
 |
 |
 |
 |
 |
Artigo
| Alkaloids from leaves of guatteria pogonopus (annonaceae) and their cytotoxicities |
|
Maria de Fátima C. SantosI,II; José Eraldo N. FontesI; Lívia M. DutraII; Larissa M. BomfimIII; Cinara O. D. CostaIII; Valéria R. S. MoraesIII; Andersson BarisonII; Milena B. P. SoaresIII,IV; Felipe Moura A. da SilvaV; Jackson R. G. da Silva AlmeidaVI; Héctor H. F. KoolenVII; Daniel P. BezerraII; Emmanoel Vilaça CostaV,*
I. Departamento de Química, Universidade Federal de Sergipe, 49100-000 São Cristóvão - SE, Brasil Recebido em: 26/03/2018 *e-mail: emmanoelvc@gmail.com The phytochemical investigation of the alkaloid-rich fraction obtained from the leaves of Guatteria pogonopus Mart. (Annonaceae) allowed the isolation and identification for the first time in this species of: (+)-nornuciferine (1), a mixture of 1 and (+)-anonaine (2), (+)-isocorydine (3), (+)-nuciferine (4), (+)-roemerine (5), (-)-tetrahydropseudocolumbamine (6), a mixture of 6, liriodenine (9) and lysicamine (10), a mixture of 1,2,9-trimethoxy-10-hydroxyaporphine (7) and bulbocapnine (8), 9, 10, and (+)-N-methyllindicarpine (11). Compounds 6, 7, 8, and 11 have not been previously reported in the family Annonaceae. Furthermore, the formerly synthetic 1,2,9-trimethoxyaporfin-10-ol (7) is described for the first time as a natural aporphine alkaloid herein. The chemical structures were established by 1D and 2D NMR as well as in comparison with data previously reported in the literature. The cytotoxic activity of the alkaloids was evaluated against tumor (B16-F10, HepG2, HL-60, and K562) and non-tumor (PBMC) cell lines. Alkaloid 1 presented significant activity against HepG2 cell lines with IC50 of 9.60 µmol L-1 while the mixture of 6, 9 and 10 displayed strong cytotoxic activity against HL-60 and K562 cell lines with IC50 values of 3.41 an 8.50 µmol L-1, respectively. INTRODUCTION Guatteria (Ruiz & Pav.) is the largest genus of the family Annonaceae with 307 species of neotropical distribution.1,2 In Brazil, about 88 Guatteria species were registered from which, 47 are considered endemic. Among the species of this genera, Guatteria pogonopus Mart. is one of the most widespread, being encountered in the Northeast and Southeastern regions, especially at the Atlantic Forest. This species shows morphological similarities with G. oligocarpa and G. ferruginea, although it can be easily identified by the observation of its large glabrous leaves and short pedicel of flowers and fruits.1,2 Plants continue to provide diverse and unique compounds with high structural diversity, which in turn are responsible by a wide range of biological activities. Previous works demonstrated a strong cytotoxic activity (in vivo and in vitro) of the essential oil from leaves of G. pogonopus against three human tumor cell lines.3 These cytotoxic properties were attributed to a synergic effect of the terpenoids in the investigated essential oil.3 Another study involving the essential oils of the species G. friesiana and G. pogonopus showed relevant trypanocidal and antimalarial activities, which were also attributed to the synergism of the terpenoids.4 A study of the chemical constituents of G. friesiana essential oil revealed significant antitumor activity and a low systemic activity associated with its α-, β- and γ-eudesmol isomers.5 In addition, studies involving the isolation of non-volatile constituents from G. latifolia revealed a leishmanicidal activity of the alkaloid oxoputerine and antimicrobial action for 9-methoxysomoschatoline, lysicamine, coreximine and isocoreximine.6 For G. hispida, O-methylmoschatoline, lysicamine and liriodenine displayed antiproliferative properties.7 Based on this context, species of Guatteria are considered as promising sources of alkaloids with different biological activities. Currently, only one previous phytochemical study of the alkaloid content of G. pogonopus was reported.8 Moving forward in the study of bioactive compounds and their chemotaxonomic significance for the Annonaceae family, we report herein a study displaying the identification of alkaloids from the leaves of G. pogonopus and their cytotoxicity against a panel of tumor cell lines.
EXPERIMENTAL General experimental procedures Optical rotations were recorded on a Jasco P-2000 polarimeter in CHCl3. 1D and 2D NMR data were acquired at 303 K in CDCl3 on a Bruker AVANCE III 600 NMR operating at 14.1 T, observing 1H and 13C at 600 and 150 MHz, respectively. The NMR spectrometer was equipped with a 5-mm multinuclear inverse detection probe (1D and 2D NMR experiments) with z-gradient. One-bond (HSQC) and long-range (HMBC) 1H-13C NMR correlation experiments were optimized for average coupling constant 1J(C,H) and LRJ(C,H) of 140 and 8 Hz, respectively. All 1H and 13C NMR chemical shifts (δ) are given in ppm related to the TMS signal at 0.00 ppm as an internal reference, and the coupling constants (J) in Hz. ESI-MS spectra were obtained in positive ion detection mode on a Thermo Scientific LTQ XL Linear Ion Trap Mass Spectrometer, equipped with an ESI source. Silica gel 60 (70-230 mesh) was used for the column chromatography (CC), while silica gel 60 F254 (Merck, 0.25 mm, aluminium) was used for analytical (0.25 mm) and preparative with thin layer chromatography (PTLC) (Merck, 1.00 mm, glass). Compounds were visualized by exposure under UV 254/365 light, by spraying with p-anisaldehyde reagent followed by heating on a hot plate, and by spraying Dragendorff's reagent. Plant material Leaves of G. pogonopus were collected in February 2012 at the Park of the Serra of Itabaiana, (geographic coordinates: 09º 57' 54" S and 37º 51' 46" W), Sergipe state, Brazil. The material was identified by PhD Ana Paula do Nascimento Prata (a plant taxonomist of the Department of Biology of the Federal University of Sergipe, Sao Cristovao, SE, Brazil). A voucher specimen (#24954#) was deposited at the Herbarium of the Federal University of Sergipe (Herbarium ASE) of the Department of Biology, UFS. The authors had authorization from the Chico Mendes Institute for Biodiversity Conservation from Brazilian Ministry of the Environment for plant collection (number 25637-1). This work was accomplished according to the special authorization for access to genetic resources in Brazil # 010240/2013-6, issued by CNPq/MCTI. Extraction and isolation Leaves of G. pogonopus (1.3 kg) were dried using an air circulating oven at 45 ºC, powdered and successively extracted with hexane (5L, 25 ºC, five times) followed by MeOH (5L, 25 ºC, five times), yielding hexane (25.30 g) and MeOH (258.60 g) extracts after solvent removal under reduced pressure. TLC analysis revealed with Dragendorff's reagent indicated a high concentration of alkaloids in the MeOH extract. Therefore, an aliquot of the MeOH extract (248.60 g) was submitted to an acid-base extraction to give an alkaloid (3.79 g) and a neutral (23.81 g) fractions. In this way, part of the alkaloid fraction (3.70 g) was subjected to silica gel column chromatography (CC) previously treated with an aqueous solution of NaHCO3 (10% v/v). Then, the CC was eluted with a gradient system consisting of increasing concentration of CH2Cl2 in hexane (95:5 to 10:90, v/v), followed by EtOAc in CH2Cl2 (95:5 to 30:70, v/v) and MeOH in EtOAc (95:5 to 50:50, v/v) to afford 274 fractions (30 mL each). The eluted fractions were evaluated and pooled according to TLC analysis yielding 20 distinct groups (GF1-GF20). Group GF3 (397.6 mg) from hexane-CH2Cl2 (60:40 to 30:70) was submitted to a new silica gel CC eluted with the same eluent system as described for initial CC, to afford 110 fractions (20 mL each) that were pooled in fifteen subgroups (GF3.1 to GF3.15) according to TLC analysis. Subgroup GF3.5 was purified by preparative TLC eluted with CH2Cl2:MeOH (95:05, v/v, two times) to yield 1 (15.3 mg), a mixture of 1 and 2 (66.0 mg) and 3 (51.2 mg). Group GF4 (342.6 mg) from hexane-CH2Cl2 (30:70 to 10:90) was also submitted to a new silica gel CC eluted with the same eluent system as described for initial CC, affording 140 fractions (20 mL each). All fractions were analyzed and pooled in sixteen subgroups (GF4.1 to GF4.16), according to TLC analysis. Subgroup GF4.1 (33.7 mg) was submitted to a preparative TLC eluted with CH2Cl2:MeOH (95:05, v/v, three times), affording 4 (6.5 mg), 5 (2.8 mg) and 6 (3.0 mg). Subgroup GF4.4 (39.9 mg) was purified to a preparative TLC eluted with CH2Cl2:MeOH (95:05, v/v, three times) to yield a mixture of 6, 9 and 10 (6.9 mg). Subgroup GF4.5 (11.1 mg) when submitted to a preparative TLC eluted with CH2Cl2:MeOH (95:05, v/v, three times) gave a mixture of 7 and 8 (2.2 mg). Subgroups GF4.6 (65.1 mg) and GF4.7 (48.1 mg) were also submitted to preparative TLC eluted with the same solvent system (two times) yielding 9 (2.5 mg) and 10 (3.0 mg), and 11 (4.1 mg), respectively. (S)-Nornuciferine (1): Brown amorphous powder (CHCl3); [α]D25 128.5º (c 0.25, CHCl3); identified by comparison with literature da (1H NMR and 13C NMR).9 Mixture of nornuciferine and anonaine (2): Brown amorphous powder (CHCl3); identified by comparison with literature data (1H NMR and 13C NMR).9,10 (S)-(+)-Isocorydine (3): Brown amorphous powder (CHCl3); [α]D25 73.4º (c 0.14, CHCl3); 1H and 13C NMR data; see Table 1. ESI-MS [M + H]+m/z 342.
(S)-(+)-Nuciferine (4): Brown amorphous powder (CHCl3); [α]D25 23.3º (c 0.46, CHCl3); 1H and 13C NMR data; see Table 1. ESI-MS [M + H]+m/z 296. (S)-(+)-Roemerine (5): Brown amorphous powder (CHCl3); [α]D25 54.4º (c 0.18, CHCl3); 1H and 13C NMR data; see Table 1. ESI-MS [M + H]+m/z 280. (S)-(-)-Tetrahydropseudocolumbamine (6): Brown amorphous powder (CHCl3); [α]D25 -110.6º (c 0.19, CHCl3); 1H and 13C NMR data; see Table 2. ESI-MS [M + H]+m/z 342.

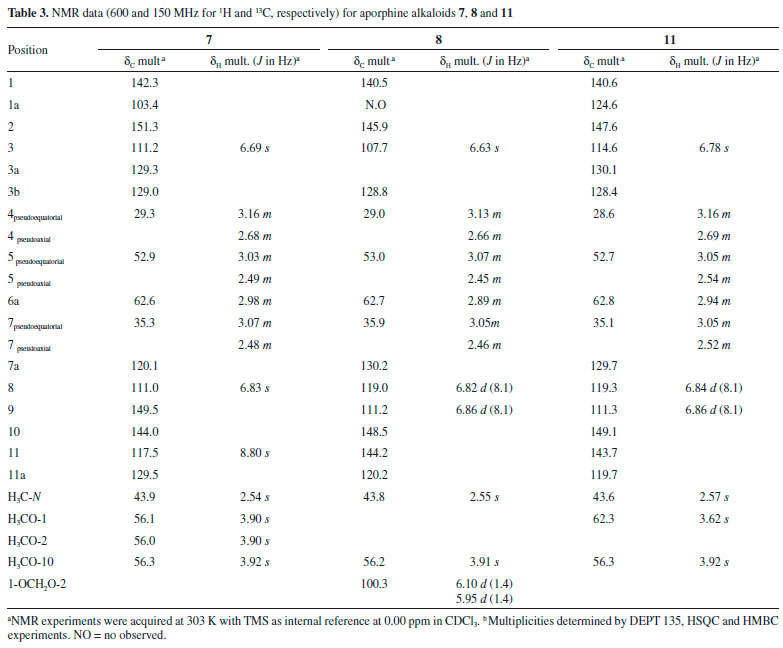
Mixture of 1,2,9-trimethoxy-10-hydroxyaporphine (7) and bulbocapnine (8): Brown amorphous powder (CHCl3); 1H and 13C NMR data; see Table 3.
Liriodenine (9): Yellow amorphous powder (CHCl3); identified by comparison with literature data (1H NMR and 13C).11-13 Lysicamine (10): Brown amorphous powder (CHCl3); identified by comparison with literature data (1H NMR and 13C).11,12 ESI-MS [M + H]+m/z 292. Tetrahydropseudocolumbamine (6), Liriodenine (9), and Lysicamine (10): Orange amorphous powder (CHCl3); identified by comparison with literature data (1H NMR and 13C NMR).11,12 EI-MS [M + H]+m/z 342, 275 and 292, respectively. (S)-(+)-N-methylindicarpine (11): Brown amorphous powder (CHCl3); [α]D25 152.3º (c 0.20, CHCl3); 1H and 13C NMR data; see Table 3. Cytotoxicity evaluation Cells Tumor cells lines B16-F10 (mouse melanoma), HepG2 (human hepatocellular carcinoma), K562 (human chronic myelocytic leukemia), and HL-60 (human promyelocytic leukemia) were donated by Hospital A.C. Camargo, Sao Paulo, SP, Brazil. Cells were maintained in Roswell Park Memorial Institute-1640 (RPMI-1640, Gibco-BRL) medium supplemented with 10% fetal bovine serum (Cultilab), 2 mM L-glutamine (Vetec Química Fina) and 50 mg µL-1 gentamycin (Novafarma). Adherent cells were harvested by treatment with 0.25% trypsin EDTA solution (Gibco-BRL). All cell lines were cultured in cell culture flasks at 37 ºC in 5% CO2 and sub-cultured every 3-4 days to maintain exponential growth. All experiments were conducted with cells in exponential growth phase. The cell lines were tested for mycoplasma with a Mycoplasma Stain Kit (Sigma-Aldrich) and found to be free from contamination. Heparinized blood (from healthy, 20-35 years old, non-smoker donors who had not taken any drug at least 15 days prior to sampling) was collected and peripheral blood mononuclear cells (PBMC) were isolated by a standard protocol by Ficoll (GE Ficoll-Paque Plus) density gradient centrifugation. PBMC were washed and resuspended at a concentration of 0.3 x 106 cells mL-1 in RPMI 1640 medium supplemented with 20% fetal bovine serum, 2 mmol L-1 glutamine, 50 mg µL-1 gentamycin at 37 °C with 5% CO2. In addition, concanavalin A (ConA, Sigma-Aldrich) was used as a mitogen to trigger cell division in T-lymphocytes. ConA (10 µg mL-1) was added at the beginning of culture and, after 24 h, cells were treated with the test drugs. The Research Ethics Committee of the Oswaldo Cruz Foundation (Salvador, Bahia, Brazil) approved the experimental protocol (number 031019/2013). All participants signed their written informed consent to participate in the study. For all experiments, cell viability was performed by Trypan Blue Exclusion (TBE) assay. Over 90% of the cells were viable at the beginning of the culture. Cytotoxic activity assay Cell viability was quantified by alamar blue method, as previously described.14 For all experiments, cells were seeded in 96-well plates (7 x 104 cells mL-1 for adherent cells or 3 x 105 cells mL-1 for suspended cells in 100 µL of medium). After 24 h, the compounds (0.19 - 25 µg mL-1) dissolved in dimethyl sulfoxide (DMSO, Sigma-Aldrich) were added to each well and incubated for 72 h. Doxorubicin (purity ≥ 95%, doxorubicin hydrochloride, Laboratory IMA S.A.I.C.) was used as positive control (0.08 - 5 µg mL-1). Negative control received the vehicle used for diluting the tested (0.5% DMSO). Four (for cell lines) or 24 (for PBMC) h before the end of the incubation, 20 µL of stock solution (0.312 mg mL-1) of the alamar blue (resazurin, Sigma-Aldrich) were added to each well. The absorbance was measured using a SpectraMax 190 multiplate reader and the drug effect was quantified as the percentage of control absorbance at 570 and 600 nm. Statistical analysis Data are presented as half maximal inhibitory concentration (IC50) values and their 95% confidence intervals (CI 95%) obtained by nonlinear regression. All statistical analyzes were performed using the GraphPad program (Intuitive Software for Science).
RESULTS AND DISCUSSION The crude MeOH extract from the leaves of G. pogonopus displayed cytotoxic activity and was subjected to successive chromatographic procedures allowing the isolation of the alkaloids (+)-nornuciferine (1),9 anonaine (2),10 (+)-isocorydine (3),11 (+)-nuciferine (4),15 (+)-roemerine (5),15 (-)-tetrahydropseudocolumbamine (6),16,17 1,2,9-trimethoxy-10-hydroxyaporphine (7),18,19 bulbocapnine (8),20 liriodenine (9),11-13 lysicamine (10),11,12 and (+)-N-methyllindicarpine (11)21 (Figure 1). Compounds 1, 2, 9, and 10 were previously found on the stem barks of this species,8 whereas compounds 6-8, and 11 are reported for the first time in Guatteria. Although the structures of the alkaloids 3-5,11,15 6-8,16-20 and 1121 have already been described a long time ago, their 1H and 13C data are incomplete or scalar coupling constants values have not been assigned and/or obtained. In this work, the complete and unequivocal 1H and 13C data were reviewed according to 1D and 2D NMR experiments.
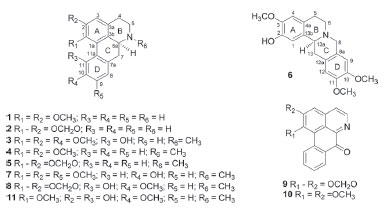 Figure 1. Alkaloids isolated from leaves of Guatteria pogonopus
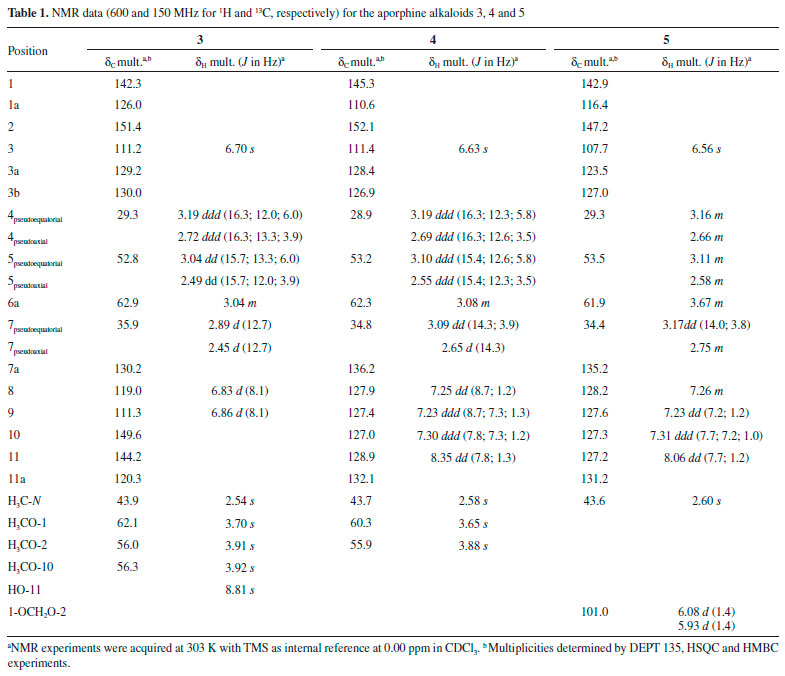
The 1H NMR spectrum of compound 3 revealed the presence of two spin systems, one consisting of the signals at δ 3.19 (1H, ddd, J = 16.3, 12.0 and 6.0 Hz, H-4 pseudoequatorial) and δ 2.72 (1H, ddd, J = 16.3, 13.3 and 3.9 Hz, H-4 pseudoaxial), as well as δ 3.04 (1H, ddd, J = 15.7, 13.3 and 6.0 Hz, H-5 pseudoequatorial) and δ 2.49 (1H, ddd, J = 15.7, 12.0 and 3.9 Hz, H-5 pseudoaxial) and the other comprising the signals at δ 3.04 (1H, m, H-6a), 2.89 (1H, d, J = 12.7 Hz, H-7 pseudoequatorial), and δ 2.45 (1H, d, J = 12.7 Hz, H-7 pseudoaxial). The singlet at δ 2.54 (3H) revealed the presence of an N-CH3 group. Furthermore, it was observed a singlet at δ 6.70 (1H, H-3), indicative of an 1,2,3,4,5-pentasubstituted benzene ring, as well as a spin system at δ 6.83 (1H, d, J= 8.1 Hz, H-8) and 6.86 (1H, d, J = 8.1 Hz, H-9), confirming the presence a disubstituted benzene ring. The signals at δ 3.70, 3.91 and 3.92 (each, 3H, s) were assigned to three methoxyl groups. The location of the methoxyl groups were established based on the long-range 1H-13C correlation map from HMBC NMR experiment (Table 1 and Figure 2). This analysis revealed that the hydrogen at δ 6.70 (H-3) had long-range 1H-13C correlation with the carbons δ 142.3 (C-1), 151.4 (C-2), 129.2 (C-3b), and 29.3 (C-4). In contrast, the hydrogen at δ 6.83 (H-8) showed correlation with carbon at δ 35.9 (C-7), 120.3 (C-11a) and 149.6 (C-10). The presence of a hydroxyl group in the molecule located in the D ring at C-10 was established on the basis of long-range 1H-13C correlation of the hydrogen at δ 6.86 (H-9) with the carbon δ 144.2 (C-11), which showed no correlation with the methoxyl group. Therefore, based on these NMR data, compound 3 was established as the aporphine alkaloid, (+)-isocorydine. This compound has been found in species of Annonaceae, such as G. oliviformis,22 G. tonduzii,22 Enantia polycarpa,23 and Gonothalamus tamirensis.24
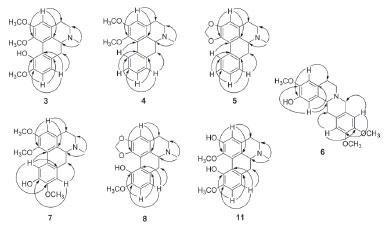 Figure 2. Key correlations observed in HMBC (arrows) of alkaloids 3-8 and 11
The 1H NMR spectrum of compound 4 revealed the presence of two spin systems, one consisting of the signals at δ 3.19 (1H, ddd, J = 16.3, 12.3 and 5.8 Hz, H-4 pseudoequatorial) and δ 2.69 (1H, ddd, J = 16.3, 12.6, 3.5 Hz, H-4 pseudoaxial) as well as δ 3.10 (1H, ddd, J = 15.4, 12.6, 5.8 Hz, H-5 pseudoequatorial) and 2.55 (1H, ddd, J = 15.4, 12.3, 3.5 Hz, H-5 pseudoaxial), and the other comprising the signals at δ 3.08 (1H, m, H-6a), δ 3.09 (1H, dd, J = 14.3 and 3.9 Hz, H-7 pseudoequatorial) and 2.65 (1H, d, J = 14.3 Hz, H-7 pseudoaxial). Similarly, to compound 3, the presence of an N-CH3 group was observed, due to the singlet at δ 2.58 (3H) with correlations in the HMBC map with the carbons at δ 53.2 (C-5) and 62.3 (C-6a). By analysis of the 1H NMR spectrum it was also possible to observe the presence of a singlet at δ 6.63 (1H, H-3). The main difference between 4 and 3 is the absence of substituents at ring D. The hydrogen H-3 showed long-range 1H-13C correlation with the carbons δ 145.3 (C-1), 152.1 (C-2), 126.9 (C-3b), and 28.9 (C-4) (Figure 2). On the other hand, a second difference between 4 and 3 was the absence of substitution at the D ring (Table 1). According with 1H and 13C NMR 1D/2D data, compound 4 was identified as the aporphine alkaloid, (+)-nuciferine. This compound has been found only in Annonaceae species, such as in G. ouregou.25 Compound 5 presented 1H NMR spectrum very similar to those of 4. The main difference is the presence of methylenedioxy group in the A ring due to the observation of dublets at δ 6.08 and 5.93 (each, 1H, d, J = 1.4 Hz, H-1 and H-2). The location of the methylenedioxy group at C-1 and C-2 was confirmed based on the long-range 1H-13C correlation map from HMBC NMR experiment. In this, the signal at δ 6.56 (H-3, s) showed long-range 1H-13C correlation with carbons δ 142.9 (C-1), 147.2 (C-2), 127.0 (C-3a), and 29.3 (C-4) (Table 1 and Figure 2). According with 1H and 13C NMR 1D/2D data, compound 5 was identified as the aporphine alkaloid, (+)-roemerine. This compound has been found in species of Annonaceae such as in Gonothalamus tamirensis,24 Guatteria sagotiana,26 and Xylopia laevigata.27 Compound 6 showed several signals in the aliphatic region of 1H NMR spectrum that along with 1H-1H correlations of the COSY map revealed four spin systems, which are typical for tetrahydropseudoprotoberberine alkaloids (Table 2). The three singlets at δ 3.84, 3.85 and 3.87 (3H, each) indicated the presence of three methoxyl groups in the structure. The complete assignments of the 1H and 13C{1H} NMR chemical shifts were established based on one-bond and long-range 1H-13C correlation map from HSQC and HMBC experiments (Figure 2). The hydrogen at δ 6.82 (H-1) showed long-range 1H-13C correlation with the carbons at δ 59.5 (C-13a), 126.0 (C-4a) and 145.1 (C-3), as well as, the signal at δ 6.59 (H-4), which displayed long-range 1H-13C correlation with the carbons at δ 29.0 (C-5), 130.7 (C-13b) and 144.1 (C-2). The hydroxyl group at C-2 was confirmed based on the long-range 1H-13C correlation of the hydrogen at δ 6.59 (H-4) with the carbon at δ 144.1 (C-2). This signal did not show any correlation with the methoxyl group. However, the singlet at δ 3.87 showed long-range correlation with the carbon at δ 145.1 (C-3), confirming the methoxyl group at A ring (Table 2). These findings suggest the presence the tetrasubstituted benzene ring. On the other hand, the singlet at δ 6.56 (H-8) showed long-range 1H-13C correlation with the carbons at δ 58.3 (C-8), 126.4 (C-12a) and 147.8 (C-11), as well as the signal at δ 6.64 (H-11), which showed long-range 1H-13C correlation with the carbons at δ 36.3 (C-13), 126.2 (C-8a) and 147.5 (C-10). The methoxyl hydrogens at δ 3.84 and 3.85 displayed long-range 1H-13C correlations with the carbons δ 147.5 and 147.8, respectively. Thus, data suggest the existence of a system tetrasubstituted at D ring of this alkaloid. Based on the 1H and 13C NMR 1D/2D data, compound 6 was identified as being a 2,3,10,11-tetrahydropseudoprotoberberine alkaloid known as (-)-tetrahydropseudocolumbamine. This compound was found for first time in Guatteria. Previously, this compound has been reported only in Chasmanthera dependens16 and Isopyrum thalictroide.17 Compounds 7 and 8 were obtained in mixture, once its NMR data indicated the presence of two set of signals with different ratios. The first set of signals in the 1H NMR spectrum displayed two spin system characteristics of aporphine alkaloids at δ 3.16 and δ 2.68 (1H each, m, H-4) and δ 3.03 and 2.49 (1H each, m, H-5), and the other, with the signals at δ 2.98 (1H, m, H-6a), and δ 3.07 and δ 2.48 (1H each, m, H-7). The signal at δ 2.54 (3H, s) revealed the presence of an N-CH3 group. The singlet at δ 6.69 (1H, H-3) indicated a tetrasubstituted benzene ring and the spin system at δ 6.83 (1H, s, H-8) and δ 8.80 (1H, s, H-11) indicating a p-substituted benzene ring. The location of the methoxyl groups C-1, C-2 and C-9 were established based on the long-range 1H-13C correlations of the HMBC map. In this, the hydrogen at δ 6.69 (H-3) showed long-range 1H-13C correlation with the carbons at δ 142.3 (C-1), 151.3 (C-2), 129.0 (C-3b), and 29.3 (C-4). On the other hand, the hydrogen at δ 8.80 (H-11) showed long-range 1H-13C correlation with carbons at δ 103.4 (C-1a), 120.1 (C-7a), and 149.5 (C-9). The hydroxyl group at C-10 was established based on the long-range 1H-13C correlation of hydrogen at δ 6.83 (H-8) with the carbon at δ 144.0 (C-10) that showed no correlation with hydrogens from methoxyl group. The overall analysis of 1D and 2D NMR experiments enabled the complete and unequivocal 1H and 13C NMR chemical shifts assignments (Table 3 and Figure 2). Considering the 1H and 13C NMR 1D/2D data, compound 7 was identified as 1,2,9-trimethoxy-10-hydroxyaporphine. This compound was only obtained by synthesis and is being reported in this communication as a natural product for the first time.18,19 The second NMR dataset in the 1H NMR showed similar spins systems for compound 7. However, in this case the methoxyl group signals were replaced by signals that suggested the presence of a methylenedioxy bridge between C-1 and C-2, which was confirmed based on the long-range 1H-13C correlation map from HMBC NMR experiment. The singlet at δ 6.63 (H-3), as well as the doublets at δ 5.95 and 6.10 of the methylenedioxy bridge hydrogens showed long-range 1H-13C correlation with carbons at δ 140.5 (C-1) and 145.9 (C-2) (Table 3). Furthermore, the doublets at δ 6.82 (H-8, J = 8.1 Hz) and 6.86 (H-9, J = 8.1 Hz) indicated a tetrasubstituted benzene ring. The methoxyl group at C-10 was established based on the long-range 1H-13C correlation of hydrogen at δ 6.82 (H-8) with the carbon at δ 148.5 (C-10). The hydroxyl group at C-11 was established on the basis of the long-range 1H-13C correlation of hydrogen at δ 6.86 (H-9) with the carbon at δ 144.2 (C-11) that showed no correlation with hydrogens from methoxyl group. The overall analysis of 1D and 2D NMR experiments enabled the complete and unequivocal 1H and 13C NMR chemical shifts assignments (Table 3 and Figure 2). Based on 1H and 13C NMR 1D/2D data, compound 8 was identified as bulbocapnine. This compound was found for the first time in Guatteria. This compound has been previously found in Menispermaceae species, such as Antizoma miersiana.20 Compound 11 displayed two spin systems in the 1H NMR spectrum, which are characteristic of an aporphine alkaloid, one consisting of the signals at δ 3.16 and δ 2.69 (1H each, m, H-4) and δ 3.05 and 2.54 (1H each, m, H-5), and the other comprising the signals at δ 2.94 (1H, m, H-6a), and δ 3.05 and 2.52 (1H, m, H-7, respectively). The signal at δ 2.57 (3H, s) was indicative of an N-CH3 group. A singlet at δ 6.78 (1H, H-3) indicated a pentasubstituted benzene ring and the spin system at δ 6.84 (1H, d, J = 8.1 Hz, H-8) and 6.86 (1H, d, J = 8.1 Hz, H-9), indicating a tetrasubstituted benzene ring. The location of the methoxyl groups at C-1 and C-10, as well as hydroxyl substituents at C-2 and C-11 were established based on the long-range 1H-13C correlations from the HMBC map (Table 3 and Figure 2). The hydrogen at δ 6.78 (H-3) showed long-range 1H-13C correlation with the carbon at δ 140.6 (C-1) which showed correlation hydrogens from the methoxyl group, as well as, the hydrogen at δ 6.84 (H-8, d, J = 8.1 Hz) showed long-range 1H-13C correlation with the carbon at δ 149.1 (C-10) revealed correlation with hydrogens from the methoxyl group. The hydroxyl group was established based on the long-range 1H-13C correlation of hydrogen at δ 6.86 (H-9, d, J = 8.1 Hz) with the carbon at δ 143.7 (C-11) that showed no correlation with hydrogens from the methoxyl group. Using the 1H and 13C NMR 1D/2D data, compound 11 was identified as (+)-N-methyllindicarpine. This compound was found for first time in Guatteria, although it has been found in species of the Papaveraceae family, such as Glacium vittelium.28 The cytotoxic activity of the isolated alkaloids was evaluated against tumor (B16-F10, HepG2, HL-60, and K562) and non-tumor (PBMC) cell lines (Table 4) using the Alamar blue assay after 72 h of incubation. Alkaloids 1, 3, 4, and 5, and the mixture of 6, 9 and 10 displayed moderate cytotoxic effects when compared to the positive control doxorubicin. In fact, some of these alkaloids have been previously reported as cytotoxic agents.27,29-31 Moreover, the cytotoxicity of the mixture 6, 9 and 10 may due to the presence of the compound 9, which has reported with a potent cytotoxic activity.31
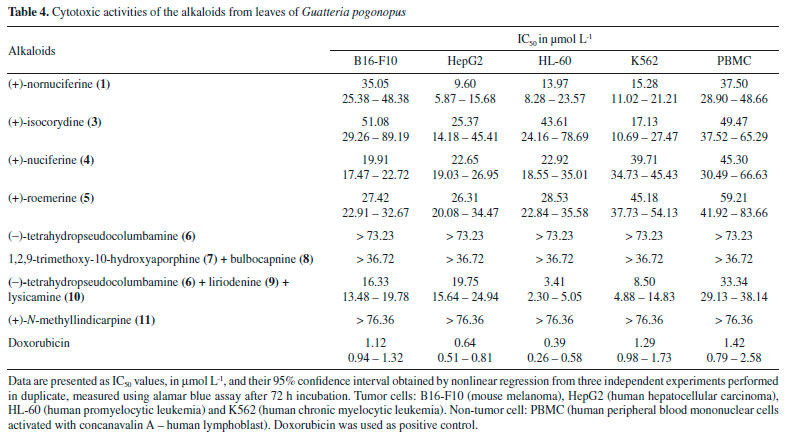
Among the alkaloids evaluated, (+)-nornuciferine (1), and the mixture of 6, 9 and 10 displayed promising cytotoxicity activities with IC50 values below to 17.0 µmol L-1 against HepG2, HL and K562. However, (+)-nornuciferine showed significant activity against HepG2 with IC50 value of 9.60 µmol L-1 while the mixture of 6, 9 and 10 demonstrated strong activity against HL-60 and K562 tumor cell lines with IC50 values of 3.41 and 8.50 µmol L-1 (Table 4). The results obtained were in accordance with Menezes et al.27 that reported the cytotoxic activity of several aporphine and tetrahydroprotoberberine alkaloids. Alkaloids 6 (IC50 >73.23 µmol/L), 11 (IC50 >76.36 µmol L-1) and the mixture of 7 and 8 (IC50 >36.72 µmol L-1) were considered inactive in any tumor cell lines tested (Table 4). The selectivity indexes (SI, SI = IC50[PBMC]/IC50[HL-60]) of 1, 3, 4, and 5 and the mixture of 6, 9 and 10, were 2.7, 1.1, 2.0, 2.1, and 9.8, respectively, for leukemia (HL-60). Doxorubicin, used as positive control, showed IC50 values ranging from 0.39 to 1.29 µmol L-1 for tumor cell lines, and a selectivity index of 3.7 for the same tumor cell line (HL-60).
CONCLUSIONS The phytochemical study of G. pogonopus displayed a structural diversity of alkaloids its leaves, which contributes to the chemotaxonomic knowledge of the genus Guatteria. Compounds 6-8 and 11 are reported for the first time in Annonaceae and 1,2,9-trimethoxy-10-hydroxyaporphine (7) is reported for the first time as a natural product. Significant cytotoxic activities recorded against human tumor cell lines have demonstrated that this species is a natural source of biologically active compounds. Strong activity against human promyelocytic leukemia (HL-60) was observed to the oxoaporphine alkaloid liriodenine (9) in a mixture with tetrahydropseudocolumbamine (6) and lysicamine (10).
ACKNOWLEDGEMENTS The authors are grateful to CNPq, CAPES, FINEP, FAPEAM, Fundaçao Araucária, FAPITEC/SE, UFAM, UFS, and UFPR for financial support and fellowship. SUPPLEMENTARY MATERIALSupplementary material containing 1D and 2D NMR data of the alkaloids isolated of the leaves of G. pogonopus are available free of charge at http://jbcs.sbq.org.br as a PDF file.
REFERENCES 1. Erkens, R. H. J.; Chatrou, L. W.; Maas, J. W.; Niet Timotheu, S. V. D., Savolainen, V.; Mol. Phylogenet. Evol. 2007, 44, 399. 2. Lobao, A. Q.; Mello-Silva, R.; Forzza, R. C.; Rodriguésia 2012, 63, 1039. 3. Fontes, J. E. N.; Ferraz, R. P.; Britto, A. C.; Carvalho, A. A.; Moraes, M. O.; Pessoa, C.; Costa, E. V.; Bezerra, D. P.; Chem. Biodiversity 2013, 10, 722. 4. Meira, C. S.; Menezes, L. R. A.; Santos, T. B.; Macedo, T. S.; Fontes, J. E. N.; Costa, E. V.; Pinheiro, M. L. B.; Silva, T. B, Guimaraes, E. T.; Soares, M. B. P.; J. Essent. Oil Res. 2017, 29, 156. 5. Britto, A. C. S.; Oliveira, A. C. A.; Henriques, R. M.; Cardoso, G. M. B.; Bonfim, D. S.; Carvalho, A. A.; Moraes, M. O.; Pessoa, C.; Pinheiro, M. L. B.; Costa, E. V.; Bezerra, D. P.; Planta Med. 2012; 78, 409. 6. Ferreira, C.; Passos, C. L. A.; Soares, D. C.; Costa, K. P.; Rezende, M. J. C.; Lobao, A. Q.; Pinto, A. C.; Hamerski, E. M.; Exp. Parasitol. 2017, 172, 51. 7. Costa, E.V.; Pinheiro, M. L.; Barison, A.; Campos, F.; Salvador, M. J.; Maia. B. H.; Cabral, E. C.; Eberlin, M. N.; J. Nat. Prod. 2010, 25, 1180. 8. Santos, M. F. C.; Dutra, L. M.; Moraes, V. R. S.; Barison, A.; Costa, E. V.; Biochem. Syst. Ecol. 2015, 60, 106. 9. Dutra, L. M.; Costa. E. V.; Moraes, V. R. S.; Nogueira, P. C. L.; Vendramin, M. E.; Barison, A.; Prata, A. P. N.; Biochem. Syst. Ecol. 2012, 41, 115. 10. Costa, E. V.; Sampaio, M. F. C.; Salvador, M. J.; Nepel, A.; Barison, A.; Quim. Nova 2015, 38, 769. 11. Chen, C.-Y.; Chang, F.-R.; Wu, Y.-C.; J. Chin. Chem. 1997, 44, 313. Teles, M. N. O.; Dutra, L. M.; Barison, A.; Costa, E. V. Biochem. Syst. Ecol. 2015, 61, 465. 12. Costa, E. V.; Marques, F. A.; Pinheiro, M. L. B.; Vaz, N. P.; Duarte, M. C. T.; Delarmelina, C.; Braga, R. M.; Maia, B. H. L. N. S.; J. Nat. Prod. 2009, 72, 1516. 13. Harrigan, G. G.; Gunatilaka, A. A.; Kingston, D. G.; Chan, G. W.; Johnson, R. K.; J. Nat. Prod. 1994, 57, 68; Costa, E. V.; Pinheiro, M. L. B.; Souza, A. D. L.; Barison, A.; Campos, F. R.; Valdez, R. H.; Ueda-Nakamura, T.; Dias Filho, B. P.; Nakamura, C. V. Molecules 2011, 16, 9714. 14. Ahmed, S. A.; Gogal, R. M.; Walsh, J. E.; J. Immunol. Methods, 1994, 170, 211. 15. Zhenjia, Z.; Minglin, W.; Daijie, W.; Wenjuan, D.; Xiao, W.; Chengcha, Z.; J. Chromatogr. B 2010, 878, 1647. 16. Moulis, C.; Stanislas, E.; Rossi, J. C.; Org. Magn. Reson. 1978, 11, 398. 17. Ohiri, F. C.; Verpoorte, R.; Svendsen, A. B.; Planta Med. 1983, 49, 17. 18. Hara, H.; Hoshino, O.; Umezawa, B.; Chem. Pharm. Bull. 1975, 8, 1921. 19. Baarschers, W. H.; Arndt, R. R.; Tetrahedron, 1965, 21, 2155. 20. Wet, H.; Heerden, F. R.; Wyk, B.-E.; Biochem. Syst. Ecol. 2005, 33, 799. 21. Johns, S. R.; Lamberton, J. A.; Aust. J. Chem. 1967, 20, 1277. 22. Lopez, J. A.; Laurito, J. G.; Brenes, A. M.; Lin, F. T.; Sharaf, M.; Wong, L. K.; Schiff Jr., P. L.; Phytochemistry 1990, 29, 1899. 23. Leboeuf, M.; Cave, A. F.; Plantes Med. Phytother. 1972, 6, 87. 24. Tran, D. T.; Mai, H. D. T.; Pham, V. C.; Nguyen, V. H.; Litaudon, M.; Gueritte, F.; Nguyen, Q. V.; Tran, T. A.; Chau, V. M.; Phytochem. Lett. 2013, 6, 79. 25. Cortes, D.; Hocquemiller, R.; Leboeuf, M.; Cave, A.; Moretti, C. J. Nat. Prod. 1986, 49, 878. 26. Rasamizafy, S.; Hocquemiller, R.; Cave, A.; Jacquemin, H.; J. Nat. Prod. 1986, 49, 1078. 27. Menezes, L. R.; Costa, C. O.; Rodrigues, A. C.; Santos, F. R.; Nepel, A.; Dutra, L. M.; Silva, F. M.; Soares, M. B.; Barison, A.; Costa, E. V.; Bezerra, D. P.; Molecules 2016, 21, 890. 28. Shafiee, A.; Ghanbarpour, A.; Lalezari, I.; Lajevardi, S.; J. Nat. Prod. 1979, 42, 174. 29. Sun, R., Jiang, H.; Zhang, W.; Yang, K.; Wang, C.; Fan, L.; He, Q.; Feng, J.; Du, S.; Deng, Z.; Geng, Z.; J. Evidence-Based Complementary Altern. Med. 2014, 580, 483. 30. Duan, X. H., Pei, L., Jiang, J. Q.; Zhongguo Zhong Yao Za Zhi 2013, 38, 4104. 31. Costa, E. V.; Pinheiro, M. L.; Maia, B. H.; Marques, F. A.; Ruiz, A. L.; Marchetti, G. M.; Carvalho, J. E.; Soares, M. B.; Costa, C. O.; Galvao, A. F.; Lopes, N. P.; Koolen, H. H.; Bezerra, D. P.; Barison, A.; J. Nat. Prod. 2016, 79, 1524. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access