
 |
 |
 |
 |
 |
Artigo
| KINETIC BEHAVIOR OF SOME AZO DYES DECOLORIZATION BY VARIATION OF ZINC OXIDE AND TITANIUM DIOXIDE CONCENTRATIONS |
|
Wallace J. C. da Silva; Natalia M. Monezi; Vanildo S. Leao Neto; Keiko Takashima*
Departamento de Química, Universidade Estadual de Londrina, 86057-970 Londrina - PR, Brasil Recebido em 04/08/2017 *e-mail: keiko.takashima@gmail.com The decolorization of three monoazo dyes (acid orange 7, direct orange 34, and methyl orange), one diazo dye (direct yellow 86) and one tetraazo dye (direct red 80) were mediated by n-type semiconductors as ZnO and TiO2 under pseudo-first order conditions at 30 ºC. The decolorization rate constants of these azo dyes were determined, varying the semiconductor concentration for the majority of them from 1.0 to 10.0 g L-1. In general, the highest rate constants were displayed for ZnO. This work elucidates that the decolorization capacity depends on the charge, structure, and adsorption of the azo dye on the semiconductor surface as well as the agglomeration of the photocatalyst particles. INTRODUCTION Azo dyes are an important class of synthetic dyes, characterized by the presence of one or more azo groups (-N=N-), bonded to a benzene or a naphthalene group. They represent about 50% of worldwide dye production and are widely used in a number of industries such as textile, paint, cosmetic, food, and plastic, with the textile industry being the largest consumer.1 Large amounts of these dyes remain in the effluent after the completion of the dyeing process. This leads to severe contamination of an aquatic system, because many of these compounds are considered carcinogenic and can produce toxic aromatic amines.2 Since these dyes are highly soluble in water, their removal from wastewater is difficult by conventional methods such as biodegradation, microfiltration, reverse osmosis and activated carbon adsorption.3-5 Heterogeneous photocatalysis, mediated by an n-type semiconductor as TiO2 and ZnO, has been considered as a cost effective alternative for the purification of dye containing wastewater.6-8 In this process, the semiconductor absorbs radiation with higher energy than band-gap, promoting the valence band electrons to the conduction band, creating an electron-hole pair. The valence band hole can either receive an electron from the adsorbed dye or oxidize the adsorbed water or a hydroxyl ion in order to convert it to hydroxyl radical, •OH. This radical is considered non-selective, presents a short lifetime and high oxidation potential (2.8 V) that can mineralize organic substances. On the other hand, the electrons promoted to the conduction band are transferred to the adsorbed oxygen molecules to convert it into ion-radical superoxide, •O2, When it is protonated, generates hydrogen peroxide and contributes to the contaminant oxidation.9 The optimization of the photocatalytic process for several organic contaminants degradation has been carried out, varying the semiconductor concentration, pH, oxidants addition such as peroxides, persulfates, among others.10-14 In general, the degradation rate constant of the contaminants increases with TiO2 concentration in heterogeneous photocatalysis. So, it has been considered that the larger the number of active sites to dye adsorption, as well the interaction with electron-hole pairs, the larger the degradation efficiency will be. Thus, the TiO2 oxidation efficiency for a fixed substrate concentration is larger, the larger the number of semiconductor particles. Conversely, when this concentration is continuously increased, the distance between particles decreases and the suspension becomes opaque. This makes the light passage difficult and decreases the decolorization rate constant.15-18 On the other hand, the decolorization rate constant of direct red 23 diazo dye enhanced when the semiconductor concentration was increased from 1.0 to 7.0 g L-1 giving a positive inflection in 5.0 g L-1, mediated by zinc oxide at 30 ºC.19 In general, the rate constant of the azo dye decolorization increases until a certain TiO2 concentration and decreases due to the agglomeration of these particles, hindering the radiation passage. Considering the results to the DR23 decolorization in ZnO medium19 and the difference with respect to TiO2,20-22 we investigated the unusual kinetic behavior in ZnO based on photocatalysis by comparing the decolorization kinetic using ZnO and TiO2 in a wide range of concentrations. Thus, we studied the decolorization of three monoazo dyes (acid orange 7, direct orange 34, and methyl orange), one diazo dye (direct yellow 86) and one tetraazo dye (direct red 80) (Figure 1) varying the concentration of ZnO and TiO2 at 30 ºC.
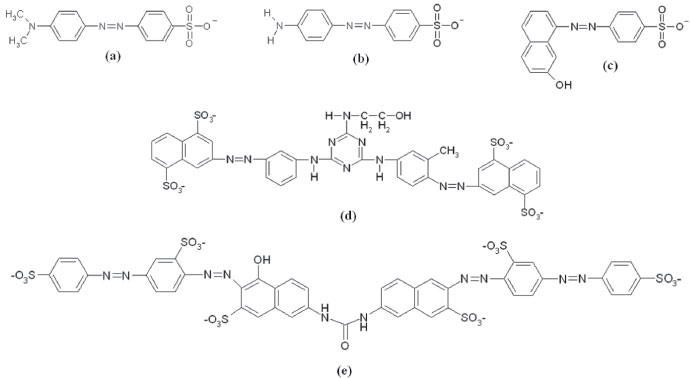 Figure 1. Structural formula of the azo dyes (a) direct orange 34, (b) methyl orange, (c) acid orange 7, (d) direct yellow 86, and (e) direct red 80
EXPERIMENTAL SECTION The monoazo dyes acid orange 7 (AO7; C.I. 15510; ε = 2.46 × 104 cm-1 mol-1 L at 483 nm) and direct orange 34 (DO34; C.I. 40215; ε = 2.60 × 104 cm-1 mol-1 L at 415 nm), direct yellow 86 diazo dye (DY86; C.I. 29325; ε = 8.51 × 104 cm-1 mol‑1 L at 370 nm) and direct red 80 tetraazo dye (DR80; C.I. 35780; ε = 2.70 × 104 cm-1 mol-1 L at 512 nm) were attained from Chimical and the methyl orange monoazo dye was purchased from Reagen (MO; CI 13025; ε = 3.15 × 104 cm-1 mol-1 L at 462 nm). The molar absorption coefficient, ε, of the azo dyes was determined experimentally and the initial concentrations were 8.0 × 10-5 mol L-1 (AO7), 2.5 × 10-5 mol L-1 (DO34), 8.0 × 10-5 mol L-1 (MO), 7.0 × 10-5 mol L-1 (DY86), and 8.0 × 10-5 mol L-1 (DR80). The initial concentration of each azo dye was determined from the molar absorption coefficient to get the initial absorbance in approximately 2.0. The adsorption and irradiation of the suspension were performed at natural pH inside a wood chamber (50 × 50 × 50 cm), covered internally with an aluminum foil and frontal part closed with a black fabric curtain, using a borosilicate glass reactor (200 mL) with a double wall for water circulation at 30.0 ± 0.1 ºC (Quimis Q214M2). The adsorption equilibrium of the azo dye in the suspension, formed by ZnO (Nuclear, PA) or TiO2 P25 (Degussa, PA) in a volume of 150 mL, was obtained leaving it in the dark in a variable time under constant stirring (600 rpm, Fisatom) at 30.0 ± 0.1 ºC (Quimis Q214M2). Aliquots of 1.0 mL were removed at predetermined times, filtered (Millipore, 0.22 µm) and UV-Vis spectra were registered (Hitachi U-3000) until the absorbance remains constant. Subsequently, Hg vapor lamp without bulb (Osram 125 W, 220 V) horizontally positioned 15 cm above the glass reactor was lighted and 1.0 mL aliquots were withdrawn as a function of irradiation time, filtered, and spectra were registered to determine the decolorization rate constant. The azo dyes photolysis was not carried out, due to their high stability under UV radiation. The decolorization rate constants, kobs, were determined under pseudo-first order conditions at 30 ºC, that is, using a large catalyst excess with respect to the azo dye, from lnAt= -kobst + lnA0, where At is the absorbance at t time and A0 at zero time.23 The decolorization of the chromophore groups and degradation of aromatic groups occurred simultaneously by the decrease of the UV-Vis bands relative to these two chemical groups. In addition, semi-empirical calculations were performed for DR80 dye using the Spartan software through the Hartree-Fock theoretical method and basis set 3-21G*,24 in order to obtain the dipole moment value and atomic charge. The characterization of ZnO (Nuclear) was carried out previously25 by X-ray diffraction (XRD), diffuse reflectance (DR) and Energy Dispersive X-Ray Diffraction (EDXRF). XRD showed a wurtzite crystalline structure; the band-gap energy of 3.16 eV was determined by DR using the Tauc model, and the purity, verified by EDX-RF. Zielinska et al. characterized TiO2 (P25, Degussa) by X-ray diffraction (XRD), diffuse reflectance (DR) and other techniques. XRD showed peaks of anatase and rutile, in which the anatase peaks presented more intensity, the band-gap energy of 3.14 eV was determined by DR according to the equation Eg = h c / λ.26
RESULTS AND DISCUSSION Three monoazo dyes were more susceptible to the oxidation by ZnO in comparison to TiO2. From these, AO7 (8.0 × 10-5 mol L-1) was more reactive in both semiconductors, followed by DO34 (2.5 × 10-5 mol L-1) and lastly, MO (8.0 × 10-5 mol L-1) as shown in Figure 2. DO34 decolorization rate constant enhanced linearly around six times (correlation coefficient, r = 0.976) from 1.65 × 10-2 to 9.36 × 10-2 min-1, when the concentration of ZnO was varied from 1.0 to 10.0 g L-1 (Figure 2a). Similarly, kobs of MO doubled from 3.15 × 10-2 to 6.36 × 10-2 min-1 (r = 0.983) (Figure 2b). This means that the reaction orders for ZnO varying from 1.0 to 10.0 g L-1 were equivalent to 0.7 and 0.3 respectively in the decolorization of DO34 (2.5 × 10-5 mol L-1) and MO (8.0 × 10-5 mol L-1) monoazo dyes. The structures of both dyes are distinguished by the functional group bonded to the ring, that is, amino group in DO34 and dimethylamino in MO (Figures 1a and 1b), suggesting a better adsorption affinity to DO34. This explains the higher reaction order in the presence of DO34 solution and consequently the larger rate constant enhancement, when mediated by ZnO. Conversely, the reaction order resulted in 0.6 when ZnO concentration was increased from 1.0 to 3.0 g L-1 in AO7 solution, enhancing kobs from 5.61 × 10-2 to 8.11 × 10-2 min-1. The observed rate constant continued to increase linearly (r = 0.978) from 3.0 to 10.0 g L-1 of ZnO with a less pronounced slope, giving a reaction order of 0.1 as shown in Figure 2c. So, the largest decolorization rate constant obtained for AO7 is attributed to the naphthalene group (Figure 1c), which could delocalize a reasonable amount of negative charge density, becoming easier the approximation on ZnO or TiO2 surface.
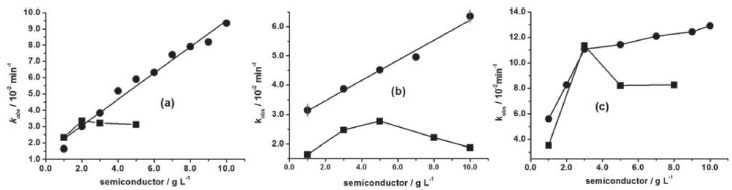 Figure 2. Decolorization rate constant, kobs, of (a) DO34 (2.5×10-5 mol L-1); (b) MO (8.0×10-5 mol L-1); (c) AO7 (8.0×10-5 mol L-1) as a function of ZnO (●) and TiO2 (■) at 30 ºC
Otherwise, when 1.0 and 2.0 g L-1 of TiO2 were used in DO34 solutions, the rate constants were equal to 2.32 × 10-2 and 3.36 × 10-2 min-1, whose values were larger with respect to ZnO medium (Figure 2). This was attributed to the larger DO34 adsorption on TiO2 surface than on ZnO, since the initial DO34 absorbance (2.5 × 10-5 mol L-1) decreased by 1.25 for TiO2 and 0.37 for ZnO after 30 min in the dark at 30 ºC. When 3.0 and 4.0 g L-1 of TiO2 were used, there was a kobs decrease trend from 3.22 × 10-2 to 3.13 × 10-2 min-1 (Figure 2a), ascribed to the obstruction of the radiation passage, caused by the agglomeration of the semiconductor particles.27 Similarly, kobs values of MO on TiO2 (Figure 2b) were lower than on ZnO, which presented analogous behavior of DO34 in TiO2 one, that is, from 1.64 × 10-2 min-1 in 1.0 g L-1 to 2.78 × 10-2 min-1 in 5.0 g L-1 and declined to 1.88 × 10-2 min-1 in 10.0 g L-1. By the same reason, this low reactivity of MO was attributed to the lower partial negative charge with respect to DO34 as mentioned above. Moreover, when TiO2 concentration was varied from 1.0 to 3.0 g L-1 the rate constant for AO7 increased three times from 3.52 × 10-2 to 11.36 × 10-2 min-1. In 3.0 g L-1 TiO2 kobs was very similar to ZnO, however, when the concentration was increased to 5.0 and 8.0 g L-1the rate constant decreased to about 8.2 × 10-2 min-1 ascribed, in this case, to the obstruction of the passage of light by the suspension. There are very few researches about the kinetic behavior of azo dyes decolorization varying the semiconductor concentrations. Danwittayakul, Jaisai, Koottatep, and Dutta28 investigated the MO decolorization of 32% after 3h UV irradiation in ZnO nanorods on polyester fiber. Alzahrani29 studied the decolorization of methyl orange (100 mg L-1) in a ZnO nanopowder (0.5 g L-1) at 25 ºC obtaining a rate constant of 2.4 × 10-3 min-1 (R2 = 0.977). Hamadanian, Sarabi, Mehra, and Jabbari30 worked with TiO2 (1.0 g L-1) doped with several Cr3+ concentrations. From these, the highest photoactivity was obtained when it was used 5% Cr3+. Daneshvar, Rasoulifard, Khataee, and Hosseinzadeh31 investigated AO7 (0.02 g L-1) decolorization in ZnO medium, varying the catalyst concentration from 0.1 to 0.2 g L-1 and measuring the decolorization percentage with no control of temperature. Zhang, Sunb, Li, and Wang32 decolorized AO7 (0.1 g L-1) in a suspension containing TiO2 0.5 g L-1 and calculated the rate constant (2.12 x 10-2 min−1) at 38 ± 1 ºC. There are few papers about the DO34 decolorization in ZnO and TiO2 media. Among them, Niehues, Scarmínio, and Takashima33 investigated the DO34 decolorization percentage in TiO2 medium at 30 ºC after 240 min UV irradiation by the 25 factorial design. Unlike monoazo dyes, the DY86 diazo dye decolorization was slower in ZnO in comparison to TiO2. For DY86, the adsorption was systematically lower in ZnO. For example, the absorbance of DY86 (7.0 × 10-5 mol L-1) decreased by 0.223 in ZnO 5.0 g L-1, whereas in TiO2 by 2.095 after 30 min adsorption in the dark at 30 ºC. Figure 3 shows that the kobs for DY86 increased gradually from ZnO 1.0 g L-1(1.85 × 10-2 min-1) to 14.0 g L-1 (3.51 × 10-2 min-1). On the other hand, kobs increased from 1.85 × 10-2 min-1 (1.0 g L-1) to 8.57 × 10-2 min-1 (5.0 g L-1) in TiO2 medium and it was three times greater than ZnO (2.56 × 10-2 min-1) in the same concentration. This kinetic behavior was attributed to the high adsorption capacity of this azo dye in TiO2. Nevertheless, in TiO2 8.0 g L-1, kobs decreased to 4.45 × 10-2 min-1, attributed to the obstruction of radiation passage through the suspension, as above mentioned. Zhang et al. reported the only work about DY86 decolorization in TiO2 suspension, using 4.0 g L-1, under pseudo-second-order conditions at 25 ºC.34 This means that the comparison is not possible with our results, since we worked at pseudo-first-order conditions at 30 ºC.
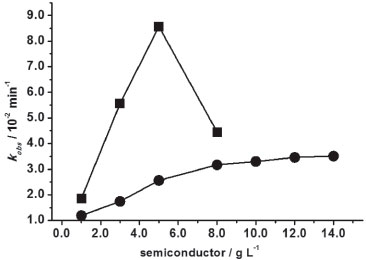 Figure 3. Decolorization rate constant, kobs, of DY86 (7.0 × 10-5 mol L-1) as a function of ZnO (●) and TiO2 (■) at 30 ºC
Similar to DY86 diazo dye, the DR80 tetraazo dye (8.0 × 10-5 mol L-1) showed higher reactivity in the TiO2 medium compared to ZnO, displaying the largest kobs values with respect to all other azo dyes. This occurs due to six sulfonate groups in DR80 (Figure 1e) that makes easier the adsorption on the surfaces of the semiconductors. According to the Figure 1d, DY86 has a triazine ring whose nitrogen atoms have p-orbitals that could generate resonance with three nitrogen atoms adjacent and stabilize the molecular structure, decreasing the reactivity. Even though four sulfonate groups are present in the structure, only two are used in the adsorption, because the other two are not in the same plane and so, the approximation becomes more difficult. Concentration of ZnO was varied from 1.0 to 10.0 g L-1 at 30 ºC in DR80 the kobs increased linearly from 3.03 × 10-2 to 20.93 × 10-2 min-1 with a positive inflection in 7.0 g L-1 with correlation coefficients of 0.994 (1.0 to 7.0 g L-1) and 0.999 (7.0 to 10.0 g L -1) respectively, as shown in Figure 4.
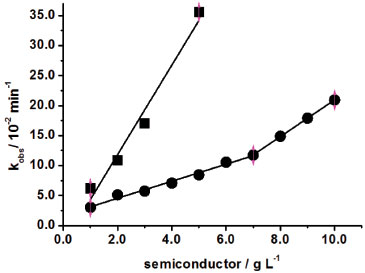 Figure 4. Decolorization rate constant, kobs, of DR80 (8.0 × 10-5 mol L-1) as a function of ZnO (●) and TiO2 (■) at 30 ºC
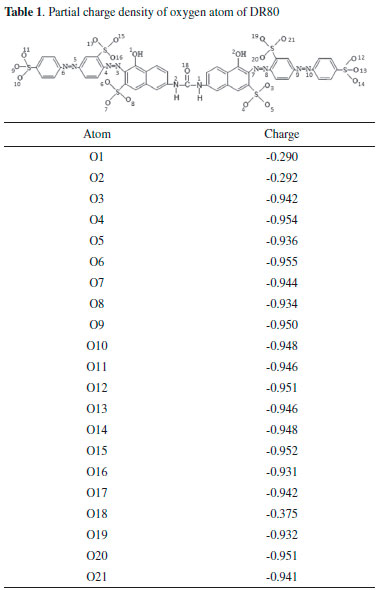
Thus, the reaction orders with respect to the ZnO concentration were equivalent to 0.7 and 1.6 in DR80 solution. Our research group found similar behavior when direct red 23 diazo dye decolorization was also mediated by ZnO, resulting in reaction orders respectively equal to 0.6 and 2.9.19 This behavior may be occurred, unlikely from TiO2, because the ZnO particles do not agglomerate even in higher concentrations, acquiring colloidal or nanoparticles characteristics, causing change in the reaction order. Therefore, the small particles of ZnO allow the radiation permeation in all the directions of the suspension. We verified this in qualitative experiments passing the radiation through the both suspensions. Due to the particles agglomeration in TiO2 medium, the radiation was absorbed in the first surface portions and the non-absorbed radiation was reflected to the out of the suspension. On the other hand, the particles did not suffer agglomeration in ZnO medium and because of this, the non-absorbed radiation passed through the suspension resulting in a higher photocatalytical efficiency. Conversely, the TiO2 concentration was varied only from 1.0 g L-1 (6.20 × 10-2 min-1) to 5.0 g L-1 (35.31 × 10-2 min-1) with r = 0.966, due to the total adsorption of DR80 (8.0 × 10-5 mol L-1) in TiO2 6.0 g L-1 for 20 min at 30 ºC in the dark. In this way, the DR80 decolorization rate constant in TiO2 5.0 g L-1 was four times higher in relation to the same conditions using ZnO (8.46 × 10-2 min-1). We investigated the decolorization rate constant for DR80 in similar conditions,25 obtaining 5.45 x 10-2 min-1 for 8.0 × 10-5 mol L-1 DR80 in 1.5 g L-1 ZnO suspension, whose value was analogous to the kobs obtained in this work. The only reference found in the literature for this dye in TiO2 was carried out by Tsuji et al that investigated the 1.0 × 10-4 mol L-1 DR80 and other several azo dyes decolorization degree in TiO2 thin films.35 In order to understand the reason of the rate constant increase with ZnO concentration enhancement, semi-empirical calculations were carried out to determine the charge densities of the constituent atoms and the molecule dipole moment.24 The oxygen atoms charge densities are shown in Table 1. The dipole moment of 7.84 D was calculated and located on the C=O bond. The negative charges densities are located in the oxygen atoms of the six sulfonate groups (O3 to O17 and O19 to O21), in OH bonded to the naphthalene rings (O1 and O2) and in the carbonyl oxygen (O18). The nitrogen atoms did not show significant negative charge density, that is, the nitrogen atoms charge is practically neutral. Theoretical results obtained for DR80 suggest that the adsorption occurs by oxygen atoms, and due the high negative charge of DR80, this leads to a higher physical adsorption on both semiconductors surface. This occurs due to the positive charge on the catalysts surface, elucidated by experimental pH of ~7.3 (ZnO) and ~4.3 (TiO2), taking into account that ZnO has a pHpzc of 11.0 and TiO2 of 6.2.
CONCLUSIONS From these results, TiO2 particles showed more facility to agglomerate in comparison to ZnO. It was verified that ZnO particles did not suffer agglomeration in the decolorization of all the azo dyes even at high concentrations like 14.0 g L-1 for DY86 and 10 g L-1 for other azo dyes as DO34, MO, AO7, and DR80 at 30 ºC. DY86 diazo dye and DR80 tetraazo dye presented a larger reactivity in TiO2 suspension, meanwhile MO, DO34, and AO7 monoazo dyes were more reactive in ZnO medium. This suggests that the structure size of the azo dye interferes in the interaction with the semiconductor type. The azo dyes decolorization rate constant was dependent on the azo dye adsorption on the photocatalyst surface and the semiconductor concentration.
ACKNOWLEDGEMENTS The authors thank to Fundaçao Araucária for financial aid (22850/2011). W. J. C. S., N. M. M., and V. S. L. N. are grateful for scholarship.
REFERENCES 1. Zollinger, H.; Color Chemistry: synthesis, properties and applications of organic dyes and pigments VCH: Weinheim, 1991. 2. Sandhu, P.; Chipman, J. K.; Mutat. Res. 1990, 240, 227. 3. Kim, S.; Park, C.; Kim, T-H.; Lee, J.; Kim, S.-W.; J. Biosci. Bioeng. 2003, 95, 102. 4. Poyatos, J. M.; Munio, M. M.; Almecija, M. C.; Torres, J. C.; Hontoria, E.; Osorio, F.; Water Air Soil Pollut. 2010, 205, 187. 5. De Jager, D.; Sheldon, M. S.; Edwards, W.; Sep. Purif. Technol. 2014, 135, 135. 6. Hoffmann, M. R.; Martin, S. T.; Choi, W.; Bahnemann, D. W.; Chem. Rev. 1995, 95, 69. 7. Konstantinou, I. K.; Albanis, T. A.; Appl. Catal., B 2004, 49, 1. 8. Gaya, U. I.; Abdullah, A. H.; J. Photochem. Photobiol., C 2008, 9, 1. 9. Legrini, O.; Oliveros, E.; Braun, A. M.; Chem. Rev. 1993, 93, 671. 10. Evgenidou, E.; Konstantinou, I.; Fytianos, K.; Poulios, I.; Albanis, T.; Catal. Today 2007, 124, 156. 11. Michael, I.; Hapeshi, E.; Michael, C.; Fatta-Kassinos, D.; Water Res. 2010, 44, 5450. 12. Chen, X.; Wang, W.; Xiao, H.; Hong, C.; Zhu, F.; Yao, Y.; Xue, Z.; Chem. Eng. J. 2012, 193, 290. 13. Singh, C.; Chaudhary, R.; Gandhi, K.; J. Renew. Sustain. Energy 2013, 5, 023124. 14. Haroune, L.; Salaun, M.; Ménard, A.; Legault, C. Y.; Bellenger, J.-P.; Sci. Total Environ. 2014, 475, 16. 15. Daneshvar, N.; Salari, D; Khataee, A. R.; J. Photochem. Photobiol., A 2003, 157, 111. 16. Ishiki, R. R.; Ishiki, H. M.; Takashima, K.; Chemosphere 2005, 58, 1461. 17. Bizani, E.; Fytianos, K.; Poulios, I.; Tsiridis, V.; J. Hazard. Mater. 2006, 136, 85. 18. Farooq, M.; Raja, I. A.; Pervez, A.; Sol. Energy 2009, 83, 1527. 19. Lucilha, A. C.; Bonancêa, C. E.; Barreto, W. J.; Takashima, K.; Spectrochim. Acta, Part A 2010, 75, 389. 20. Parra, S.; Olivero, J.; Pulgarin, C.; Appl. Catal., B 2002, 36, 75. 21. Daneshvar, N.; Salari, D.; Khataee, A. R.; J. Photochem. Photobiol., A 2003, 157, 111. 22. Bizani, E.; Fytianos, K.; Poulios, I.; Tsiridis, V.; J. Hazard Mater. 2006, 136, 85. 23. Atkins, P.; De Paula, J.; Physical Chemistry, 10th ed., OUP Oxford: Oxford, 2010. 24. Wavefunction; Spartan v.06, Program for Structure Optimization, Inc. Irvine, USA, 2006. 25. Da Silva, W. J. C.; Da Silva, M. R.; Takashima, K.; J. Chil. Chem. Soc. 2015, 60, 2749. 26. Zielinska, B.; Grzechulska, J.; Grzmil, B.; Morawski, A. W.; Appl. Catal., B 2001, 35, L5. 27. Li, G.; Lv, L.; Fan, H.; Ma, J.; Li, Y.; Wan, Y.; Zhao, X. S.; J. Colloid Interface Sci. 2010, 348, 342. 28. Danwittayakul, S.; Jaisai, M.; Koottatep, T.; Dutta, J.; Ind. Eng. Chem. Res. 2013, 52, 13629. 29. Alzahrani, E.; Curr. Anal. Chem. 2016, 12, 465. 30. Hamadanian, M.; Sarabi, A. S.; Mehra, A. M.; Jabbari, V.; Mater. Sci. Semicond. Process. 2014, 21, 161. 31. Daneshvar, N.; Rasoulifard, M. H.; Khataee, A. R.; Hosseinzadeh, F.; J. Hazard. Mater. 2007, 143, 95. 32. Zhang, X.; Sunb, D. D.; Li, G.; Wang, Y; J. Photochem. Photobiol., A 2008, 199, 311. 33. Niehues, E.; Scarmínio, I. S.; Takashima, K.; J. Chil. Chem. Soc. 2010, 55, 320. 34. Zhang, Q.; Jing, Y. H.; Shiue, A.; Chang, C-T.; Chen, B-Y.; Hsueh, C.-C.; J. Taiwan Inst. Chem. Eng. 2012, 43, 760. 35. Tsuji, E.; Taguchi, Y.; Aoki, Y.; Hashimoto, T.; Skeldon, P.; Thompson, G. E.; Habazaki, H; Appl. Surf. Sci. 2014, 301, 500. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access