
 |
 |
 |
 |
 |
Nota Técnica
| Synthesis of gold nanoparticles by tetrachloroaurate reduction with cyclodextrins |
|
Yuri S. Pestovsky*; Agustino Martínez-Antonio#
Genetic Engineering Department, Center for Research and Advanced Studies of the National Polytechnic Institute, 36824 Irapuato, Guanajuato, Mexico Recebido em: 05/03/2018 *e-mail: iurii.pestovskii@cinvestav.mx One-step synthesis of gold nanoparticles functionalized with α-, β- and γ-cyclodextrins was carried out using a synthetic method of direct tetrachloroaurate reduction in alkaline solutions. The two experimental conditions studied were direct heating and microwave irradiation. This is the first publication comparing the characteristics of gold nanoparticles produced using three cyclodextrins under different conditions. The resulting nanoparticles were characterized by absorbance spectra, FTIR spectra, dynamic light scattering data and transmission electron microscopy images. The reducing ability of cyclodextrins increased in the following order: β-cyclodextrin < α-cyclodextrin < γ-cyclodextrin. Therefore, γ-cyclodextrin was the most active reductant and the only cyclodextrin that was able to reduce tetrachloroaurate at room temperature. In this case, nanoparticles were observed only after 4 days of storage. The nanoparticle size was dependent on the cyclodextrin type and correlated with the difference in the reducing ability. Using microwave irradiation, most of nanoparticles were found to be free nanoparticles. Using direct heating, aggregates formed due to the high ionic strength of the solution after prolonged boiling. Therefore, nanoparticles synthesized by direct heating were less stable during storage than those synthesized by microwave irradiation. INTRODUCTION Cyclodextrins, naturally occurring cyclic oligosaccharides composed of α-(1-4)-linked D-glycosyl residues, are widely used for various applications including drug delivery.1-3 A promising approach is capturing the drug in nanoparticulated forms of cyclodextrins. Unless chemically crosslinked,2 cyclodextrins cannot form nanoparticles. Although spontaneous formation of cyclodextrin inclusion complex nanoparticles has been reported,3 it is very rare. Gold nanoparticles have emerged as attractive nanomaterials for drug delivery. They can be used as multifunctional platforms for both therapeutic and diagnostic purposes.4 If a drug is released from nanoparticles, it can only be non-covalently conjugated with nanoparticles, making it difficult to achieve prolonged release. Therefore, functionalization of gold nanoparticles with cyclodextrins that are capable of encapsulating a drug appears to be a more promising approach. The most obvious way to immobilize cyclodextrins onto the surface of gold nanoparticles is by conjugation of thiolated cyclodextrins with gold nanoparticles during their preparation, as reported for β-cyclodextrin,5,6 and for α- and γ-cyclodextrins.6 The multi-step synthesis of thiolated cyclodextrins requires various chemicals, and it is difficult to avoid trace amounts of them in the final product. Moreover, this synthesis can be carried out only in a rather advanced laboratory. Therefore, the possibility of synthesizing cyclodextrin-capped gold nanoparticles using direct tetrachloroaurate reduction by cyclodextrins was investigated. This synthesis was reported for α-7 and β-cyclodextrins 8 as well as for hydroxypropyl-β-cyclodextrin,9 but not for γ-cyclodextrin. γ-cyclodextrin-capped gold nanoparticles could be used for drug (or pesticide) delivery involving molecules too large for forming a complex with α- and β-cyclodextrins, e.g. chloramphenicol, as shown for silver nanoparticles.10 Gold nanoclusters coated with an inclusion complex of γ-cyclodextrin with dihydrolipoic acid can also be used as an intracellular pH-sensitive fluorescent probe.11 Cyclodextrin-capped gold nanoparticles synthesized using cyclodextrins as reductants demonstrate an additional advantage of having high stability without applying stabilizers.7,8 This study explored the possibility of tetrachloroaurate reduction by γ-cyclodextrin compared to α- and β-cyclodextrins.
EXPERIMENTAL Synthesis of gold nanoparticles by microwave irradiation 500 µL of solution containing 0.002 mol L-1 cyclodextrin, 0.1% (w/w) HAuCl4, and 0.98 g L-1 KOH was placed in a 2-mL plastic test tube, vortexed (Vortex-Genie 2 G-560, Scientific Industries, Inc., USA) at maximal rate for 10 minutes, and then heated in a microwave oven (Panasonic NN-ST253W, USA) at high power for 5 minutes. Then the microwave oven door was opened and the solution was allowed to cool down to room temperature for about 15 minutes. The tubes remained open during heating and subsequent cooling. The solutions were then stored at 4 °C. Synthesis of gold nanoparticles by direct heating The solution containing cyclodextrin, 0.1% (w/w) HAuCl4 and NaOH in a glass bulb was vortexed at speed 1 for 10 min, and then its pH was measured and adjusted as necessary. Then the bulb was placed on a hot plate (Corning PC-620, USA) and heated at power 4. In this heating regime, the solution temperature was close to the boiling point. The plate was preheated in the same regime to a constant temperature. During the heating, the 200-µL aliquots (for spectrophotometry) or 2-mL (for hydrodynamic size and pH measurements) of the solution were taken for analysis at different time points. These aliquots were immediately placed into a refrigerator and stored at 4 °C. The following concentrations of each cyclodextrin were tested: 0.001 mol L-1, 0.002 mol L-1 and 0.004 mol L-1. Although the volume of every solution was 11 mL, only 7 mL were used for the synthesis of nanoparticles because 2 mL were consumed for pH adjustment (see below), and another 2 mL were taken for dynamic light scattering analysis as the zero time point. pH measurement and adjustment Before nanoparticle synthesis by direct heating, a 2-mL aliquot was taken from the solution. Its pH was measured using a Pinnacle series M 540P pH meter (SCHOTT Instruments, Germany). If the pH reading was below 10.56, it was adjusted to be in the range of 10.56-11.15 using 0.4 mol L-1 NaOH. The amount needed to adjust the pH of the remaining solution was calculated from the amount of NaOH consumed to adjust the pH of the aliquot. This was done in order to avoid contacting the electrode of the pH meter with the solution used for the synthesis of nanoparticles. Back adjustment of pH using an acid was never used. The pH of the solution of synthesized nanoparticles was measured after dynamic light scattering analysis. Characterization of nanoparticles Absorbance spectra were acquired by means of a microplate reader BioTek Synergy 2 (USA). Hydrodynamic size and zeta potential of the nanoparticles were measured using Microtrac ZetatracTM (USA). The solutions of the nanoparticles formed during the synthesis were used for measurements without any further preparation. The solutions without HAuCl4 served as blanks for the nanoparticles synthesized using microwave irradiation. Water was used as a blank for those synthesized using direct heating. FTIR spectra were acquired using Thermo Nicolet 6700 (USA) and analyzed using Spekwin32 1.72.0 software.12 The samples of gold nanoparticles were centrifuged (12000 rpm, 20 min) using a centrifuge 5430 with a rotor FA-45-30-11 (Eppendorf, Germany). Then samples were resuspended in 1 mL of water, lyophilized using centrifugal evaporator MAXI Dry Lyo (Heto Holten, Denmark) and stored on a silica gel prior to FTIR spectra acquisition. The sample was used in the form of powder without applying pressure and without any additives. Transmission electron microscopy (TEM) images were acquired by means of Philips FEI Morgagni 268D microscope (USA). The solutions were dropped onto 200 mesh formvar/carbon-coated copper grids (Ted Pella, Inc., USA), and the remaining liquid was collected with filter paper after 20 minutes of incubation under a Petri dish.
RESULTS AND DISCUSSION The four day storage of the reaction mixture containing α- or β-cyclodextrins that was not subjected to heating or microwave irradiation failed to produce nanoparticles in the concentration that could be detected by spectrophotometry (Figure 1). But for γ-cyclodextrin, a small peak around 560 nm could be seen (Figure 1) (i.e. gold nanoparticles were present in the solution in a very small amount). As more time passed, the concentration of nanoparticles increased and became detectable in TEM images (Figure 2A).
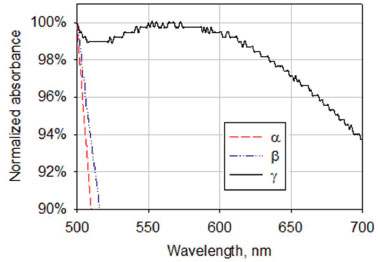 Figure 1. Absorbance spectra of the reaction mixtures containing KOH, HAuCl4 and cyclodextrins after 4 days of storage at room temperature
 Figure 2. TEM images of the sample containing KOH, HAuCl4 and γ-cyclodextrin after 3 weeks of storage at 4 °C (a); of samples containing KOH, HAuCl4 and α- (b), β- (c) or γ-cyclodextrins (d) after microwave irradiation; of the sample containing only KOH and HAuCl4 after microwave irradiation (e) and of a similar sample that was not exposed to either microwave irradiation or heating (f)
The absorbance spectra of the reaction mixture after microwave irradiation for 5 minutes showed a clear absorbance peak around 520 nm (Figure 3), indicating the formation of nanoparticles with the three cyclodextrins. Hydrodynamic size distribution diagrams of the nanoparticles are presented in Figure 4.
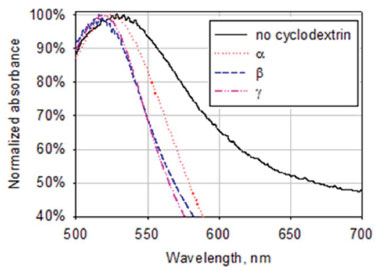 Figure 3. Absorbance spectra of the reaction mixtures containing KOH, HAuCl4 and cyclodextrins after microwave irradiation
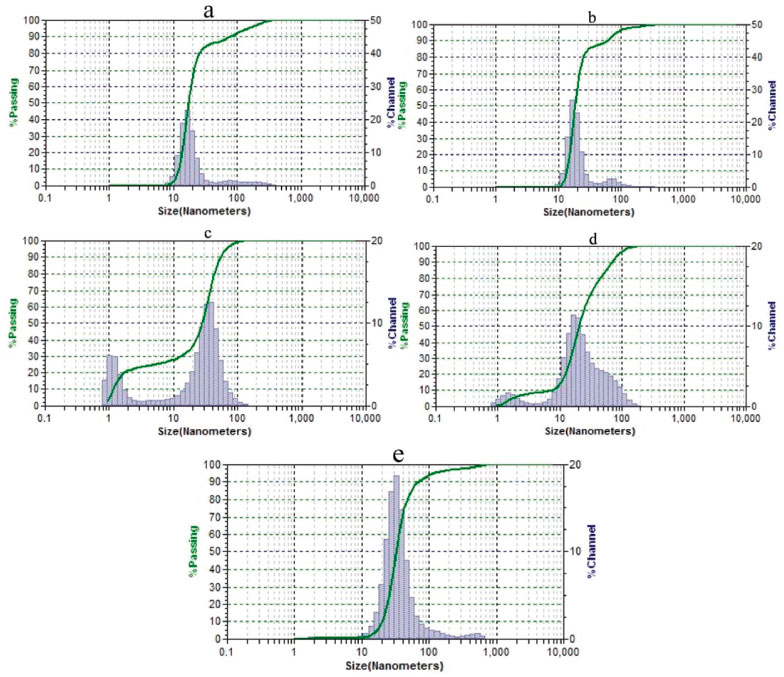 Figure 4. Hydrodynamic size distribution diagrams of nanoparticles. Sample containing KOH, HAuCl4 and γ-cyclodextrin without microwave irradiation (a); samples containing KOH, HAuCl4, and α- (b), β- (c) or γ-cyclodextrins (d) after microwave irradiation; and sample containing only KOH and HAuCl4 after microwave irradiation (e)
TEM analysis revealed the presence of the nanoparticles in the case of each cyclodextrin (Figure 2). Extensive aggregation observed in the case of β-cyclodextrin possibly occurred during microscopy sample preparation, because it was not seen in the absorbance spectrum (Figure 3) or in the dynamic light scattering data (Figure 4C). All of the samples showed dark red colors. The only exception was the solution of nanoparticles prepared without cyclodextrin which had a faint red color. This sample contained a fraction of aggregates (Figure 4E) due to the absence of a stabilizer. A similar non-irradiated sample was colorless and did not have the characteristic peak of nanoparticles in the spectrum. TEM analysis of this sample also did not reveal the presence of nanoparticles (Figure 2F). The spectra of the reaction mixture without tetrachloroauric acid did not show this absorbance peak independently of microwave irradiation. The solutions produced by microwave heating could be stored for at least 21 months without any sign of precipitation at 4 °C. The solution produced without cyclodextrins could be stored for at least 11 months. A high stability was confirmed in most samples by high zeta potential values as follows: 119.09, 0.92, 110.77, 130.05 and 13.67 mV (given in the same order as in Figure 4). The FTIR spectra shown in Figures 1S-3S confirmed the presence of cyclodextrins on the surface of the nanoparticles. The broad absorption band centering around 3300 cm-1 that decreased in intensity corresponds to a hydroxyl group. A band around 1600 cm-1 corresponding to a carboxyl group increased in intensity when compared to that of free cyclodextrin. Therefore, the cyclodextrins underwent oxidation by tetrachloroaurate at the time of nanoparticle formation.7 The intensity of the band at 2360 cm-1 was always higher for the nanoparticle samples than for the free cyclodextrins. In addition, the band position was close to that reported for carbon dioxide encapsulated in α-cyclodextrin (about 2338 cm-1).13 These data might reflect autocatalytic decarboxylation of the oxidized cyclodextrin, as seen in encapsulated carboxylic acids,14 with a subsequent carbon dioxide complex formed with cyclodextrin.13,15 The intensities of the bands around 945 cm-1 (skeletal vibration involving α-1,4 linkage), 754 cm-1 (ring 'breathing' vibration), 704 cm-1 and 577 cm-1 (pyranose ring vibration) decreased in the gold nanoparticle spectra that suggested a 'fixed' condition of cyclodextrins 16 and confirmed their immobilization onto the nanoparticles' surfaces. The synthesis of gold nanoparticles by tetrachloroauric acid decomposition in water without reducing agents was reported,17 and the following mechanisms were proposed:
The global standard redox potential of the reaction was -0.227 V.17 However, it did not seem to be the cause of the formation of nanoparticles in the presence of cyclodextrins. The gold nanoparticle solutions prepared in the presence of cyclodextrins had a higher absorbance and therefore a higher nanoparticle concentration compared with those without cyclodextrins. The cyclodextrin oxidation occurring simultaneously with the nanoparticle formation was further proof of their participation in tetrachloroaurate reduction, because tetrachloroaurate was the only oxidant present in the reaction system. Moreover, the morphology of the nanoparticles produced without cyclodextrins (Figure 2E) was very different from the morphology of those produced in the presence of cyclodextrins (Figures 2A-2D). Irregularly shaped nanoparticles formed during tetrachloroauric acid decomposition in the absence of stabilizers were previously reported.17 In the case of γ-cyclodextrin, the nanoparticles were spherical independently of microwave irradiation (Figures 2A, 2D). Claiming tetrachloroauric acid decomposition in a thiol-containing solution to be reductant-free 19 might be controversial, because thiols used as stabilizers could reduce tetrachloroaurate.20 Nevertheless, tetrachloroaurate decomposition without the participation of cyclodextrin cannot be ruled out as a side-reaction. Reduction potentials of cyclodextrins probably differ, although there is not any literature data for reduction potentials of cyclodextrins or a comparative study of their oxidation kinetics. Only the kinetics of β-cyclodextrin oxidation by permanganate had been sufficiently studied previously and provided the first evidence of the reducing nature of β-cyclodextrin. The oxidation mechanism was revealed to be rather complex with an autocatalytic effect. The kinetics remained unaffected by increasing the ionic strength of the medium, but pH dependence was shown.21 The oxidation kinetics of various starches in the pH range of 7.5 to 11.0 was reported to be similar. There was no preference in the oxidation between amylose and amylopectin.22 Therefore, the different oxidation kinetics of cyclodextrins could not be explained by their unequal molecular weights. The C-2-OH group of one glucopyranoside unit could form a hydrogen bond with the C-3-OH group of the adjacent glucopyranose unit. A complete secondary belt formed in β-cyclodextrin with six hydrogen bonds, making it a rather rigid structure. This belt was incomplete in α-cyclodextrin while γ-cyclodextrin had a noncoplanar, more flexible structure.23 Taking the dissociation energy of these hydrogen bonds into account, the reducing ability of cyclodextrins was expected to increase in the following order: β-cyclodextrin < α-cyclodextrin < γ-cyclodextrin. The same trend was observed for the solubility of cyclodextrins due to an increase in the dissolution entropy caused by dissociation of water-water hydrogen bonds during cyclodextrin hydration.24 For the case of rubredoxin, an iron-containing protein, changes in the six hydrogen bond strengths caused a reduction potential change of up to 126 mV. By varying the length of a single hydrogen bond, the reduction potential could vary by nearly 80 mV.25 β-cyclodextrin had the least average glucopyranose ring twist of the three cyclodextrins26 which might be another reason for its smaller reducing potential. In general, the greater the power of the reducing agent, the smaller the resultant particles would be.27 Therefore, the proposed order of the reducing ability of cyclodextrins was consistent with the experimental data (average diameters of the nanoparticles calculated from hydrodynamic size distribution data in parentheses): β-cyclodextrin (26.52 nm) < α-cyclodextrin (25.42 nm) < γ-cyclodextrin (20.15 nm). Although the nanoparticles formed carry oxidized cyclodextrins with carboxyl groups giving them a negative charge, the acidification of the medium during the nanoparticle formation should be also taken into account. This acidification occurred when potassium hydroxide was consumed during tetrachloroaurate reduction, thus producing a rather strong effect in the unbuffered solution. The pH values of the nanoparticle solutions were as follows: 7.6; 8.3; 9.0; 8.1 and 11.6 (given in the same order as in Figure 4). Before the reaction, pH values of the solutions were in the range of 11.6 to 11.7. Lower pH values after the formation of nanoparticles in the presence of cyclodextrins demonstrated that more potassium hydroxide had been consumed compared with the formation of nanoparticles without cyclodextrins. Moreover, the order of cyclodextrins in terms of decreased pH was the same as that for expected reducing ability (β-cyclodextrin < α-cyclodextrin < γ-cyclodextrin). These data provided additional proof that the main reductant was cyclodextrin instead of water. Unreacted hydroxyl groups cannot be ionized at the observed pH, because they do not deprotonate at pH < 12.0 with a pKa1,2 13.5 ± 0.2 as shown for β-cyclodextrin.28 But the carboxyl groups must still be ionized at the observed pH because pKa, eff of oxidized starch, although depending on the degree of oxidation, is always less than 4.5.29 It should be noted that adsorbed cyclodextrins might lose some of their carboxyl groups because of autocatalytic decarboxylation, as discussed above. Zeta potential is a drop of the potential across the mobile part of the double layer 30 and is therefore not equal to the surface charge of the nanoparticle. The role of associated ions should also be considered because of the abundance of K+ (added as 0.98 g L-1 KOH) and Cl-. Electrolytes, especially NaCl, were shown to decrease the module of negative zeta potential of silica nanoparticles 31,32 by compressing the Stern layer.31 However, a sign change of zeta potential was not reported for the case of NaCl. β-cyclodextrin was shown to form complexes with metal cations using computational methods. The interaction energy for Na+ in the case of the most stable conformers is -304.4 kJ mol-1 in vacuum and -65.3 kJ mol-1 in water.33 Three complexes of α-cyclodextrin with K+ having free energy 0, 16 and 30 kJ mol-1, and seven complexes with Na+ having free energy 0, 2, 8, 11, 64, 72 and 132 kJ mol-1 in the gas phase were also shown.34 For Na+, Mg2+, Cu+, Zn2+, but not Al3+,33 electrostatic energy was shown to be the main source of complex stabilization. There was also a significant amount of Cl- in the solution, and this anion was known to form a complex with cyclodextrins. It was assumed that the electrolyte existed as an ion-pair inside or at the entrance of the cyclodextrin cavity.35 For this reason, the formation of complex of immobilized cyclodextrin with Cl- was not expected to make the zeta potential of the nanoparticles negative. The Ka of the complex of α-cyclodextrin with KCl at 298 K was 5 L mol-1 and with NaCl was 3 L mol-1.35 The formation of β-cyclodextrin-anion-cation mixed complexes was also very probable. The Ka of the complexes of β-cyclodextrin with NaCl and KCl at 298 K was close to zero,36 but these data were contradicted in later publications (Ka = 1.3 L mol-137 or 42.1 L mol-138 at 298 K for the complex with KCl). KAuCl4 was shown to form inclusion complexes with all three cyclodextrins, wherein tetrachloroaurate anions acting as bridges were located inside the cyclodextrin channels. They were supported between the adjacent cyclodextrin rings by hydrogen bonds, thus forming superstructures. In the case of α- and β-cyclodextrins, K+ ions acted as a linker between adjacent cyclodextrin dimers. In the case of γ-cyclodextrin, K+ ions resided outside the channel and were disordered. But in all cases, they were exposed to water molecules that kept these complexes solvated.39 Tetrachloroaurate could be reduced to zero valent metal via two distinct pathways:40 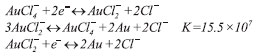 The first stage of tetrachloroaurate reduction is non-catalytic,41 but monovalent gold reduction is autocatalytic. The reaction proceeds by forming complexes of gold ions with a reductant onto the surface of the gold particle.41,42 Because cyclodextrin can form inclusion complexes with anions, cyclodextrin might bind with an intermediate AuCl2- to facilitate the formation of nanoparticles. The formation of nanoparticles during direct heating of the reaction mixture using a hot plate (Figures 5, 4S-12S) seemed to proceed differently and slower compared to microwave-assisted synthesis. As shown in the spectra of Figures 13S-21S, the absorbance maximum slowly shifted to longer wavelengths with time. Together with an increase in absorbance, these data showed that the particle size increased.43 An opposite scenario was observed when the nucleation occurred simultaneously with the growth of the existing particles. The simultaneous precipitation of very large particles made them invisible on the spectra which resulted in a decrease of solution absorbance. Dynamic light scattering data supported this conclusion (Figures 4S-12S) with peaks of smaller nanoparticles compared to those registered at the previous time point. The appearance of nanoparticles at time point zero (i.e. before heating) produced another interesting finding. After nucleation occurred upon mixing the reagents, nanoparticles grew during heating and then aggregated and precipitated with the two latter stages being accompanied by nucleation.
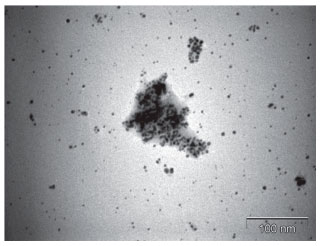 Figure 5. TEM image of a sample containing KOH, HAuCl4 and 0.002 mol L-1 β-cyclodextrin after heating for 20 minutes
Nucleation could proceed either by detaching gold nanoclusters from the surface of the growing nanoparticles44 or independently of already existing particles, possibly by the disproportionation of AuCl2- ions. Another mechanism has been proposed in order to explain the decrease of the diameter of gold nanoparticles during tetrachloroauric acid reduction by citrate.45,46 This mechanism assumes aggregation of small nanoparticles, growth of aggregated nanoparticles and subsequent breakdown of the aggregates. This mechanism was proposed only for the early stages of the reaction. However, the smaller particles also appeared at later stages in our experiments. Based on the decrease of the nanoparticle concentration in the solution as seen in the absorbance spectra and observed black precipitate, we concluded that the observed aggregation was irreversible. Nevertheless, this mechanism contributed to the overall process. The formation of gold nanoparticles in the present case differed from that described by classical LaMer theory. This theory assumed burst nucleation with subsequent growth of the size of the nuclei without further nucleation.47 Aggregation is probably caused by the increase of the ionic strength of the solution that, in turn, is caused by extensive water evaporation during prolonged heating.48 The aggregation caused some samples to show broad peaks in the absorbance spectra,43 to display peaks corresponding to hydrodynamic sizes of several hundreds of nanometers (i.e. the peaks of aggregates) and to have a dark blue color. The aggregation significantly lowered the storage stability of samples. The particles were completely precipitated after 3 days at 4 °C. The solutions became colorless, although some nanoparticle concentrations were recovered by shaking. The only exceptions were the nanoparticles produced at 0.004 M α-cyclodextrin and at all three concentrations of γ-cyclodextrin. They could be stored at least for 4 months without any sign of precipitation. Hydrodynamic size distribution data of these samples (Figures 6S, 10S, 11S, 12S) reflected significant fractions of small nanoparticles that might be less prone to aggregation. As expected, the most active cyclodextrin in the case of direct heating was γ-cyclodextrin. The activity of α- and β-cyclodextrins was comparable and much smaller than that of γ-cyclodextrin. In general, the average particle size decreased with increasing concentrations of cyclodextrin (Figures 4S-12S) as was consistent with earlier reports dealing with tetrachloroaurate reduction by sodium citrate.49 In the present case, this dependence was more complex due to multimodal size distribution of the nanoparticles. The pH change caused by the nanoparticle formation increased with the concentration of cyclodextrin, because it caused more sodium hydroxide to be consumed. For example, the pH change grew with the concentration of γ-cyclodextrin as follows: 0.7, 0.78 and 0.92. For the other two cyclodextrins, the pH change for the highest cyclodextrin concentration was also greater than those of lower concentrations. As in microwave-associated synthesis, the zeta potential of the nanoparticles synthesized using direct heating was positive (data provided with Figures 4S-12S) in most cases, but negative values were registered in some cases. This behavior might be explained by competitive adsorption of the stabilizers onto the surface of the nanoparticles. It was shown that the zeta potential sign of liposomes depended on the ionic composition of the medium, because different ions had unequal binding energy.50 Besides cyclodextrin, AuCl4- could strongly adsorb onto the gold surface.45 Moreover, bulk gold surfaces could adsorb chloride (and bromide) ions to a large extent with the resulting charge about 100 µC cm-2.51 The presence of a chloride ion in the solution must be considered, because HAuCl4 was used as a solution in dilute hydrochloric acid. Hence, Stern layer compression by electrolytes 31 did not make the data of the present experiment incomparable, because the portion of chloride ions generated from tetrachloroaurate was minute in comparison to the overall concentration of Cl-. The concentration of hydrochloric acid was not specified by the manufacturer, but we noticed that when looking at the addition of NaOH calculated from HAuCl4 without taking HCl into account, the pH of the solution was 2.64 instead of expected 10.5 (i.e. the concentration of HCl was relatively high). Therefore, the charge of the nanoparticles synthesized by tetrachloroaurate reduction with cyclodextrins was a rather complex phenomenon strongly depending on the reaction conditions which determined the following features: the surface concentration of cyclodextrin bound to the nanoparticles, the degree of oxidation of the bound cyclodextrin, the degree of its decarboxylation, the extent of consumed AuCl4_ and the behavior of ions (to bind to the surface of the nanoparticle directly or to form complex with the bound cyclodextrin), among other factors.
CONCLUSIONS Gold nanoparticles were synthesized by direct tetrachloroaurate reduction with α-, β-, and γ-cyclodextrins in a microwave oven making the process quick and simple. Tetrachloroaurate reduction by cyclodextrins did not require seeding. One main advantage was that the particles formed were already functionalized by cyclodextrin. Cyclodextrins underwent oxidation during the formation of the nanoparticles. The size of the nanoparticles was found to be dependent on the type of cyclodextrin, because it determined its reducing potential. Particularly, γ-cyclodextrin with more volume to encapsulate molecules in its cavity showed good performance for nanoparticle formation under this method of tetrachloroaurate reduction. Microwave irradiation of the alkaline solution of tetrachloroaurate without cyclodextrin also led to nanoparticles, but their concentration was significantly lower, and aggregates were present. The use of a heating plate instead of microwave irradiation led to unstable nanoparticles prone to aggregation because of the high ionic strength of the resulting solution. Nucleation occurred after mixing the reagents (i.e. even before heating) as well as during the growth of the nanoparticles.
ACKNOWLEDGEMENTS Authors thank Dr. Sánchez-Segura for his assistance in the acquisition of TEM images; A. L. Hernández Orihuela for her assistance with materials; and D. Partida Gutiérrez for zeta potential measurements, dynamic light scattering data and FTIR spectra acquisition. Authors also thank Carolyn Smith of Peace Corps Response for editing the manuscript. SUPPLEMENTARY MATERIAL FTIR spectra of cyclodextrins and of the corresponding gold nanoparticles, hydrodynamic size distribution and absorbance spectra of the nanoparticles synthesized by direct heating are available online with free access at http://quimicanova.sbq.org.br in the form of a PDF file.
REFERENCES 1. Khodaverdi, E.; Gharechahi, M.; Alibolandi, M.; Mirzazadeh Tekie, F. S.; Khashyarmanesh, B. Z.; Hadizadeh, F.; Int. J. Pharm. Invest. 2016, 6, 78. 2. Zhu, W.; Zhang, K.; Chen, Y.; Xi, F.; Langmuir 2013, 29, 5939. 3. Abu-Dahab, R.; Odeh, F.; Ismail, S. I.; Azzam, H.; Al Bawab, A.; Pharmazie 2013, 68, 939. 4. Lévy, R.; Shaheen, U.; Cesbron, Y.; Sée, V.; Nano Reviews (2010), doi:10.3402/nano.v1i0.4889. DOI: http://dx.doi.org/10.3402/nano.v1i0.4889 5. Crespo-Biel, O.; Jukovic, A.; Karlsson, M; Reinhoudt, D. N.; Huskens, J.; Isr. J. Chem. 2005, 45, 353; Liu, X.; Chen, C.; Liu, X.; Shi, J.; Wang, G.; Zhu, L.; Guo, L.; Glennon, J. D.; Scully, N. M.; Doherty, B. E.; Electrophoresis 2010, 31, 1697. 6. Liu, J.; Ong, W.; Román, E.; Lynn, M. J.; Kaifer, A. E.; Langmuir 2000, 16, 3000. 7. Huang, T.; Meng, F.; Qi, L.; J. Phys. Chem. C 2009, 113, 13636. 8. Aswathy, B.; Avadhani, G. S.; Suji, S.; Sony, G.; Frontiers of Materials Science 2012, 6, 168; Pande, S.; Ghosh, S. K.; Praharaj, S.; Panigrahi, S.; Basu, S.; Jana, S.; Pal, A.; Tsukuda, T.; Pal, T.; J. Phys. Chem. C 2007, 111, 10806. 9. Celebioglu, A.; Uyar, T.; RSC Adv. 2013, 3, 10197. 10. Gannimani, R.; Ramesh, M.; Mtambo, S.; Pillay, K.; Soliman, M. E.; Govender, P.; J. Inorg. Biochem. 2016, 157, 15. 11. Das, T.; Poria, D. K.; Purkayastha, P.; Nanomed. Nanotechnol. Biol. Med. 2016, 12, 1105. 12. Menges, F.; Spekwin32; Optical Spectroscopy Software; Universität Konstanz, Germany, 2015. 13. Neoh, T. L.; Yoshii, H.; Furuta, T.; J. Inclusion Phenom. Macrocyclic Chem. 2006, 56, 125. 14. Cramer, F.; Kampe, W.; J. Am. Chem. Soc. 1965, 87, 1115. 15. Ho, T. M.; Howes, T.; Bhandari, B. R.; Food Chem. 2015, 187, 407; Ho, T. M.; Howes, T.; Bhandari, B. R.; Food Chem. 2016, 203, 348. 16. Huang, Y.; Li, D.; Li, J.; Chem. Phys. Lett. 2004, 389, 14. 17. Vargas-Hernandez, C.; Mariscal, M. M.; Esparza, R.; Yacaman, M. J.; Appl. Phys. Lett. 2010, 96, 213115. 18. Vaks, V. L.; Domrachev, G. A.; Rodygin, Yu. L.; Selivanovskii, D. A.; Spivak, E. I.; Radiophys. Quantum Electron. (Engl. Transl.) 1994, 37(1), 85. 19. Cai, X.; Chen, H.-H.; Wang, C.-L.; Chen, S.-T.; Lai, S.-F.; Chien, C.-C.; Chen, Y.-Y.; Kempson, I. M.; Hwu, Y.; Yang, C. S.; Margaritondo, G.; Anal. Bioanal. Chem. 2011, 401, 809. 20. Pavlov, V.; Xiao, Y.; Willner, I.; Nano Lett. 2005, 5(4), 649; Love, J. C.; Estroff, L. A.; Kriebel, J. K.; Nuzzo, R.G.; Whitesides, G. M.; Chem. Rev. (Washington, DC, U. S.) 2005, 105, 1103. 21. Manhas, M. S.; Mohammed, F.; Khan, Z.; Colloids Surf., A 2007, 295, 165. 22. Tomasik, P.; Schilling, C. H.; Adv. Carbohydr. Chem. Biochem. 2004, 59, 175. 23. Szejtli, J.; Chem. Rev. (Washington, DC, U. S.) 1998, 98, 1743. 24. Sabadini, E.; Cosgrove, T.; Egídio, F. C.; Carbohydr. Res. 2006, 341, 270. 25. Lin, I-J.; Gebel, E. B.; Machonkin, T. E.; Westler, W. M.; Markley, J. L.; Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 14581. 26. French, A. D.; Johnson, G. P.; Carbohydr. Res. 2007, 342, 1223. 27. Hermanson, G. T.; Bioconjugate Techniques, 3rd ed., Academic Press: Amsterdam, Boston, Heidelberg, London, New York, Oxford, Paris, San Diego, San Francisco, Singapore, Sydney, Tokyo, 2013. 28. Gaidamauskas, E.; Norkus, E.; Butkus, E.; Crans, D. C.; Grincienė, G.; Carbohydr. Res. 2009, 344, 250. 29. Li, Y.; de Vries, R.; Slaghek, T.; Timmermans, J.; Cohen Stuart, M. A.; Norde, W.; Biomacromolecules 2009, 10, 1931. 30. IUPAC; Compendium of Chemical Technology (the "Gold Book"), 2nd ed., Blackwell Scientific Publications: Oxford, 1997. 31. Brown, M. A.; Goel, A.; Abbas, Z.; Angew. Chem. 2016, 128, 3854. 32. Chibowski, S.; Grządka, E.; Patkowski, J.; Croat. Chem. Acta 2009, 82, 623. 33. Stachowicz, A.; Styrcz, A.; Korchowiec, J.; Modaressi, A.; Rogalski, M.; Theor. Chem. Acc. 2011, 130, 939. 34. Gámez, F.; Hurtado, P.; Hortal, A. R.; Martínez-Haya, B.; Berden, G.; Oomens, J.; ChemPhysChem 2013, 14, 400. 35. Sanemasa, I.; Fujiki, M.; Deguchi, T.; Bull. Chem. Soc. Jpn. 1988, 61, 2663. 36. Buvári, A.; Barcza, L.; Inorg. Chim. Acta 1979, 33, L179. 37. Yu, Z.; Zhao, C.; Huang, Z.; Chen, H.; Yu, J.; Phys. Chem. Chem. Phys. 1999, 1, 441. 38. Santos, C. I. A. V.; Ribeiro, A. C. F.; Veríssimo, L. M. P.; Lobo, V. M. M.; Esteso, M. A.; J. Chem. Thermodyn. 2013, 57, 220. 39. Liu, Z.; Frasconi, M.; Lei, J.; Brown, Z. J.; Zhu, Z.; Cao, D.; Iehl, J.; Liu, G.; Fahrenbach, A. C.; Botros, Y. Y.; Farha, O. K.; Hupp, J. T.; Mirkin, C. A.; Stoddart, J. F.; Nat Commun. 2013, 4, 1855. 40. Pérez-Juste, J.; Liz-Marzán, L. M.; Carnie, S.; Chan, D. Y. C.; Mulvaney, P.; Adv. Funct. Mater. 2004, 14, 571. 41. Tatarchuk, V. V.; Bulavchenko, A. I.; Russ. J. Inorg. Chem. 2004, 49, 1225. 42. Bulavchenko, A. I.; Arymbaeva, A. T.; Tatarchuk, V. V.; Russ. J. Phys. Chem. A 2008, 82, 801. 43. Pestovsky, Y. S.; All-Russian journal of scientific publications 2013, 5, 6. (In Russian) 44. Zayats, M.; Baron, R.; Willner, I.; Nano Lett. 2005, 5, 21. 45. Biggs, S.; Chow, M. K.; Zukoski, C. F.; Grieser, F.; J. Colloid Interface Sci. 1993, 160, 511. 46. Chow, M. K.; Zukoski, C. F.; J. Colloid Interface Sci. 1994, 165, 97. 47. LaMer, V. K.; Dinegar, R. H.; J. Am. Chem. Soc. 1950, 72, 4847. 48. Pestovsky, Y. S.; International Journal Of Applied And Fundamental Research 2016, 4, 1077. (In Russian) 49. Frens, G.; Nature Physical Science 1973, 241, 20. 50. Šegota, S.; Vojta, D.; Kendziora, D.; Ahmed, I.; Fruk, L.; Baranović, G.; J. Phys. Chem. B 2015, 119, 5208. 51. Paik, W.; Genshaw, M. A.; Bockris, J. O'M.; J. Phys. Chem. 1970, 74, 4266. |
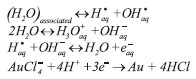
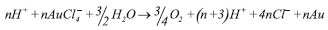
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access