TSk2
Relative energies for optimized structures in all pathways have been calculated in Table 3 by considering electronic plus zero point energy (E), enthalpy (H) and Gibbs free energy (G) of reactants (NTCOOH+TPZ) equal to zero (See supplementary material for absolute energies). The activation energy (Ea), activation enthalpy (ΔH‡) and activation Gibbs free energy (ΔG‡) for k1 step are 185.68 kJ mol-1, 185.32 kJ mol-1 and 198.80 kJ mol-1, respectively (Table 3). The total rate constant for overall reaction (COOH pathway) is equal to k1 × K1, so the total activation energy (Ea(COOH path) = Ea(k1 step) + ΔE(K1 step)), the total activation enthalpy (ΔH‡(COOH path) = ΔH‡(k1 step) + ΔH(K1 step)) and total activation Gibbs free energy (ΔG‡(COOH path) = ΔG‡(k1 step) + ΔG(K1 step)) for COOH mechanism are 150.93 kJ mol-1, 151.70 kJ mol-1 and 206.49 kJ mol-1, respectively (Table 3). In room temperature, the energy barrier related to COOH mechanism is too high to occur.
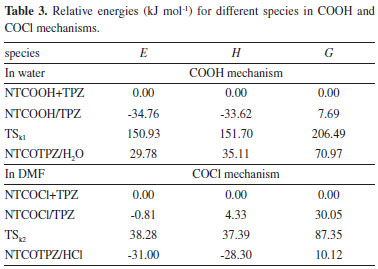
The other reaction for the covalent functionalization of TPZ onto COCl functionalized carbon nanotube is shown in Scheme 2 (COCl mechanism).70 In this reaction, NTCOOH was firstly converted into alkyl chloride using SOCl2 (NTCOCl). TPZ then reacts with NTCOCl to form covalent bond. COCl mechanism begins with the attack of amino group of TPZ to Cl in the NTCOCl to form product NTCOTPZ/HCl (Figure 4).
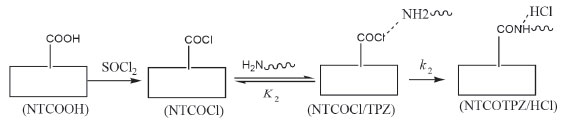 Scheme 2. COCl Mechanism of covalent functionalization
Using NTCOCl/TPZ and NTCOTPZ/HCl, a transition state is optimized which we call TSk2 (Figure 5). Considering Figures 3-5, Cl-H and C-N bond lengths decrease (increase) from 2.81 Å and 4.87 Å (1.34 Å and 1.36 Å) for NTCOCl/TPZ (NTCOTPZ/HCl) to 2.44 Å and 3.70 Å for TSk2, respectively. Ea, ΔH‡ and ΔG‡ for k2 step are 39.10 kJ mol-1, 33.06 kJ mol-1 and 57.30 kJ mol-1, respectively (Table 3). The total activation energy, the total activation enthalpy and the total activation Gibbs free energy for COCl mechanism are 38.28 kJ mol-1, 37.39 kJ mol-1 and 87.35 kJ mol-1, respectively (Table 3). The total activation energy, enthalpy and Gibbs free energy for COCl mechanism are lower than COOH mechanisms by 112.65 kJ mol-1, 114.31 kJ mol-1 and 119.14 kJ mol-1, respectively. Considering activation parameters, calculations suggest that covalent functionalization can take place at room temperature through COCl mechanism.
CONCLUSIONS Possible configurations of noncovalent interactions of drug tirapazamine (TPZ) onto NT, NTCOOH and NTCOCl were studied. In NT/TPZ systems, the parallel configuration is preferred over the perpendicular orientation due to enhanced π - π stacking interaction, where di-N-oxide and NH2 groups of TPZ are in close proximity to the NT. The binding energy related to NTCOCl is lower than that related to NTCOOH, indicating NTCOOH/TPZ configuration is stabilized. The global hardness and HOMO-LUMO energy gap of NT/TPZ1-2 and NTCOOH/TPZ configuration are higher than that of NTCOCl/TPZ configuration, showing the reactivity of the tirapazamine increases in the presence of NTCOCl and its stability decreases. Mechanisms of covalent functionalization of tirapazamine onto NTCOOH (COOH mechanism) and NTCOCl (COCl mechanism) have been investigated in detail. The activation parameters related to COOH mechanism are higher than those related to COCl mechanism. Therefore, for covalent functionalization, COCl functionalized single wall carbon nanotube is more suitable.
ACKNOWLEDGEMENTS We thank the Research Center for Animal Development Applied Biology for allocation of computer time.
SUPPLEMENTARY MATERIAL Cartesian coordinates, sum of electronic and zero-point energies (E), sum of electronic and thermal enthalpies (H) and Gibbs free energies (G) for the calculated structures are available at http://www.quimicanova.sbq.org.br in PDF format with free access.
REFERENCES 1. Lavall, R. L.; Sales, J.; Borges, R. S.; Calado, H. D.; Machado, J. C.; Windmoller, D.; Silva, G. G.; Lacerda, R. G.; Ladeira, L. O.; Quim. Nova 2010, 33, 133. 2. Lai, P.; Chen, S.; Lin, M.-F.; Physica E 2008, 40, 2056. 3. De Volder, M. F.; Tawfick, S. H.; Baughman, R. H.; Hart, A. J.; Science 2013, 339, 535. 4. de Souza Filho, A. G.; Fagan, S. B.; Quim. Nova 2007, 30, 1695. 5. Maiti, U. N.; Lee, W. J.; Lee, J. M.; Oh, Y.; Kim, J. Y.; Kim, J. E.; Shim, J.; Han, T. H.; Kim, S. O.; Adv. Mater. 2014, 26, 40. 6. Pimenta, M.; Gomes, A.; Fantini, C.; Cançado, L.; Araujo, P.; Maciel, I.; Santos, A.; Furtado, C.; Peressinotto, V.; Plentz, F.; Physica E 2007, 37, 88. 7. Rungnim, C.; Arsawang, U.; Rungrotmongkol, T.; Hannongbua, S.; Chem. Phys. Lett. 2012, 550, 99. 8. Adeli, M.; Soleyman, R.; Beiranvand, Z.; Madani, F.; Chem. Soc. Rev. 2013, 42, 5231. 9. Mundra, R. V.; Wu, X.; Sauer, J.; Dordick, J. S.; Kane, R. S.; Curr. Opin. Biotechnol. 2014, 28, 25. 10. Hirsch, A.; Angew. Chem., Int. Ed. 2002, 41, 1853. 11. Chen, R. J.; Zhang, Y.; Wang, D.; Dai, H.; J. Am. Chem. Soc. 2001, 123, 3838. 12. Nikitin, A.; Ogasawara, H.; Mann, D.; Denecke, R.; Zhang, Z.; Dai, H.; Cho, K.; Nilsson, A.; Phys. Rev. Lett. 2005, 95, 225507. 13. Georgakilas, V.; Tagmatarchis, N.; Pantarotto, D.; Bianco, A.; Briand, J.-P.; Prato, M.; Chem. Commun. 2002, 3050. 14. Khabashesku, V. N.; Billups, W. E.; Margrave, J. L.; Acc. Chem. Res. 2002, 35, 1087. 15. Banerjee, S.; Wong, S. S.; J. Phys. Chem. B 2002, 106, 12144. 16. Liu, Z.; Cai, W.; He, L.; Nakayama, N.; Chen, K.; Sun, X.; Chen, X.; Dai, H.; Nat. Nanotechnol. 2007, 2, 47. 17. Lacerda, L.; Bianco, A.; Prato, M.; Kostarelos, K.; Adv. Drug Deliv. Rev. 2006, 58, 1460. 18. Kalinke, A. H.; Zarbin, A. J.; Quim. Nova 2014, 37, S1. 19. Lin, Y.; Allard, L. F.; Sun, Y.-P.; J. Phys. Chem. B 2004, 108, 3760. 20. Chang, C. M.; Tseng, H. L.; de Leon, A.; Posada-Amarillas, A.; Jalbout, A. F.; J. Comput. Theor. Nanosci. 2013, 10, 521. 21. Silva, I. R. d.; Barreto, P. L.; Bellettini, I. C.; Quim. Nova 2013, 36, 5. 22. Ji, X.-T.; Ge, Z.-X.; Bu, J.-H.; Liu, Q.; Wang, W.; Xu, C.; Chin. Chem. Lett. 2014, 25, 292. 23. Georgakilas, V.; Kordatos, K.; Prato, M.; Guldi, D. M.; Holzinger, M.; Hirsch, A.; J. Am. Chem. Soc. 2002, 124, 760. 24. Chen, J.; Liu, H.; Weimer, W. A.; Halls, M. D.; Waldeck, D. H.; Walker, G. C.; J. Am. Chem. Soc. 2002, 124, 9034. 25. Pompeo, F.; Resasco, D. E.; Nano Lett. 2002, 2, 369. 26. Basiuk, V. A.; Nano Lett. 2002, 2, 835. 27. Huang, W.; Taylor, S.; Fu, K.; Lin, Y.; Zhang, D.; Hanks, T. W.; Rao, A. M.; Sun, Y.-P.; Nano Lett. 2002, 2, 311. 28. Pennock, G. D.; Dalton, W. S.; Roeske, W. R.; Appleton, C. P.; Mosley, K.; Plezia, P.; Miller, T. P.; Salmon, S. E.; J. Natl. Cancer Inst. 1991, 83, 105. 29. Lindley, C.; McCune, J. S.; Thomason, T. E.; Lauder, D.; Sauls, A.; Adkins, S.; Sawyer, W. T.; Cancer Pract. 1999, 7, 59. 30. Prato, M.; Kostarelos, K.; Bianco, A.; Acc. Chem. Res. 2007, 41, 60. 31. Allen, T. M.; Cullis, P. R.; Science 2004, 303, 1818. 32. Tomalia, D.; Reyna, L.; Svenson, S.; Biochem. Soc. Trans. 2007, 35, 61. 33. Karimi, M.; Solati, N.; Ghasemi, A.; Estiar, M. A.; Hashemkhani, M.; Kiani, P.; Mohamed, E.; Saeidi, A.; Taheri, M.; Avci, P.; Expert. Opin. Drug Deliv. 2015, 12, 1089. 34. Franchi, L. P.; Santos, R. A.; Matsubara, E. Y.; de Lima, J. C.; Rosolen, J. M.; Takahashi, C. S.; Quim. Nova 2012, 35, 571. 35. Zhang, H.; Hou, L.; Jiao, X.; Ji, Y.; Zhu, X.; Li, H.; Chen, X.; Ren, J.; Xia, Y.; Zhang, Z.; Curr. Pharm. Biotechnol. 2014, 14, 1105. 36. Unnati, S.; Shah, R.; Int. J. Pharm. Technol. 2011, 3, 927. 37. Flahaut, E.; Toxicity and Environmental Impact of Carbon Nanotubes. Carbon Nanotubes for Biomedical Applications, Springer: Berlin, 2011. 38. Ajima, K.; Yudasaka, M.; Murakami, T.; Maigné, A.; Shiba, K.; Iijima, S.; Mol. Pharm. 2005, 2, 475. 39. Zheng, Y. B.; Kiraly, B.; Huang, T. J.; Nanomedicine 2010, 5, 1309. 40. Linko, V.; Ora, A.; Kostiainen, M. A.; Trends Biotechnol. 2015, 33, 586. 41. Szymański, W.; Beierle, J. M.; Kistemaker, H. A.; Velema, W. A.; Feringa, B. L.; Chem. Rev. 2013, 113, 6114. 42. Denny, W. A.; Curr. Med. Chem. Anticancer Agents 2004, 4, 395. 43. Zeman, E. M.; Brown, J. M.; Lemmon, M. J.; Hirst, V. K.; Lee, W. W.; Int. J. Radiat. Oncol. Biol. Phys. 1986, 12, 1239. 44. Rungnim, C.; Rungrotmongkol, T.; Hannongbua, S.; Okumura, H.; J. Mol. Graphics Modell. 2013, 39, 183. 45. Becke, A. D.; Phys. Rev. A 1988, 38, 3098. 46. Becke, A. D.; J. Chem. Phys. 1993, 98, 5648. 47. Lee, C.; Yang, W.; Parr, R. G.; Phys. Rev. B 1988, 37, 785. 48. Gaussian 09; Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Montgomery, Jr., J. A.; Vreven, T.; Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G. A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Gonzalez, C.; Pople, J. A.; Gaussian, Inc., Wallingford CT, 2009. 49. Zhao, Y.; Schultz, N. E.; Truhlar, D. G.; J. Chem. Theory Comput. 2006, 2, 364. 50. Zhao, Y.; Truhlar, D. G.; J. Chem. Phys. 2006, 125, 194101. 51. Zhao, Y.; Truhlar, D. G.; J. Chem. Theory Comput. 2007, 3, 289. 52. Zhao, Y.; Truhlar, D. G.; Theor. Chem. Acc. 2008, 120, 215. 53. Hooman Vahidi, S.; Morsali, A.; Ali Beyramabadi, S.; Comp. Theor. Chem. 2012, 994, 41. 54. Akbari, A.; Hoseinzade, F.; Morsali, A.; Ali Beyramabadi, S.; Inorg. Chim. Acta 2013, 394, 423. 55. Morsali, A.; Hoseinzade, F.; Akbari, A.; Beyramabadi, S. A.; Ghiasi, R.; J. Solution Chem. 2013, 42, 1902. 56. Mohseni, S.; Bakavoli, M.; Morsali, A.; Prog. React Kinet. Mec. 2014, 39, 89. 57. Beyramabadi, S. A.; Eshtiagh-Hosseini, H.; Housaindokht, M. R.; Morsali, A.; Organometallics 2007, 27, 72. 58. Gharib, A.; Morsali, A.; Beyramabadi, S.; Chegini, H.; Ardabili, M. N.; Prog. React Kinet. Mec. 2014, 39, 354. 59. Morsali, A.; Int. J. Chem. Kinet. 2015, 47, 73. 60. Ardabili, M. N.; Morsali, A.; Beyramabadi, S. A.; Chegini, H.; Gharib, A.; Res. Chem. Intermed. 2015, 41, 5389. 61. Cammi, R.; Tomasi, J.; J. Comput. Chem. 1995, 16, 1449. 62. Tomasi, J.; Persico, M.; Chem. Rev. 1994, 94, 2027. 63. Dapprich, S.; Komáromi, I.; Byun, K. S.; Morokuma, K.; Frisch, M. J.; J. Mol. Struc.: THEOCHEM 1999, 461, 1. 64. Saikia, N.; Deka, R. C.; J. Mol. Model. 2013, 19, 215. 65. Rajarajeswari, M.; Iyakutti, K.; Kawazoe, Y.; J. Mol. Model. 2012, 18, 771. 66. Das, A.; Sood, A.; Maiti, P. K.; Das, M.; Varadarajan, R.; Rao, C.; Chem. Phys. Lett. 2008, 453, 266. 67. Parr, R. G.; Szentpaly, L. v.; Liu, S.; J. Am. Chem. Soc. 1999, 121, 1922. 68. Saikia, N.; Deka, R. C.; Chem. Phys. Lett. 2010, 500, 65. 69. Raissi, H.; Mollania, F.; Eur. J. Pharm. Sci. 2014, 56, 37. 70. Lin, T.; Bajpai, V.; Ji, T.; Dai, L.; Aust. J. Chem. 2003, 56, 635. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access