
 |
 |
 |
 |
 |
Artigo
| New degraded quinone diterpenoid from the stems of Byrsonima coccolobifolia Kunth. (Malpighiaceae) |
|
Lorena R. F. de SousaI,II; Marcos H. F. SantosII; Vanessa G. P. SeverinoII,III; Richele P. SeverinoII; Paulo C. VieiraI,IV,*
I Departamento de Química, Universidade Federal de São Carlos, 13565-905 São Carlos - SP, Brasil Recebido em 13/09/2017 *e-mail: dpcv@ufscar.br A chemical investigation of two specimens of Byrsonima coccolobifolia collected in the southeast cerrado and from central Brazil was performed. A new degraded diterpenoid, byrsonimaquinone, was isolated from the stems along with known compounds. This is the first study on the roots of B. coccolobifolia, and several triterpenes, such as α-amyrin, β-amyrin, oleanolic acid, and glochidonol, along with a mixture of stigmasterol, β-sitosterol and campesterol, were identified. These compounds were identified by spectroscopic analysis techniques, including 1D and 2D NMR, GC-MS and high-resolution mass spectrometry. INTRODUCTION The genus Byrsonima, a member of the Malpighiaceae family, contains 150 species and is widespread in tropical America. In Brazil, species of the genus Byrsonima are known as “muricis” and can be found in north, northeast, southeast and central regions of the cerrado (savannah).1 The cerrado is the second largest Brazilian biome and a global hotspot of biodiversity, which makes further understanding the chemistry of these organisms quite valuable in view of increasing deforestation.2 Byrsonima coccolobifolia Kunth. is a medicinal plant used to treat stomach disorders and as an antidiarrheal.1,3 Previously, the extracts of the leaves have been shown to have antibacterial and molluscicidal activities and the ability to inhibit arginase, a molecular target of Leishmania amazonensis.3-5 The compounds most commonly isolated from this genus are flavonoids, terpenes, gallic acids and quinic acid derivatives.1 Among the constituents, flavonoids (flavanones, bioflavonoids, flavonols and procyanidins), triterpenes (with oleanolic and ursolic skeletons) and modified triterpenoids (steroids) are commonly found in B. basiloba,6,7 B. bucidaefolia,8 B. crassa,9,10 B. crassifolia,11-14 B. fagifolia,15-17 B. intermedia,18 B. microphylla,19,20 and B. verbascifolia.21,22 Previous studies on the leaves of B. coccolobifolia revealed the presence of flavonoids, gallic acid and xanthones.23,24 Recently, we investigated the ethyl acetate extracts of the leaves and stems of this species, and guided by arginase inhibition, we isolated flavonoids and tannins with potent inhibitory activities against arginase (IC50 values ranging from 0.13 to 4.8 µmol L -1).4,5 In our on-going studies of the chemistry of B. coccolobifolia, we have investigated two specimens collected in the southeast (stems and leaves) and central (roots) cerrado of Brazil. As part of our analysis of the sample collected in the central cerrado, from its roots, we only isolated triterpenes and steroids such as α-amyrin, β-amyrin, oleanolic acid and glochidonol (Figure 1). Our investigation of its leaves and stems led to the isolation of a new diterpene derivative, byrsonimaquinone, in addition to other known compounds that we have described before.4,5
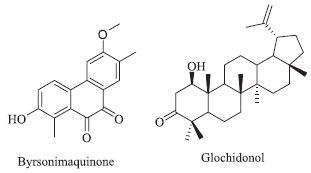 Figure 1. Byrsonimaquinone, a new degraded quinone diterpenoid isolated from B. coccolobifolia stems. Glochidonol, a triterpenoid described for the first time in a member of the Byrsonima genus, was isolated from B. coccolobifolia roots
EXPERIMENTAL SECTION General experimental procedures 1H-NMR and 2D NMR spectra were obtained on a Bruker model ARX 100 MHz and DRX-400 NMR spectrometer. Me4Si was used as an internal standard (J in Hz), and CDCl3, CD3OD and DMSO-d6 were used as solvents. MS spectra (m/z) were acquired using a Bruker Daltonics, Micromass TOF - Q II - ESI - TOF, high-resolution mass spectrometer. For GC-MS analysis, an Agilent GC-7820A model gas chromatograph - mass spectrometer coupled to an MSD 5975 was used. Experimental conditions were as follows: 10 µL of sample was injected in the GC-MS in the splitless mode; starting at 120 °C (2 min), then increasing 10 °C/min until 250 °C, then increasing at 2 °C/min until 275 °C, and then 35 °C/min until 310 °C (10 min); the split was 20:1; the flow rate was 1 mL/min; the column was an HP-5 (30 m x 250 µm and film 0.25 µm; Agilent); and the data were collected in the electron impact ionization mode at 70 eV. For chromatography, silica gel (SiO2) (Merck, 60-200 mesh; and Macherey-Nagel, 70-230 and 230-400 mesh) and Sephadex LH-20 (Amersham Pharmacia Biotech AB) were used. The solvents were ethanol (EtOH), methanol (MeOH), hexane, dichloromethane, and ethyl acetate (EtOAc), and they were purchased from Vetec. Thin-layer chromatography (TLC) was carried out with F254, φ = 0.2 mm, pre-coated aluminum silica 60 (20 x 20 cm) (Whatman, Fluka and Merck). TLC plates were visualized using UV254/366 light as well as a sulfuric vanillin solution. Plant material B. coccolobifolia materials were collected in distinct regions. Stems were collected from the cerrado at the Federal University of Sao Carlos (UFSCar), Sao Carlos - SP, Brazil, in July 2011. This species was identified by Dr. Maria Inês Salgueiro Lima and deposited at the Herbarium of the Botany Laboratory (HUFSCar) at UFSCar (Voucher nº 8367). The roots were collected in central Brazil in the Federal District - Gama - DF, Brazil, in December 2010. The sample of B. coccolobifolia was identified by Dr. Helder Nagai Consolaro and deposited at the EMBRAPA Herbarium - Genetic Resources and Biotechnology (CEN) (Voucher nº BW 6029). Extraction and isolation The crude extract of the roots (65 g) of B. coccolobifolia was obtained after percolating the dried powder three times in ethanol for six days each time. Ethanolic extracts from the stems (30.5 g) were obtained from dried powder macerated with EtOH at room temperature over two weeks as described previously.4,5 Both extracts were concentrated by rotatory evaporation at 40 °C. The crude extract from the roots was suspended in a mixture of MeOH/H2O (3:7), and the crude extract from the stems was suspended in EtOH/H2O (1:3). Both suspensions were sequentially partitioned in hexane and EtOAc. From the roots, this procedure generated 2.5 g of material from the hexane layer (BcRH), 24.1 g of material from the EtOAc layer (BcRA), and 38.5 g of material from the water-alcoholic layer (BcRW). The liquid-liquid partitioning of the crude stem extract led to 1.3 g of material from the hexane layer (BcSH), 10.0 g of material from the EtOAc layer (BcSA), and 19.0 g of material from the water-alcoholic layer (BcSW). Compounds isolation from stem extracts The EtOAc extract from the stems, BcSA (10.0 g), was chromatographed on a column with a height (h) of 12.0 cm and a diameter (φ) of 5.0 cm (SiO2, 60-200 mesh; isocratic mode with hexane/acetone (9:1)) and yielded four fractions. Fraction 4 (4.0 g) was chromatographed several times as follows: Sephadex LH-20, h = 7.0 cm, φ = 5.0 cm, isocratic mode with MeOH; fraction four from the Sephadex column (55.0 mg) was further purified over silica gel (SiO2, 230-400 mesh, h = 26.0 cm, φ = 2.0 cm, isocratic mode with CH2Cl2/MeOH using 1% of MeOH); fraction fourteen (15 mg) from the silica column was fractionated over Sephadex LH-20, h = 54.0 cm, φ = 1.6 cm, isocratic mode with MeOH; and fraction 8 from that column contained the diterpene byrsonimaquinone (1.8 mg). MS and 1D and 2D NMR were performed, and the results were compared to the literature data.25 Byrsonimaquinone. TOF - Q II - ESI - TOF - MS: [2 M + Na] + m/z: 587.1678; [M + Na] + m/z: 305.0780 and M of m/z: 282.0898 (C17H14O4, calcd 282.0892); 1H and 2D NMR (Table 1).
The compounds 5-O-galloylquinic acid, 3,5-di-O-galloylquinic acid, 5-O-(3-methylgalloyl)-quinic acid, 3,4,5-tri-O-galloylquinic acid, gallic acid, (+)-catechin, (-)-epicatechin, quercitrin, isoquercitrin, (+)-syringaresinol and trigonostemone were isolated from the leaves and stems and were characterized in our previous work.4,5 Compounds isolated from the root extracts BcRH (2.5 g) was subjected to column chromatography (CC) (SiO2, 70-230 mesh; h = 11.0 cm, φ = 5.5 cm; gradient mode with hexane/EtOAc and EtOAc/MeOH). The second fraction (1.6 g) obtained from this column was repurified by CC (SiO2, 70-230 mesh; h = 12.0 cm, φ = 5.0 cm; gradient mode with hexane/EtOAc and EtOAc/MeOH) and afforded five fractions after TLC analysis (F2-1, F2-2, F2-3, F2-4 and F2-5), and the second of those fractions (0.8 g) was a mixture of α- and β-amyrin. The third fraction, F2-3 (0.1 g), was further purified using CC (SiO2, 230-400 mesh; h = 23.0 cm, φ = 2.5 cm; gradient mode with hexane/EtOAc and EtOAc/MeOH). From the four fractions afforded by that column (F3-1, F3-2, F3-3, F3-4), F3-2 (0.1 g) was further purified using CC (SiO2, 230-400 mesh; h = 30.0 cm, φ = 1.7 cm; gradient mode with hexane/CH2Cl2 and CH2Cl2/EtOAc), which lead to the isolation of glochidonol (3.0 mg). Fractions F4-4 and F4-5 contained a mixture of phytosterols (5.0 mg), namely, stigmasterol, β-sitosterol and campesterol. The EtOAc extract BcRA (24.1 g) was fractionated using CC with SiO2, and after seven chromatography steps, oleanolic acid (9.7 mg) was obtained. Briefly, the CC conditions for the isolation of oleanolic acid are as follows: the first CC of BcRA (24.1 g) was SiO2, 70-230 mesh, h = 15.0 cm, φ = 5.5 cm, gradient mode with hexane/EtOAc and EtOAc/MeOH; fraction two from first column (4.6 g) was used for the second column, which was SiO2, 70-230 mesh, h = 9.0 cm, φ = 5.5 cm, gradient mode with hexane/EtOAc and EtOAc/MeOH; fraction one from the second column (4.6 g) was subjected to a third column using SiO2, 70-230 mesh, h = 12.0 cm, φ = 5.5 cm, gradient mode with hexane/EtOAc and EtOAc/MeOH; fraction one from the third CC (1.7 g) was subjected to a fourth CC using SiO2, 230-400 mesh, h = 24.5 cm, φ = 2.5 cm, gradient mode with hexane/EtOAc and EtOAc/MeOH; fraction three from the fourth column (0.2 g) was fractionated again over SiO2, 230-400 mesh, h = 21.0 cm, φ = 2.5 cm, gradient mode with hexane/EtOAc and EtOAc/MeOH; fraction two of the fifth column (53.8 mg) was purified again using SiO2, 230-400 mesh, h = 26.0 cm, φ = 1.7 cm, gradient mode with hexane/EtOAc and EtOAc/MeOH; and fraction two from the sixth column (18 mg) was fractionated over SiO2, 230-400 mesh, h = 21.0 cm, φ = 1.5 cm, gradient mode with hexane/EtOAc and EtOAc/MeOH. Fraction two of the seventh column contained oleanolic acid. The compounds were identified by 1H-NMR and 2D NMR and GC/MS, and the results were compared to the literature data.26-30
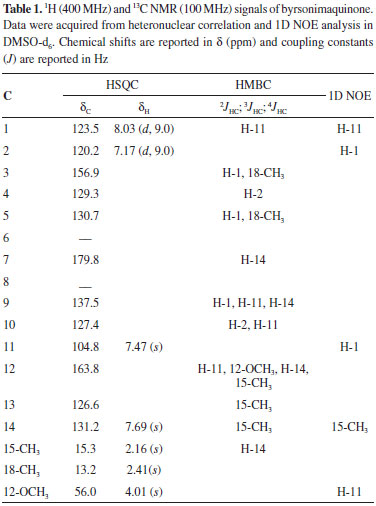
RESULTS AND DISCUSSION The EtOAc extracts of B. coccolobifolia stems and leaves were fractionated, and the compounds isolated include 5-O-galloylquinic acid, 3,5-di-O-galloylquinic acid, 5-O-(3-methylgalloyl)-quinic acid, 3,4,5-tri-O-galloylquinic acid, gallic acid, (+)-catechin, (-)-epicatechin, quercitrin, isoquercitrin, (+)-syringaresinol and trigonostemone.4,5 A new degraded diterpenoid purified from stems was named byrsonimaquinone (Figure 1). In a second study, the hexane and EtOAc extracts of the roots of the specimen collected in the central Brazilian cerrado were submitted to several chromatographic columns, which eventually lead to the identification of the triterpenes α-amyrin, β-amyrin,26 oleanolic acid,27 glochidonol,28 and the mixture of stigmasterol, β-sitosterol and campesterol.29,30 The new quinone diterpene derivative was identified by 1H NMR and 2D NMR experiments, such as HSQC, HMBC, and 1D NOE (Table 1), as well high-resolution mass spectrometry. The mass spectrum was obtained in the positive mode showing an ion signal at m/z 587.1678 corresponding to the sodiated dimer, [2 M + Na] +, of the diterpene. The base peak, observed at m/z 305.0780, can be attributed to the sodiated diterpene molecular ion, which indicates a molecular formula of C17H14O4 (calc. 282.0892). In the 1H NMR spectrum of the diterpene, there are signals from aromatic hydrogens at δH 8.03 (d; J = 9.0 Hz) and 7.17 (d; J = 9.0 Hz), indicating an ortho relationship between hydrogens H-1 and H-2. H-1 is more deshielded due to its meta relationship to the hydroxyl group instead of the ortho relationship observed in manniorthoquinone.25 The chemical shifts of the singlets at δH 7.69 and 7.47 indicate hydrogens H-14 and H-11 on the second aromatic ring moiety are para to each other. The proton at δH 4.01 (s) was attributed to the methoxyl group bound to an aromatic ring and the two singlets at δH 2.41 and 2.16 (18-CH3 and 15-CH3) are characteristic of methyl groups bound to aromatic rings, and they are bound to C-4 and C-13, respectively. The HSQC indicated that the aromatic hydrogens at δH 8.03 (d; J = 9.0 Hz) and 7.17 (d; J = 9.0 Hz) are directly correlated (1JHC) with C-1 (δC 123.5 ppm) and C-2 (δC 120.2 ppm), respectively. The hydrogens on the second aromatic ring, δH 7.69 (s) and 7.47 (s), are coupled to C-14 (δC 131.2 ppm) and C-11 (δC 104.8), respectively. The methyl group hydrogens of 18-CH3 correlate with C-18 (δC 13.2), and the hydrogens of 15-CH3 correlate with C-15 (δC 15.3). The hydrogens of the 12-OCH3 methoxyl group at δH 4.01 (s) correlate with the carbon at δC 56.0 ppm. In comparing the data obtained here for isolated byrsonimaquinone with those reported in the literature for manniorthoquinone,25 the differences were primarily related to the positioning of the substituents around the aromatic ring as seen in the different chemical shifts, multiplicities and coupling constants in the NMR spectra. Hydroxyl groups are inductively withdrawing and therefore deshield C-3 (δC 156.9). However, hydroxyl groups are electron donating by resonance and therefore shield C-4 (δC 129.3). In byrsonimaquinone, C-5 is meta to the hydroxyl group, so it is more deshielded (δC 130.7) than in manniorthoquinone25 where it is para to the hydroxyl group. The heteronuclear long-range couplings and 1D NOE experiments together with all the NMR and MS data allowed us to propose the molecular structure of the new degraded diterpenoid (Figure 2). The substitution arrangement on the aromatic rings was established based on HMBC correlations (Figure 2). The 3JHC of H-14 (δH 7.69) with C-9 (δC 137.5), C-12 (δC 163.8), 15-CH3 (δC 15.3) and C-7 (δC 179.8) provided evidence that the methoxyl group was attached to this ring and that the carbonyl is next to H-14. In addition, the hydrogens of the methoxyl group (δH 4.01) correlate to C-12 (δC 163.8), and the hydrogens of the 15-CH3 methyl (δH 2.16) correlate with C-12 (δC 163.8), C-13 (δC 126.6) and C-14 (δC 131.2).
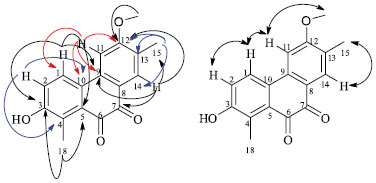 Figure 2. Heteronuclear correlations. Long-range couplings in the HMBC spectrum of byrsonimaquinone:  ; 1D NOE interactions of 1: ; 1D NOE interactions of 1: 
The 2JHC cross-peaks between H-11(δH 7.47) with C-9 (δC 137.5) and C-12 (δC 163.8), as well the 3JHC of H-11 to C-10 (δC 127.4) are in agreement with those reported for manniorthoquinone.25 A 4JHC coupling between H-11(δH 7.47) and C-1 (δC 123.5) was also observed. For the second aromatic ring, the spectrum showed 3JHC correlations of H-1 (δH 8.03) with C-3 (δC 156.9), C-5 (δC 130.7) and C-9 (δC 137.5), which provides evidence that the hydroxyl group is attached at the more deshielded carbon (C-3). H-2 (δH 7.17) correlates to C-4 (δC 129.3) and C-10 (δC 127.4), and the 18-CH3 methyl protons (δH 2.41) showed cross-peaks with C-3 (δC 156.9) and C-5 (δC 130.7), which confirm the hydroxyl group is adjacent to this methyl group. The methyl (18-CH3) is bound to C-4, and long-range correlation (4JHC) with carbonyl (C-6) was not observed. The presence of a carbonyl at C-6 can be confirmed by the high-resolution mass spectrometry (molecular formula: C17H14O4) and NOE experiments. Additionally, the common correlations observed in the HMBC spectrum of H-1, H-11, H-14 with C-9 (δC 137.5); H-2 and H-11 with C-10 (δC 127.4); H-1 and the 18-CH3 methyl hydrogens with C-5 (δC 130.7); as well as H-11, H-14, the 15-CH3 methyl hydrogens, and the 12-OCH3 methoxyl hydrogens with C-12 (δC 163.8) confirm their proximity. To confirm the assignment of the arrangement of the functional groups in the proposed structure, the nuclear Overhauser effects were analyzed after the straightforward process of decoupling the protons and acquiring the spectrum (Table 1). The selective irradiation of the resonance frequency of H-14 (δH 7.69) generated an NOE for the signal of 15-CH3 at δH 2.16. Additionally, selective irradiation of the resonance frequency of H-11 (δH 7.47) caused an enhancement of the signals at δH 8.03 and δH 4.01 (H-1 and 12-OCH3, respectively), which indicated that H-11 is close in space to the methoxyl group and to H-1. When H-1 (δH 8.03) was irradiated, an NOE was also observed at δH 7.47, confirming the proximity of H-1 and H-11. Selective irradiation of the resonance frequency of H-2 (δH 7.17) intensified a single signal at δH 8.03, which corresponds to the signal of H-1. These results allowed us to propose a structure for this degraded diterpenoid. Byrsonimaquinone can be considered a terpenoid derived from ortho-naphthoquinone. Naphthoquinones can be formed either from acetate/malonate or shikimate/2-oxoglutarate/isoprenoid pathways. Naphthoquinones, their derivatives, xanthones, and terpene derivatives have been isolated previously from members of this genus.4,19,20,23 Quinones are derived from the oxidation of phenolic compounds; the oxidation of catechols (1,2-dihydroxybenzenes) leads to ortho-quinones, and the oxidation of quinols (1,4-dihydroxybenzenes) leads to para-quinones. Accordingly, terpenoid quinones are built up from shikimate and terpenoid pathways. Furthermore, a terpenoid quinone biosynthetic pathway is suggested for this compound, a combined metabolic pathway of shikimate-derived (quinone derivatives) and terpenoid pathways (diterpenoid moiety).31 All the compounds isolated from the roots of the second specimen were identified through comparison of their spectral data with the literature.26-30
CONCLUSION This work contributed to our knowledge of the chemistry of the genus Byrsonima. Of the compounds isolated from the roots, the triterpenoids α-amyrin, β-amyrin, and oleanolic acid, and the mixture of stigmasterol, β-sitosterol and campesterol have been described before for other species, most of which have been isolated from B. crassifolia11-14 and B. verbascifolia,21,22 except for glochidonol which has not been described in the genus so far. The phytochemical investigation of the leaves and stems afforded the flavonoids (+)-catechin, (-)-epicatechin, quercitrin and isoquercitrin, which are frequently found in Byrsonima species including B. basiloba,6,7 B. bucidaefolia,8 B. crassa,9,10 B. crassifolia,11-14 B. fagifolia,15-17 B. intermedia,18 B. microphylla,19,20 and B. verbascifolia;21,22 the investigation also afforded the tannins 3,5-di-O-galloylquinic acid, 5-O-galloylquinic acid, 5-O-(3-methylgalloyl)-quinic acid, 3,4,5-tri-O-galloylquinic acid and gallic acid, which have been previously described in B. crassa9,10 and B. fagifolia.15-17 Among the compounds isolated from the stems of B. coccolobifolia, (+)-syringaresinol and trigonostemone are new in the genus Byrsonima. Additionally, the degraded diterpenoid byrsonimaquinone, obtained from stems of B. coccolobifolia, is described for the first time in the literature.
SUPPLEMENTARY MATERIAL The high-resolution mass spectrometry, TOF - Q II - ESI - TOF, 1H NMR, HSQC, HMBC and 1D NOE spectra for isolated compounds (Figures 1S-14S) are freely available at http://quimicanova.sbq.org.br in PDF file.
ACKNOWLEDGMENTS We thank N. P. Lopes for his contribution to this work with high-resolution mass spectrometry experiments. This research project received financial support from the State of Sao Paulo Research Foundation (FAPESP, Fundaçao de Amparo à Pesquisa do Estado de Sao Paulo) Proc. 2010-52326-9, the State of Goiás Research Foundation (FAPEG, Fundaçao de Amparo à Pesquisa do Estado de Goiás) Proc. 200910267000366, INBEQMEdi, and Proc. 563286/2010-5, the National Council for Scientific and Technological Development (CNPq, Conselho Nacional de Pesquisa e Desenvolvimento), Brazil.
REFERENCES 1. Guilhon-Simplicio, F.; Pereira, M. M.; Quim. Nova 2011, 34, 1032. 2. Françoso, R. D.; Brandao, R.; Nogueirac, C. C.; Salmona, Y. B.; Machado, R. B.; Colli, G. R.; Natureza & Conservaçao 2015, 13, 35. 3. Sannomiya, M.; Fonseca, V. B.; da Silva, M. A.; Rocha, L. R. M.; dos Santos, L. C.; Hiruma-Lima, C. A.; Souza Brito, A. R. M.; Vilegas, W.; J. Ethnopharmacol. 2005, 97, 1. 4. de Sousa, L. R. F.; Ramalho, S. D.; Burger, M. C. M.; Nebo, L.; Fernandes, J. B.; da Silva, M. F. G. F.; Iemma, M. R. C.; Corrêa, C. J.; de Souza, D. H. F; Lima, M. I. S.; Vieira, P. C.; J. Nat. Prod. 2014, 77, 392. 5. de Sousa, L. R. F.; Ramalho, S. D.; Fernandes, J. B.; da Silva, M. F. das G. F.; Iemma, M. R. da C.; Corrêa, C. J.; de Souza, D. H. F.; Lima, M. I. S.; Vieira, P. C.; J. Braz. Chem. Soc. 2014, 25, 1832. 6. Lira, W. M.; Santos, F. V.; Sannomiya, M.; Rodrigues, C. M.; Vilegas, W.; Varanda, E. A.; J. Med. Food 2008, 11, 111. 7. Figueiredo, M. E.; Michelin, D. C.; Sannomiya, M.; Silva, M. A.; Santos, L. C.; Almeida, L. F. R.; Salgado, H. R. N.; Vilegas, W.; Rev. Bras. Cienc. Farm. 2005, 41, 79. 8. Castillo-Avila, G. M.; García-Sosa, K.; Peña-Rodríguez, L. M.; Nat. Prod. Commun. 2009, 4, 83. 9. Sannomiya, M.; Rodrigues, C. M.; Coelho, R. G.; Santos, L. C.; Hiruma-Lima, C. A.; Brito, A. R. M. S.; Vilegas, W.; J. Chromatogr. A 2004, 1035, 47. 10. Sannomiya, M.; Fonseca, V. B.; Silva, M. A.; Rocha, L. R.; Santos, L. C.; Hiruma-Lima, C. A.; Souza Brito, A. R.; Vilegas, W.; J. Ethnopharmacol. 2005, 97, 1. 11. Geiss, F.; Heinrich, M.; Hunkler, D.; Rimpler, H.; Phytochemistry 1995, 39, 635. 12. Rastrelli, L.; De Tommasi, N.; Berger, I.; Caceres, A.; Saravia, A.; De Simone, F.; Phytochemistry 1997, 45, 647. 13. Amarquaye, A.; Che, C.; Bejar, E.; Malone, M. H.; Fong, H. H. S.; Planta Med. 1994, 60, 85. 14. Bejar, E.; Amarquaye, A.; Che, C.; Malone, M. H.; Fong, H. H. S.; Int. J. Pharmacogn. 1995, 33, 25. 15. Lima, Z. P.; Santos, R. C.; Torres, T. U.; Sannomiya, M.; Rodrigues, C. M.; Santos, L. C.; Pellizzon, C. H.; Rocha, L. R. M.; Vilegas, W.; Brito, A. R. M. S.; Cardoso, C. R. P.; Varanda, E. A.; Moraes, H. P.; Bauab, T. M.; Carli, C.; Carlos, I. Z.; Hiruma Lima, C. A.; J. Ethnopharmacol. 2008, 120, 149. 16. Higuchi, C. T.; Sannomiya, M.; Pavan, F. R.; Leite, S. R. A.; Sato, D. N.; Franzblau, S. G.; Sacramento, L. V. S.; Vilegas, W.; Leite, C. Q. F.; J. Evidence-Based Complementary Altern. Med. 2011, ID 128349. 17. Sannomiya, M.; Santos, L. C.; Carbone, V.; Napolitano, A.; Piacente, S.; Pizza, C.; Souza-Brito, A. R. M.; Vilegas, W.; Rapid Commun. Mass Spectrom. 2007, 21, 1393. 18. Sannomiya, M.; Cardoso, C. R. P.; Figueiredo, M. E.; Rodrigues, C. M.; Santos, L. C.; Santos, F. V.; Serpeloni, J. M.; Colus, I. M.; Vilegas, W.; Varanda, E. A.; J. Ethnopharmacol. 2007, 112, 319. 19. Aguiar, R. M.; David, J. P.; David, J. M.; Phytochemistry 2005, 66, 2388. 20. Rocha, J. H.; Cardoso, M. P.; David, J. P.; David, J. M.; Biosci., Biotechnol., Biochem. 2006, 70, 2759. 21. Gottlieb, O. R.; Mendes, P. H.; Magalhaes, M. T.; Phytochemistry 1975, 14, 1456. 22. Dosseh, C.; Morreti, C.; Tessier, A. M.; Delaveau, P.; Plant. Med. Phytother. 1980, 14, 136. 23. Lorenzi, K. C.; Rodrigues, C. M.; Sannomiya, M.; de Almeida, L. F. R.; Brito, A. R. M. S.; Vilegas, W.; Resumos da 29ª Reuniao Anual da Sociedade Brasiliera de Química, Aguas de Lindóia, Brasil, 2006. 24. Lorenzi, K. C.; Rodrigues, C. M.; Sannomiya, M.; Rinaldo, D.; Brito, A. R. M. S.; Vilegas, W.; Resumos da 30ª Reuniao Anual da Sociedade Brasileira de Química, Aguas de Lindóia, Brasil, 2007. 25. Tene, M.; Tane, P.; Tamokou, J. de D.; Kuiate, J. R.; Cnnolly, J. D.; Phytochem. Lett. 2008, 1, 120. 26. Vieira-Junior, G. M.; Souza, C. M. L.; Chaves, M. H.; Quim. Nova 2005, 28, 183. 27. Seebacher, W.; Simic, N.; Weis, R.; Saf, R.; Kunert, O.; Magn. Reson. Chem. 2003, 41, 636. 28. Puapairoj, P.; Naengchomnong, W.; Kijjoa, A.; Pinto, M. M.; Pedro, M.; Nascimento, M. S. J.; Silva, A. M. S.; Herz, W.; Planta Med. 2005, 71, 208. 29. Garg, V. K.; Nes, W. R.; Phytochemistry 1984, 23, 2925. 30. Costa, E. V.; Sampaio, M. F. C.; Salvador, M. J.; Nepel, A.; Barison, A.; Quim. Nova 2015, 38, 769. 31. Dewick, P. M.; Medicinal Natural Products: A Biosynthetic Approach, 2nd ed., John Wiley & Sons, Ltd: New York, 2002. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access