
 |
 |
 |
 |
 |
Artigo
|
|
| Avaliação do papel do óxido de grafeno (GO) na geração fotocatalítica de hidrogênio em sistemas binários (GO-CdS) e ternários (Pt-GO-CdS) Evaluation of the graphene oxide (GO) role in the photocatalytic generation of hydrogen in binary (GO-CdS) and ternary (Pt-GO-CdS) systems |
|
Cristiane Gomes AlmeidaI, Tuany Nascimento dos Santos TrindadeI, Marcus Vinicius Santos da SilvaIII e Luciana Almeida SilvaI,II,*
I. Instituto de Química, Universidade Federal da Bahia, 40170-115 Salvador – BA, Brasil Recebido em 06/02/2018 *e-mail: las@ufba.br Water splitting is a promising process to produce hydrogen from friendly feedstock and solar energy. In this work we have evaluated binary (GO-CdS) and ternary (Pt-GO-CdS) hybrid photocatalysts for hydrogen production assisted by visible light irradiation. Cadmium sulfide and composites with GO were prepared by sonochemical and thermal methods. GO addition took place by different strategies: during the synthesis or by mechanical mixture. A variety of configurations was tested and the best performance in hydrogen production among all materials was the ternary photocatalyst named Pt(GO/CdSTT), whose hydrogen production rate was 651 µmol gcat-1 h-1. Such material was obtained by thermal method with GO addition during the synthesis. Additionally, the XRD and Raman analyses have confirmed the GO photoreduction during photocatalytic hydrogen evolution. INTRODUÇAO Recentemente, o hidrogênio tornou-se foco de uma verdadeira corrida científica no sentido de torná-lo, em definitivo, um vetor que irá compor a matriz energética em substituiçao aos derivados de petróleo. Diversas alternativas de produçao já foram propostas e, constantemente, tecnologias de conversao de hidrogênio em diferentes formas de energia sao aprimoradas, a exemplo das células a combustível. No entanto, ainda cerca de 95% do hidrogênio produzido sao provenientes de combustíveis fósseis via reforma a vapor do gás natural.1,2 Neste cenário, é necessário o desenvolvimento de novas tecnologias que permitam produzir hidrogênio em larga escala utilizando fontes primárias renováveis e inesgotáveis, tais como energia solar, água e biomassa; de modo que as necessidades energéticas da humanidade sejam supridas com um combustível, de fato, limpo. A decomposiçao direta da água em hidrogênio e oxigênio usando luz solar com auxílio de um semicondutor é um meio de imitar a fotossíntese das plantas e se configura como processo promissor com grande potencial de aplicaçao em larga escala. Esse processo é capaz de captar e distribuir energia solar armazenada na ligaçao H-H. O princípio da fotodecomposiçao da água já é bem conhecido desde a década de setenta quando Honda e Fujishima3 conseguiram produzir hidrogênio e oxigênio usando uma célula fotoeletroquímica, tendo TiO2 como fotoanodo e platina como fotocatodo. Uma partícula semicondutora com uma pequena quantidade de nanopartículas de platina depositada, ou outro metal adequado, é essencialmente uma célula fotoeletroquímica em miniatura, onde a água, ou outro doador de elétrons (hole scavenger), é oxidada na parte exposta do semicondutor (fotoanodo) e é reduzida na superfície das nanopartículas do metal (fotocatodo, que ainda atua como cocatalisador para a formaçao de hidrogênio molecular), resultando na decomposiçao fotocatalítica da água. Sob irradiaçao com fótons de energia superior à energia de bandgap (Eg, energia que separa a banda de valência (BV) da banda de conduçao (BC) do semicondutor/fotocatalisador), elétrons na BV sao excitados para a BC, gerando o par elétron-lacuna que participa de reaçoes redox.4-7 Para realizar a fotodecomposiçao da molécula de água, é necessário que o limiar da banda de conduçao do semicondutor esteja posicionado em potenciais mais negativos que o potencial de reduçao da H2O a H2 (Er° = -0,83 V vs EPH em pH básico, ou Er° (H+/H2) = 0 V em pH = 0), enquanto que o limiar da banda de valência deve estar posicionado em potenciais mais positivos que aquele de oxidaçao de H2O a O2 (Er° = +1,23 V vs EPH, pH = 0 ou Er° (OH-/O2) = +0,40 V em pH básico). Termodinamicamente, a decomposiçao da água é uma reaçao nao espontânea (ΔG° = +238 kJ mol-1; ΔE° = -1,23 V), sendo necessário o suprimento de energia que coincide com fótons de energia de λ ≤ 1000 nm (i.e., 1,23 eV ≈ energia de fótons de 1000 nm). Isto significa que a energia necessária para a produçao fotocatalítica de hidrogênio a partir da água pode ser suprida pelo Sol. Desde a década de 1980 o sulfeto de cádmio é um dos fotocatalisadores mais estudados na decomposiçao fotocatalítica da água sob irradiaçao de luz visível,8-20 que constitui a maior porçao da radiaçao solar. Mas problemas associados à rápida recombinaçao de cargas fotogeradas, baixa estabilidade química em meio aquoso e os desafios no sentido de melhorar sua atividade fotocatalítica justificam o interesse constante em explorar esse material.21 O CdS é um semicondutor do tipo n, com energia de bandgap de 2,41 eV, portanto, ativo com luz visível, e potenciais de banda adequados para conduzir as reaçoes de oxidaçao e reduçao da água (E°BV = +1,41 V e E°BC = -1,0 V). Em geral, problemas associados à fotocorrosao de CdS sao minimizados com o emprego de doadores de elétrons, especialmente, os sistemas contendo o par S2-/SO32- em meio básico,14 ou adiçao de um eletrólito quando o reagente de sacrifício é um álcool.22 Já problemas associados à recombinaçao de cargas sao contornados com a introduçao de átomos à rede de sulfeto formando soluçoes sólidas23 ou com a associaçao a outro material em um compósito para a formaçao de heterojunçao.24 Estudos apontam que sulfeto de cádmio em fase hexagonal apresenta maior atividade fotocatalítica na geraçao de hidrogênio comparada à fase cúbica.8,14,24,25 Geralmente, a fase hexagonal é obtida com o tratamento térmico de CdS comercial em atmosfera de nitrogênio e, recentemente, nós desenvolvemos um método sonoquímica para preparar nanoesferas de CdS hexagonal.25 Diversos metais podem ser empregados como fotocatodo na decomposiçao fotocatalítica da água, tais como Pt, Rh, Ru, Au, Pd e Ag, sendo a platina o mais usado.26,27 Nesses sistemas, os elétrons fotogerados com a excitaçao do semicondutor migram para as nanopartículas do metal, através da heterojunçao, até o alinhamento dos níveis de Fermi, gerando a barreira Schottky na interface semicondutor/metal.28 A formaçao dessa barreira funciona como uma armadilha eletrônica impedindo a recombinaçao de cargas elétron-lacuna, evitando perda de energia na forma de calor. Nas reaçoes de geraçao fotocatalítica de hidrogênio, os elétrons aprisionados sao transferidos para prótons adsorvidos à superfície do metal que irao promover sua reduçao a hidrogênio molecular, enquanto que as lacunas fotogeradas no semicondutor irao oxidar a água ou um reagente de sacrifício.27 Deste modo, o semicondutor atua como anodo e o cocatalisador atua como catodo, semelhante a uma célula fotoeletroquímica. Nos últimos anos, o grafeno tem despertado interesse de diversos grupos de pesquisa ao redor do mundo, inclusive os dedicados à produçao fotocatalítica de hidrogênio, cujo objetivo é desenvolver metodologias capazes de obter compósitos de semicondutores com óxido de grafeno (GO) ou óxido de grafeno reduzido (RGO) de modo que estes desempenhem papel similar ao da platina ou atuem de forma sinérgica.29-41 O grafeno tem grande área superficial específica e elevada mobilidade eletrônica (10.000 cm2 V-1 s-1),35 propriedades que o tornam perfeito para atuar como aprisionador reversível e transportador de elétrons fotogerados em processos fotocatalíticos. Desta forma, é desejável que nesses sistemas prevaleça a forma reduzida (RGO), visto que a forma oxidada (GO) pode aprisionar irreversivelmente os elétrons fotogerados.36 Em geral, os compósitos semicondutor/RGO sao preparados a partir do GO obtido em etapas preliminares de oxidaçao do grafite e posterior esfoliaçao, com subsequente etapa de reduçao. A reduçao química é preferencialmente empregada que métodos alternativos, tais como térmico, eletroquímico e micro-ondas, por causar menos distorçoes e defeitos na regeneraçao da rede π conjugada de átomo de carbono sp2.37 Recentemente, diferentes estratégias vêm sendo testadas para preparar compósitos GO/CdS empregando diferentes métodos de síntese, tais como solvotérmico, coprecipitaçao e reaçao gás-sólido.36-41 No presente trabalho, foram empregados dois diferentes métodos de síntese, sonoquímico e térmico, para preparar compósitos de CdS hexagonal com óxido de grafeno, assim como foram feitas misturas mecânicas desses componentes, para avaliar a influência do método de preparaçao na atividade fotocatalítica para geraçao de hidrogênio em presença do par de agentes redutores S2-/SO32-. A possibilidade de reduçao simultânea do óxido de grafeno, que elimina uma etapa prévia de reduçao no procedimento de preparo dos fotocatalisadores, também foi avaliada empregando técnicas de difratometria de raios X e espectroscopia Raman.
PARTE EXPERIMENTAL Materiais Os reagentes utilizados nas sínteses e testes fotocatalíticos sao de grau analítico, exceto as soluçoes, e todos foram utilizados sem etapa prévia de purificaçao. Grafite, permanganato de potássio, sulfeto de sódio nonahidratado, etilenoglicol e soluçoes de ácido hexacloroplatínico 8% (m/m) e peróxido de hidrogênio 30% (m/m) foram adquiridos da Sigma Aldrich. Cloreto de cádmio, sulfeto de cádmio, sulfeto de sódio e tiossulfato de sódio foram adquiridos da Merck. O ácido sulfúrico usado foi da Qhemis, enquanto que brometo de cetiltrimetilamônio (CTBA) da VETEC. Síntese do óxido de grafite (GrO) O óxido de grafite foi preparado pelo método de Hummers modificado, que consiste na oxidaçao do grafite por permanganato de potássio, KMnO4, seguindo protocolo descrito por Mehl e colaboradores.42 Síntese de CdS Método sonoquímico O procedimento de síntese sonoquímica do sulfeto de cádmio hexagonal consistiu na dissoluçao de 0,9599 g de CdCl2, 1,5055 g de Na2S2O3 e 0,20 g de brometo de cetiltrimetilamônio (CTAB) em um béquer contendo 50 mL de etilenoglicol. Em seguida, a soluçao foi exposta à radiaçao ultrassônica usando uma sonda ultrassônica desruptora de célula R2D091109 da Unique, com os seguintes parâmetros: potência de 80 W, ponteira de titânio diretamente imersa até cerca de 80% da mistura no béquer por 15 minutos. Ao final da reaçao, um precipitado amarelo foi obtido, separado por centrifugaçao e lavado, na sequência, duas vezes com água deionizada, duas vezes com etanol e seco à temperatura ambiente por 48 horas. Método térmico O sulfeto de cádmio em fase hexagonal também foi obtido por tratamento térmico do CdS comercial em atmosfera dinâmica de nitrogênio a 700 ºC por 1 h. Preparo dos compósitos Os compósitos foram preparados com adiçao de quantidades apropriadas de GrO, de modo que o teor final de GO fosse de 2% (m/m), seguindo diferentes procedimentos; i) adiçao de 20 mg de óxido de grafite (GrO) à mistura reacional para obtençao de 1 g de sulfeto de cádmio sonoquímico, amostra denominada de GO/CdSson; ii) adiçao de 20 mg de GrO a 1 g de CdS comercial em 50 mL de água e a suspensao sonicada por 15 minutos; em seguida, o sólido foi separado por centrifugaçao, seco e submetido a tratamento térmico em atmosfera de nitrogênio por 1 h. Esta amostra foi denominada de GO/CdSTT; iii) misturas mecânicas de GrO com CdS obtido pelo método sonoquímico e GrO com CdS obtido pelo método térmico, seguidas de tratamento com radiaçao ultrassônica para esfoliaçao; amostras denominadas respectivamente de GO+CdSson e GO+CdSTT. Nestes casos, 3 mg de GrO, pesados em uma microbalança Mettler Toledo, modelo MX5, d = 1 µg, foram misturados mecanicamente a 147 mg de CdS; iv) adiçao de soluçao de H2[PtCl6].6H2O 8% (m/m) à mistura submetida ao teste fotocatalítico, em que Pt(IV) é fotorreduzida a Pt0 in situ. Nestes casos, as amostras foram denominadas de Pt(GO/CdSson), Pt(GO+CdSson), Pt(GO/CdSTT) e Pt(GO+CdSTT), além das amostras sem GO, denominadas Pt/CdSson e Pt/CdSTT. Caracterizaçao Todos os materiais preparados foram caracterizados por difratometria de raios X, empregando um equipamento da Shimadzu modelo XRD 6000, radiaçao CuKα e filtro de níquel. A velocidade de varredura programada foi de 2º 2θ min1, na regiao 2θ de 5-80º, em 35 kV e 15 mA. Os difratogramas obtidos foram comparados com o banco de dados de padroes ICSD (Inorganic Crystal Structure Database) e dados da literatura para identificar as fases cristalinas das amostras. Os materiais ante e após irradiaçao foram caracterizados por Espectroscopia Raman, empregando um espectrômetro Raman dispersivo da Jasco NRS-5100, com resoluçao 0,4 cm-1. Testes fotocatalíticos Os testes fotocatalíticos foram conduzidos empregando 150 mg do fotocatalisador em 100 mL de soluçao aquosa de NaOH 1 mol L-1, contendo 0,1 mol L-1 de Na2S.9H2O e 0,02 mol L-1 de Na2SO3, adicionados a um reator com capacidade de 180 mL. Nos testes contendo platina, esta foi fotodepositada in situ com adiçao do 40 µL do precursor ácido hexacloroplatínico hexahidratado (H2[PtCl6].6H2O 8% m/m). O reator empregado é equipado com entrada e saída de gás, conectadas em linha a um cromatógrafo para a retirada periódica de alíquotas da fase gasosa com auxílio de uma bomba de gás SCHARZER, modelo PN SP625EC. Antes do início da irradiaçao, para todos os sistemas, foi feita purga com argônio ultrapuro por 1 h a fim de eliminar o ar atmosférico para que a reaçao aconteça em meio anaeróbico. Oxigênio deve ser removido do meio, visto que este concorre com a água na reaçao com elétrons fotogerados para formar radicais superóxido. As reaçoes fotocatalíticas foram conduzidas em atmosfera de argônio, à temperatura ambiente, com uma ventoinha posicionada na direçao do reator para evitar variaçoes significativas de temperatura. O reator foi irradiado com luz visível, empregando uma lâmpada de arco xenônio da Newport, com potência fixada em 500 W e área de exposiçao de 19,6 cm2. Para seleçao da faixa espectral foram utilizados filtros de corte de radiaçao ultravioleta (λ < 400 nm) e radiaçao infravermelha (filtro de água Newport) com refrigeraçao por meio de um banho termostático Quimis. Foram realizadas injeçoes no cromatógrafo de 1 em 1 h, em um total de 5 horas de irradiaçao. O hidrogênio molecular produzido nas reaçoes fotocatalíticas foi analisado em um cromatógrafo a gás, Shimadzu CG-2014, com detecçao de condutividade térmica (TCD), empregando coluna empacotada de peneira molecular, com as seguintes condiçoes cromatográficas: argônio como gás de arraste a uma vazao de 10 mL min-1 e temperatura de detecçao de 200 °C. Nestas condiçoes, o tempo de retençao do hidrogênio é de 3 minutos, o qual foi quantificado por comparaçao com padrao de calibraçao de 5% H2 em argônio.
RESULTADOS E DISCUSSAO A Figura 1 mostra os padroes de difraçao de raios X do grafite comercial (a) e dos materiais obtidos após sua oxidaçao (b) e esfoliaçao com tratamento ultrassônico por 15 minutos em água (c) e em soluçao aquosa de S2-/SO32- em meio básico (d), reproduzindo as condiçoes de reaçao dos testes fotocatalíticos sem o fotocatalisador (CdS). Além disso, apresenta as amostras irradiadas com luz visível sem platina (e) e com platina (f) após o tratamento com radiaçao ultrassônica. O difratograma de raios X da amostra oxidada (b) apresenta o pico centrado em 26,68º (2θ) referente ao conjunto de planos (002) do grafite, que sofre alargamento em funçao do processo de oxidaçao. Além desse pico, surge o pico em 10º (2θ) associado ao conjunto de planos (002) do óxido de grafite (GrO). A esfoliaçao em água (c) resultou em um sólido com padrao de difraçao semelhante à amostra oxidada. O padrao de difraçao de raios X para o óxido de grafeno quando a esfoliaçao é bem sucedida apresenta um aumento na intensidade do pico em 10º e o desaparecimento do pico referente ao grafite (~27º), indicando que as lâminas do óxido de grafite sofreram desagregaçao para formar o GO.42-44 Nesse trabalho, o tratamento com radiaçao ultrassônica em meio redutor resultou na formaçao de fases segregadas nao identificadas, provavelmente de compostos de enxofre com o material carbonáceo. Quando irradiado na ausência de platina, surgem picos adicionais e aumento nas intensidades dos já existentes. Já quando o GrO é esfoliado e irradiado nas mesmas condiçoes e com adiçao de platina, o número de picos referentes a fases segregadas diminui, assim como suas intensidades. O pico referente ao conjunto de planos (002) do grafite é regenerado e deslocado para 25,96º (2θ), indicando aumento na distância interplanar, assim como o alargamento do pico que pode estar associado à diminuiçao do grau de empilhamento, resultando no agregado de um número pequeno de lâminas. Esses resultados constatam os já relatados,45,46 indicando que a reduçao fotoquímica do GO pode ocorrer mesmo sem o fotocatalisador, resultado da geraçao do par elétron-lacuna com a fotoexcitaçao do próprio GO. Os elétrons fotogerados atuam na reduçao do GO, enquanto que as lacunas oxidam o agente de sacrifício formado pelo par S2-/SO32-.
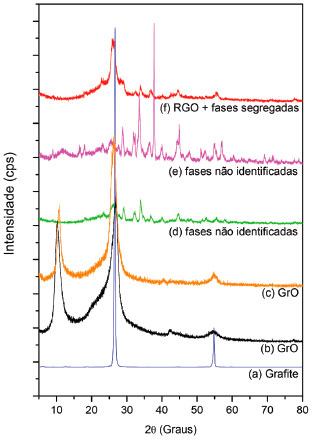 Figura 1. Difratogramas de raios X das amostras de grafite (a), óxido de grafite, GrO, (b), GrO sonicado em água (c), GrO sonicado em soluçao aquosa contendo o par redox S2-/SO32- em meio básico (d), GrO esfoliado (em sol. S2-/SO32-) e irradiado sem platina (e), GrO esfoliado (em sol. S2-/SO32-) e irradiado em presença de platina (f)
Os difratogramas de raios X das amostras de CdS preparadas pelos métodos sonoquímico (CdSson) e térmico (CdSTT), assim como os dos compósitos GO/CdSson e GO/CdSTT sao apresentados na Figura 2, acompanhados do padrao difraçao de CdS em fase hexagonal (pdf 01-075-1545-41-1049) para comparaçao. Em todos os casos é possível identificar os conjuntos de picos característicos dessa fase, porém, as amostras obtidas pelo método térmico apresentam picos estreitos e bem resolvidos, evidenciando elevada cristalinidade, característico de materiais obtidos por via térmica. Nos DRX das amostras de CdS térmico também é possível identificar uma fase segregada minoritária de CdO (pdf 01-073-2245-5-640), formada, provavelmente, devido à presença de traços de oxigênio durante o tratamento térmico em atmosfera de nitrogênio. Já os difratogramas das amostras de CdS obtidos pelo método sonoquímico nao apresentam picos referentes à fase segregada, mas os picos associados à fase hexagonal sao alargados em funçao da diminuiçao do tamanho dos cristalitos (uma discussao completa acerca da caracterizaçao das nanopartículas de CdS obtidas por via sonoquímica pode ser encontrada na referência 25). Os difratogramas dos compósitos GO/CdSson e GO/CdSTT nao apresentam diferenças significativas quando comparados aos de CdS puro obtido pelo respectivo método de síntese, nao sendo possível identificar picos associados aos materiais carbonáceos tais como grafite, óxido de grafeno ou óxido de grafeno reduzido.
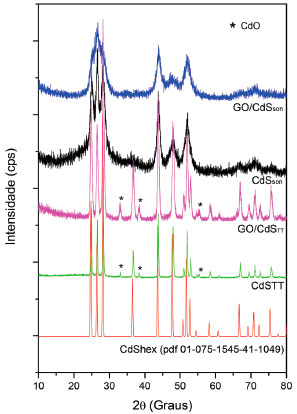 Figura 2. Difratograma de raios X de CdS puro obtido pelos métodos sonoquímico (CdSson) e térmico (CdSTT) e na forma de compósitos com GO (GO/CdSson e GO/CdSTT) e padrao de difraçao de CdS em fase hexagonal (pdf # 01-075-1545-41-1049)
Os diâmetros médios de cristalitos das amostras de CdS puro e na forma de compósitos foram determinados a partir de dados de raios X. A equaçao de Scherrer (equaçao 1) foi empregada nos cálculos, considerando as partículas esféricas; onde D é o diâmetro médio dos cristalitos, λ é o comprimento de onda dos raios X (1,541 Å), B é a largura a meia altura de um pico de difraçao em 2θ.  O pico centrado em torno de 43,8° (2θ) foi selecionado para os cálculos por ser o de melhor resoluçao com maior intensidade nas quatro amostras. Os resultados encontram-se na Tabela 1 e constatam que o método sonoquímico é capaz de produzir cristalitos com diâmetro médio menor que o método térmico, como previsto.25 Além disso, a adiçao de GO às amostras de CdS durante a síntese contribui para reduçao do tamanho de partícula. No caso específico de GO/CdSTT, essa reduçao no diâmetro de cristalitos pode estar associada, também, à etapa de tratamento ultrassônico com o GO durante o preparo do compósito.

Todas as amostras foram empregadas como fotocatalisadores na reaçao de geraçao de hidrogênio sob irradiaçao de luz visível (λ > 400 nm), usando o sistema S2-/SO32- como reagentes de sacrifício em meio básico. Neste sistema, o sulfeto age como doador de elétrons, sendo oxidado a polissulfetos, enquanto o sulfito atua como agente regenerador dos íons sulfeto, indo a tiossulfato, de acordo com as equaçoes 2 e 3.14 A regeneraçao do sulfeto por íons sulfito reestabelece a transparência da soluçao que se torna amarelada com a presença dos polissulfetos, já o meio básico evita a perda de sulfeto como H2S. 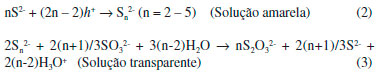 A Figura 3 mostra os perfis de evoluçao de hidrogênio em funçao do tempo de irradiaçao para as amostras com CdS hexagonal obtido pelo método sonoquímico (CdSson, GO/CdSson, GO+CdSson, Pt(GO/CdSson), Pt(GO+CdSson) e Pt/CdSon).
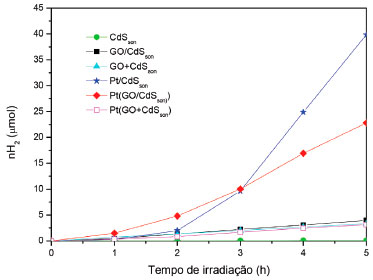 Figura 3. Produçao fotocatalítica de hidrogênio (λ > 400 nm) empregando CdS puro e compósitos com GO e/ou Pt em diferentes configuraçoes obtidos pelo método sonoquímico
Pode-se notar que o CdS puro obtido pelo método sonoquímico apresenta atividade fotocatalítica negligenciável quando irradiado com luz visível na presença dos reagentes de sacrifício S2-/SO32-. A adiçao do óxido de grafeno contribui para promover um pequeno aumento na atividade fotocatalítica dos sistemas binários GO/CdSson e GO+CdSson, mas nao há distinçao entre os métodos de adiçao de GO na preparaçao dos compósitos. No entanto, o método de adiçao de GO é determinante na atividade fotocatalítica dos sistemas ternários Pt(GO/CdSson) e Pt(GO+CdSson), com um aumento significativo da atividade fotocatalítica quando GO é adicionado durante a síntese sonoquímica do CdS, o que proporciona sua maior dispersao no compósito. Das amostras avaliadas dentro desse grupo, a que apresentou maior atividade fotocatalítica foi a amostra Pt/CdSson, indicando que platina é um cocatalisador mais ativo que o GO nesse sistema e a introduçao do óxido de grafeno na formaçao do compósito com CdS obtido pelo método sonoquímico nao contribui para melhorar a fotoatividade na produçao de hidrogênio. Os perfis de evoluçao de hidrogênio em funçao do tempo para as amostras contendo CdS hexagonal obtido pelo método térmico sao mostrados na Figura 4. Os resultados revelam que a adiçao apenas de GO por mistura mecânica inibe a atividade fotocatalítica do CdS térmico (amostra GO+CdSTT), mas quando GO é disperso com radiaçao ultrassônica e tratado termicamente junto com CdS comercial (GO/CdSTT) esta é melhorada. A adiçao de platina aumenta significativamente a atividade fotocatalítica de ambos, especialmente de Pt(GO/CdSTT) que tem a fotoatividade melhorada em mais de dez vezes quando comparada ao mesmo material sem a platina e em cerca de três vezes quando comparada ao material sem GO e com platina.
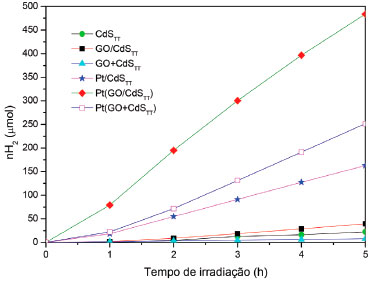 Figura 4. Produçao fotocatalítica de hidrogênio (λ > 400 nm) empregando CdS puro e compósitos com GO e/ou Pt em diferentes configuraçoes obtidos pelo método térmico
A Tabela 2 compara as taxas de evoluçao de hidrogênio para amostras obtidas pelo método sonoquímico e pelo método térmico e revela que os fotocatalisadores obtidos pelo segundo método sao mais ativos que aqueles obtidos pelo primeiro. Pode-se concluir que a maior fotoatividade na geraçao de hidrogênio está associada à maior cristalinidade dos materiais obtidos pelo método térmico, à maior dispersao do GO no compósito quando inserido durante a síntese e à presença da platina que atua como cocatalisador, além de auxiliar no processo de reduçao do óxido de grafeno como sugerem os resultados de difratometria de raios X apresentados na Figura 1. Sólidos mais cristalinos apresentam menor número de defeitos, geralmente, relacionados ao fenômeno de recombinaçao de cargas fotogeradas com a excitaçao do semicondutor, o que resulta em perda de energia na forma de calor. Esse fenômeno diminui a quantidade de portadores de carga e limita sua transferência na interface fotocatalisador/soluçao para promover as reaçoes redox. A forma de adiçao do GO também é um fator que interfere na fotoatividade, visto que a adiçao de GO durante o processo de síntese, em geral, resulta em maiores taxas de produçao de hidrogênio quando comparada à mistura mecânica, provavelmente, devido à maior dispersao do GO no compósito.

A amostra Pt(GO/CdSTT) foi a que apresentou maior fotoatividade, com uma taxa de produçao de hidrogênio de 651,6 µmol gcat-1 h-1. Esse resultado é superior aos obtidos por Leo e colaboradores36 e Singh e colaboradores37 que avaliaram a atividade fotocatalítica de compósitos GO/CdS obtidos pelo método solvotérmico, seguido de reduçao química, e pelo método de reaçao gás-sólido, respectivamente, empregando o mesmo par de reagentes de sacrifício. Outros trabalhos38-41 apresentaram taxas de produçao de hidrogênio superiores, especialmente quando o método solvotérmico é empregado na síntese dos fotocatalisadores; porém, nesses casos, foram feitas modificaçoes químicas no GO, tais como inserçao de dopantes, e outros reagentes de sacrifício foram usados. Para avaliar a restauraçao da rede π conjugada de átomos de carbono sp2 pelo processo de fotorreduçao do óxido de grafeno (GO), os compósitos foram coletados ao final das 5 horas de irradiaçao para análise por difratometria de raios X e espectroscopia Raman e comparados com os materiais antes da irradiaçao. Os fotocatalisadores pós irradiados foram submetidos à análise de difraçao de raios X para identificar picos referentes aos materiais carbonáceos. Nestes casos, as amostras pós irradiadas foram renomeadas substituindo GO por RGO, visto que há indícios de reduçao do óxido de grafeno durante as reaçoes fotocatalíticas. A Figura 5 mostra os difratogramas de raios X das amostras de CdS e compósitos após irradiaçao e compara com as amostras de CdS puro e compósito GO/CdS antes da irradiaçao para os dois métodos de síntese: (a) sonoquímico e (b) térmico. Os DRX das amostras GO/CdSson e GO/CdSTT (antes da irradiaçao) apresentam apenas picos referentes ao CdS em fase hexagonal e é semelhante ao padrao de difraçao do CdS puro obtido pelo respectivo método de síntese, nao sendo possível identificar o pico em 10º (2θ) associado ao conjunto de planos (002) do GO. Entretanto, após irradiaçao, todas as amostras, exceto RGO/CdSTT e Pt(RGO/CdSTT), apresentaram um pico largo em 21,04º (2θ), associado ao óxido de grafeno reduzido.42-44 A regeneraçao do pico referente ao conjunto de planos (002) do grafite, originalmente em 26,68º (2θ), agora deslocado para 21,04º (2θ), indica um aumento significativo da distância interplanar, de 3,34 Å no grafite para 4,22 Å no óxido de grafeno reduzido presente nos compósitos após irradiaçao, assim como o alargamento do pico está associado a diminuiçao do grau de empilhamento das lâminas de grafeno. Os DRX das amostras RGO/CdSTT e Pt(RGO/CdSTT), embora apresentem o pico referente ao conjunto de planos (002) deslocados para menor ângulo em relaçao grafite, o deslocamento foi menor comparado ao das demais amostras, assim como sua intensidade, provavelmente devido à maior agregaçao das lâminas, como consequência do tratamento térmico no processo de obtençao desses fotocatalisadores.
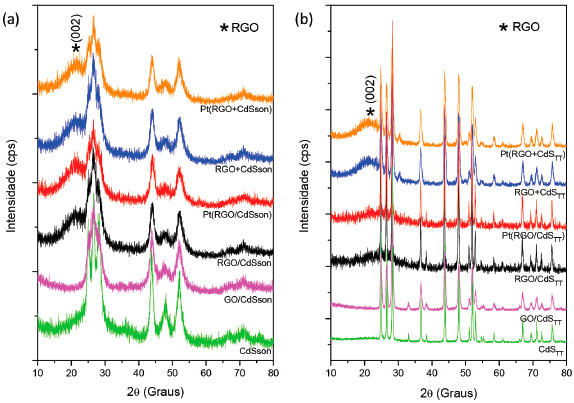 Figura 5. difratogramas de raios X de CdS e compósitos, antes e depois da irradiaçao, obtidos pelos dois métodos de síntese: (a) sonoquímico e (b) térmico
O processo de reduçao do óxido de grafeno também foi acompanhado por espectroscopia Raman. Esta técnica aplicada à análise do grafeno fornece informaçoes únicas em termos estrutural, eletrônico e vibracional pelo fato da maioria das transiçoes serem ressonantes. Essas transiçoes no grafeno ocorrem entre as regioes dos pontos de alta simetria, Γ, K, M, K', que limitam a chamada primeira zona de Brillouin,47,48 da célula unitária do grafeno. Além disso, as bandas de energia do material apresentam valores de mínimo e máximo em torno desses pontos, formando cones onde podem ocorrer diversas transiçoes, tanto internas (intravale) quanto externas (intervale). As medidas Raman foram utilizadas no monitoramento das intensidades da banda G (1585 cm-1), associada a modos vibracionais de primeira ordem do fônon E2g dos átomos de carbono sp2,49,50 bem como da banda D (1350 cm-1), associada a defeitos da estrutura. Esta última só é visível em estruturas carbonáceas com defeitos devido ao espalhamento inelástico e recombinaçao de portadores de carga excitados na rede.48,51 No entanto, a medida absoluta dessas intensidades nao é tarefa fácil. Por conta disso, a razao ID/IG é largamente utilizada para avaliar o grau de desordem do material.48,52 De igual forma, foi possível monitorar a banda G' (2718 cm-1),53 que apresenta dois picos (2D1 e 2D2) resultantes da interaçao de dois planos vizinhos, e da banda G* (2430 cm-1),53 fruto de um processo intervale de segunda ordem originário de transiçoes envolvendo um fônon longitudinal acústico (LA) e outro transversal óptico (iTO) na regiao do ponto Γ, centro da primeira zona de Brillouin.47 A Figura 6 compara os espectros dos compósitos preparados pelo método sonoquímico (a) e pelo método térmico (b), com introduçao de GO no processo de síntese, antes e após a irradiaçao. O perfil dos espectros revela o efeito da irradiaçao no processo de reduçao do GO. Observa-se uma melhora na estrutura do material carbonáceo nos compósitos após irradiaçao, seja pela diminuiçao na razao ID/IG, seja pelo surgimento da banda G', indicando um menor grau de desordem. A melhor eficiência no processo de reduçao de GO é verificada para o compósito preparado pelo método térmico, no qual a razao ID/IG diminui consideravelmente. No caso do compósito preparado pelo método sonoquímico, apesar do pequeno aumento da razao ID/IG, há também um aumento nas intensidades tanto da banda G quanto da G' revelando também uma melhora estrutural do RGO nesse compósito.
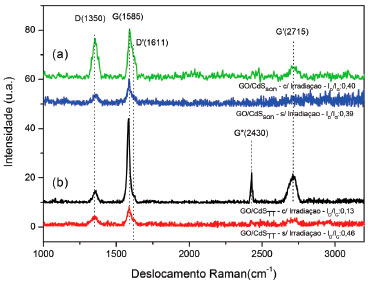 Figura 6. Espectros Raman dos compósitos GO/CdS, antes e após a irradiaçao, obtidos pelas vias sonoquímica (a) e térmica (b)
Uma vez confirmado o efeito positivo da irradiaçao no processo de reduçao do GO, o passo seguinte foi comparar os espectros dos compósitos preparados pelo método sonoquímico (a) e pelo método térmico (b), com introduçao de GO no processo de síntese ou por mistura mecânica nas diferentes configuraçoes após irradiaçao, como mostra a Figura 7 (a e b).
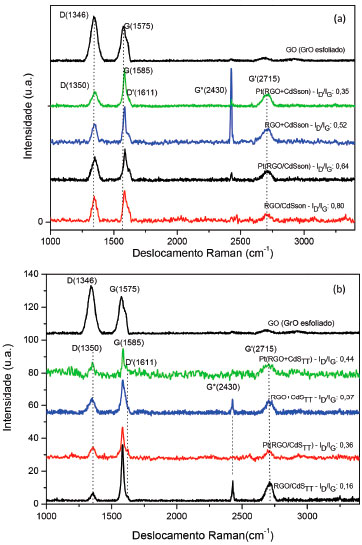 Figura 7. Espectros Raman do óxido de grafite esfoliado e dos compósitos de CdS pós irradiados obtidos por vias sonoquímica (a) e térmica (b)
Os espectros Raman (Figura 7) confirmam a reduçao do GO após irradiaçao nos demais compósitos produzidos com CdS pelas vias sonoquímica e térmica, conforme indicado anteriormente na Figura 6, bem como para os diferentes métodos de introduçao do GO: reacional e mecânico. Os espectros que apresentam uma menor desordem sao aqueles das amostras produzidas pelo método térmico (Figura 7b), em que sao observadas menores razoes ID/IG, tanto para a adiçao de GO durante à síntese quanto na mistura mecânica, corroborando os resultados de difraçao de raios X de uma melhor cristalinidade para esses compósitos. A banda G', associada à quantidade de camadas de grafeno,54,49 indica, em todos os espectros, estruturas de multicamadas, visto que a intensidade desta banda é bastante atenuada. No entanto, pela posiçao e forma do pico, há uma indicaçao de poucas camadas (~ 3 - 5), confirmada pela pequena largura (FWHM) do pico G*.53,54 Verificam-se, ainda nos espectros, dois picos de dupla ressonância: a transiçao D' (~1620 cm-1),53,55 relacionada ao processo intravale no cone do ponto K, e que também está associada à desordem,53 mas com menor intensidade que a banda D; e o pico G* (exp ~2430 cm-1),56 que apresenta uma dispersao negativa,48,52 ou seja, a posiçao do pico se desloca para valores menores à medida que a energia do laser de excitaçao aumenta.57 Comparando os espectros Raman das amostras Pt(RGO/CdSTT) e Pt(RGO+CdSTT), pós irradiadas, ambas obtidas pelo método térmico (Figura 7b), é possível observar que a razao ID/IG menor e a ausência da banda G* no espectro da primeira representam uma melhor eficiência na reduçao do GO quando introduzido durante a síntese. Os resultados de análise por espectroscopia Raman indicam que o processo de fotorreduçao do óxido de grafeno é mais eficiente quando o compósito com CdS é obtido pelo método térmico comparado ao método sonoquímico, o que justifica maior atividade do fotocatalisador Pt(RGO/CdSTT) em relaçao aos demais. Para o método térmico de síntese de CdS e compósitos, os sistemas ternários mostraram-se mais ativos que os binários, com destaque para Pt(RGO/CdSTT), fotocatalisador mais ativo dentre todos os testados nesse trabalho, indicando que platina e RGO, neste caso, atuam de forma sinérgica na geraçao fotocatalítica de hidrogênio, como ilustrado no esquema da Figura 8.
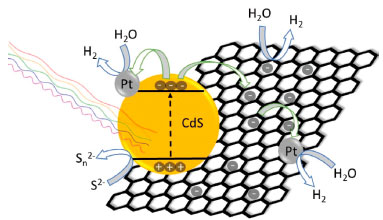 Figura 8. Ilustraçao dos processos primários de geraçao e transferência de carga no sistema ternário Pt(RGO/CdSTT) para a produçao fotocatalítica de hidrogênio
Durante a reaçao fotocatalítica tanto Pt(IV) quanto o óxido de grafeno sao reduzidos, com possibilidade de fotodeposiçao de Pt0 na superfície de CdS e na do próprio RGO. O óxido de grafeno reduzido atua como aprisionador de elétrons que podem ser transferidos diretamente para moléculas de água adsorvidas à sua superfície ou podem ser transferidos para nanopartículas de platina que também promoverao a reduçao de H2O a H2. Além disso, a reduçao ainda pode se dar na superfície do CdS, na qual também ocorre a oxidaçao do sulfeto a polissulfetos pelas lacunas fotogeradas.
CONCLUSOES A atividade fotocatalítica dos compósitos de CdS com GO na geraçao de hidrogênio quando o par S2-/SO32- é usado como reagentes de sacrifício é dependente do método de síntese e da forma como GO é introduzido. O método térmico de síntese resultou em compósitos mais fotoativos na geraçao de hidrogênio, especialmente se a platina está presente. Tanto as análises por difratometria de raios X quanto por espectroscopia Raman indicam que o óxido de grafeno nos compósitos é reduzido in situ durante a geraçao fotocatalítica de hidrogênio. Os resultados de DRX indicam que o RGO nos compósitos obtidos via rota sonoquímica apresenta maior distância interplanar e menor grau de empilhamento; já os resultados do estudo com espectroscopia Raman indicam que o RGO nos compósitos obtidos pelo método térmico apresenta menos defeitos e o processo de fotorreduçao do óxido de grafeno é mais eficiente quando comparado aos compósitos obtidos pelo método sonoquímico. Desta forma, o melhor desempenho do compósito Pt(RGO/CdSTT) está associado à maior cristalinidade do CdS hexagonal obtido pelo método térmico, a maior dispersao do GO quando introduzido durante a síntese, a maior eficiência no processo de fotorreduçao de GO a RGO em compósitos obtidos pelo método térmico e a atuaçao da platina como cocatalisador e auxiliar na fotorreduçao de GO. A combinaçao desses fatores resultou em um efeito sinérgico da platina com o óxido de grafeno reduzido, rendendo uma taxa de produçao de hidrogênio de 651,6 mmol gcat-1 h-1.
AGRADECIMENTOS Os autores sao gratos ao CNPq pelo suporte financeiro (Processo 442840/2014-4), à CAPES pela bolsa de Doutorado concedida a C. G. Almeida e ao Laboratório Multi-Usuário de Microscopia Eletrônica da UFBA (LAMUME) pelas análises de espectroscopia Raman.
REFERENCIAS 1. Gallego, G. S.; Mondragón, F.; Barrault, J.; Tatibouët, J.-M.; Batiot-Dupeyrat, C.; Appl. Catal., A 2006, 311, 164. 2. Kappen, P.; Grunwaldt, J.-D.; Hammershoi, B. S.; Tröger, T.; Clausen, B. S.; J. Catal. 2001, 198, 56. 3. Fujishima, A.; Honda, K.; Nature 1972, 238, 37. 4. Hoffmann, M. R.; Martin, S. T.; Choi, W.; Bahnemann, D. W.; Chem. Rev. 1995, 95, 69. 5. Wang, C.; Pagel, R.; Dohrmann, J. K.; Bahnemann, D. W.; C. R. Chimie 2006, 9, 761. 6. Melo, M. O.; Silva, L. A.; J. Braz. Chem. Soc. 2011, 22, 1399. 7. Marques, F. C.; Stumbo, A. M.; Canela, M. C.; Quim. Nova 2017, 40, 561. 8. Matsumura, M.; Furukawa, S.; Saho, Y.; Tsubomura, H.; J. Phys. Chem. 1985, 89, 1327. 9. Matsumura, M.; Ohnishi, H.; Hanafusa, K.; Tsubomura, H.; Bull. Chem. Soc. Jpn. 1987, 60, 2001. 10. Nosaka, Y.; Yamaguchi, K.; Kuwabara, A.; Miyama, H.; Baba, R.; Fujishima, A.; J. Photochem. Photobiol., A 1992, 64, 375. 11. Jin, Z. S.; Li, Q. L.; Zheng, X. H.; Xi, C. J.; Wang, C. P.; Zhang, H. Q.; Feng, L. B.; Wang, H. Q.; Chen, Z. S.; Jiang, Z. C.; J. Photochem. Photobiol., A 1993, 71, 85. 12. Janet, C. M.; Viswanath, R. P.; Nanotechnology 2006, 17, 5271. 13. Jang, J. S.; Li, W.; Oh, S. H.; Lee, J. S.; Chem. Phys. Lett. 2006, 425, 278. 14. Silva, L. A.; Ryu, S. Y. Choi, J.; Choi, W.; Hoffmann, M. R.; J. Phys. Chem. C. 2008, 112, 12069. 15. Souza E. A.; Silva, L. A.; J. Environ. Chem. Eng. 2016, 4, 2114. 16. Trindade, T. N. S.; Silva, L. A.; J. Alloys. Compd. 2018, 735, 400. 17. Rahmawati, F.; Yuliati, L.; Alaih, I. S.; Putri, F. R.; J. Environ. Chem. Eng. 2017, 5, 2251. 18. El-Maghrabi, H. H.; Barhoum, A.; Nada, A. A.; Moustafa, Y. M.; Seliman, S.; M.; Youssef, A. M.; Bechelany, M.; J. Photochem. Photobiol., A 2018, 351, 26 19. Chang, Y.; Yu, K.; Zhang, C.; Yang, Z.; Feng, Y.; Hao, H.; Jiang, Y.; Lou, L-L.; Zhou, W.; Liu, S.; Appl. Catal., B 2017, 215, 74. 20. Peng, Q.-X.; Xue, D.; Zhan, S.-Z.; Ni, C.-L.; Appl. Catal., B 2017, 219, 353. 21. Ai, Z.; Zhao, G.; Zhong, Y.; Shao, Y.; Huang, B.; Wu, Y.; Hao, X.; Appl. Catal., B 2018, 221, 179. 22. Bastos, S. A. L.; Santos, F. N.; Lopes, P. A. L.; Silva; L. A.; Int. J. Hydrogen Energy 2014, 39, 14588. 23. Lopes, P. A. L.; Mascarenhas, A. J. S.; Silva; L. A.; J. Alloy. Compd. 2015, 649, 332. 24. Melo, M. O.; Silva, L. A.; J. Photochem. Photobiol., A 2011, 226, 36. 25. Lopes, P. A. L.; Santos, M. B.; Mascarenhas, A. J. S.; Silva, L. A.; Mater. Lett. 2014, 136, 111. 26. Acar, C.; Dincer, I.; Zamfirescu, C.; Int. J. Energy Res. 2014; 38, 1903. 27. Maeda, K.; J. Photochem. Photobiol., C 2011, 12, 237. 28. Wang, G.; Ling, Y.; Wang, H.; Lu, X.; Li, Y.; J. Photochem. Photobiol., C 2014, 19, 35. 29. Xue, C.; Yan, X.; An, H.; Li, H.; Wei, J.; Yang, G.; Appl. Catal., B 2018, 222, 157. 30. Chen, F.; Zhang, L.; Wang, X.; Zhang, R.; Appl. Surf. Sci. 2017, 422, 962. 31. Ali, M. B.; Jo, W.-K.; Elhouichet, H.; Boukherrou, R.; Int. J. Hydrogen Energy 2017, 42, 16449. 32. Kumar, D. P.; Hong, S.; Reddy, D. A.; Kim, T. K.; Appl. Catal., B 2017, 212, 7. 33. Siqi, L.; Zhang, C. Nan, Zhang; Zi-Rong, T.; Yi-Jun, X.; J Phys. Chem. C 2013; 117, 8251. 34. Li, X.; Shen, R.; Ma, S.; Chen, X.; Xie, J.; Appl. Surf. Sci. 2018, 430, 53. 35. Zhou, T.; Chen, F.; Liu, K.; Deng, H.; Zhang, Q.; Feng, J.; Fu, Q.; Nanotechnology 2011, 22, 045704. 36. Leo, M.; Soto, E.; Vaquero, F.; Mota, N.; Navarro, R. M.; Fierro, J. L. G.; Int. J. Hydrogen Energy 2017, 42, 13691. 37. Singh, S.; Sinha, A. S. K.; Appl. Surf. Sci. 2018, 430, 184. 38. Hong, Y.; Shi, P.; Wang, P.; Yao, W.; Int. J. Hydrogen Energy 2015, 40, 7045. 39. Xu, J.; Wang, L.; Cao, X.; Chem. Eng. J. 2016, 283, 816. 40. Lei, Y.; Yang, C.; Hou, J.; Wang, F.; Min, S.; Ma, X.; Jin, Z.; Xu, J.; Lu, G.; Huang, K-W.; Appl. Catal., B 2017, 216, 59. 41. Khan, M. E.; Khan, M. M.; Cho, M. H.; J. Colloid Interface Sci. 2016, 482, 221. 42. Mehl, H.; Matos, C. F.; Neiva, E. G. C.; Domingues, S. H.; Zarbin, A. J. G.; Quim. Nova 2014, 37, 1639. 43. Stobinski, L.; Lesiak, B; Malolepszy, A.; Mazurkiewicz, M.; Mierzwa, B.; Zemek, J.; Jiricek, P.; Bieloshapk, I.; J. Electron Spectrosc. 2014, 195, 145. 44. Mauro, M.; Cipolletti, V.; Galimberti, M.; Longo, P.; Guerra, G.; J. Phys. Chem. C 2012, 116, 24809. 45. Cho, H-W.; Wu, J-J.; J. Colloid Interface Sci. 2015, 438, 291. 46. Li, X. H.; Chen, J. S.; Wang, X.; Schuster, M. E.; Schlogl, R.; Antonietti, M.; ChemSusChem 2012, 5, 642. 47. Dartora, C. A.; Jimenez, M. J. S.; Zanella, F.; Revista Brasileira de Ensino de Física, 2015, 37, 3301. 48. Childres, I.; Jauregui, L. A.; Park, W.; Cao, H.; Chen, Y. P. In New Developments in Photon and Materials Research; Jang, J. I., ed.; Nova Science Publishers: New York, 2013, Cap. 19. 49. Yoon, D.; Moon, H.; Cheong, H.; Choi, J. S.; Choi, J. A.; Park, B. H.; J. Korean Phys. Soc. 2009, 55, 1299. 50. Ferrari, A. C.; Solid State Commun. 2007, 143, 47. 51. Maultzsch, J.; Reich, S.; Thomsen, C.; Requardt, H.; Ordejon, P.; Phys. Rev. Lett. 2004, 92, 075501. 52. Dresselhaus, M. S.; Jorio, A.; Filho, A. G. S.; Saito, R.; Philos. Trans. R. Soc. 2010, 368, 5355. 53. Nemanich, R. J.; Solin, S. A.; Phys. Rev. B 1970, 15, 392. 54. Ferrari, A. C.; Meyer, J. C.; Scardaci, V.; Casiraghi, C.; Lazzeri, M.; Mauri, F.; Piscanec, S.; Jiang, D.; Novoselov, K. S.; Roth, S.; Geim, A. K.; Phys. Rev. Lett. 2006, 97, 187401 55. Ni, Z. H.; Wang, Y. Y.; Yu, T.; You, Y.; Shen, Z. X.; Phys. Rev. B 2008, 77, 235403. 56. Ado, J.; International Scholarly Research Network, 2012, 1, 234216. 57. Popov, V. N.; J. Phys. Conf. Ser. 2016, 682, 012013. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access