
 |
 |
 |
 |
 |
Artigo
| Influence of low and high glycerol concentrations on wettability and flexibility of chitosan biofilms |
|
Erveton P. PintoI,#,‡; Walter de S. TavaresII; Robert S. MatosI,#; Adriana M. FerreiraII; Rodrigo P. MenezesIII; Marcelo Eduardo H. M. da CostaIII; Tiago M. de SouzaIV; Irlon M. FerreiraI,§,*; Franscisco Fábio O. de SousaII; Robert Ronald M. ZamoraI
I. Departamento de Ciências Exatas e Tecnológicas, Universidade Federal do Amapá, 68903-419 Macapá - AP, Brasil Recebido em 16/04/2018 *e-mail: irlon.ferreira@gmail.com The use of chitosan biofilm in biomedical fields has a vast therapeutic potential due to useful properties such as biodegradability and antimicrobial activity, however, to achieve successful application some physicochemical properties need to be improved. In this context, the addition of glycerol to this biofilm is an alternative to improve its wettability and flexibility. Thus, low and high concentrations of plasticizer were added to this biomaterial for later characterization regarding its wettability, microstructure, mechanical and chemical properties. Glycerol addition to chitosan resulted in biofilms with more homogeneous surfaces, increased wettability, and increased flexibility, without significant changes in its chemical structure. INTRODUCTION In the last decades of the 20th century there was a paradigm change, biomedical fields researchers who used to worry only about biostable materials, now seek to study biodegradable ones, once useful applications were attributed to it. Biopolymers such as alginates, hyaluronic acid, cellulose, collagen, chitosan and gelatin, for example, have great potential for application in skin regeneration.1-3 In this context, chitosan stands out due to its large applications number and biological properties including biocompatibility, biodegradability, non-toxicity, physiological inertia and antimicrobial activity.2-4 Darmadji e Izumimoto,5 for example, reported that chitosan at 0.1% concentration is enough to inhibit Escherichia Coli growth. In alignment with it, Kumar6 has verified through in vitro tests that chitosan biofilms can inhibit the growth of Escherichia coli and Helminthosporium sp. due to a probable bonding of the biopolymer's protonated amine groups with anionic groups present in these microorganisms. Chitosan is a water-insoluble cationic polysaccharide obtained by deacetylation of chitin (present in shrimps, crabs and insects exoskeletons),7 which is the substitution of acetyl groups (-COCH3) by free amine groups (-NH2). This amine groups can be protonated in an acid medium (-NH3+).8 Thus, chitosan is structurally constituted by repetitions of the monomeric unit shown in figure 1(a) and can vary according to its degree of deacetylation.9,10
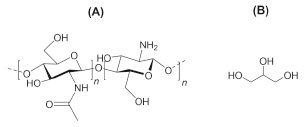 Figure 1. Chemical structure of the main reagents: (a) chitosan's monomeric unit and (b) glycerol molecule
Moreover, Shi et al.11 observed that chitosan oligosaccharides have a stimulatory effect on macrophages, performing in vitro and in vivo tests they reported that both chitosan and chitin attract neutrophils, this process called chemotaxis has a crucial role in wound healing. Other studies have shown that chitosan biofilms are not cytotoxic when tested with fibroblasts12 or keratinocytes.13 In addition, clinical studies show that human wounds treated with chitosan biofilms heal faster compared to traditional treatments.14 Escárcega-Galaz et al.,15 for example, evaluated chitosan topical gel and biofilm treatment in individuals with ulcerations and diabetes-related wounds. Initially, most lesions presented some degree of infection, tissue injury and ulceration. At the end of the treatment, all patients had a significant improvement of wound conditions, developed granulation tissue and a healthy skin cover. Among the application methods of chitosan, biofilms are the most used, allowing this polymer to be used in the encapsulation of controlled release systems, as a chelating agent in water treatment and as an antimicrobial agent. This curative biofilm produced from chitosan is already being marketed in the United States by the company HemCon®, known by the name of Chitoflex, with high therapeutic potential.1 However, chitosan biofilms are semi-crystalline materials with little flexibility and reduced wettability due to little interaction with water.16,17 Rotta,17 for example, used deionized water to study the wettability of chitosan biofilms (using sorbitol at 10% as plasticizer) by measurement of contact angle, obtaining an angle of about 92°. While Hsu et al.,18 Hamilton et al.19 e Tsai e Wang20 using distilled water, found values close to 80°. The randomness in contact angle values of chitosan biofilms is due to the fast absorption by these biofilms; this hampers the measurements accuracy of contact angle.21 However, in general, results show that chitosan biofilms have low hydrophilicity since contact angles lower than 90° indicates a hydrophilic surface, where the closer the angle is to zero the more significant will be sample's hydrophilicity.16-21 Thus, aiming to improve some physicochemical properties of this biofilm, glycerol was used as a plasticizer. Considering that glycerol molecule has a hydrophilic nature (Figure 1 (b)), it can be used to increase both the flexibility and wettability of biofilms.22 A fixed plasticizer concentration is often used to improve materials mechanical properties,17-26 although it may influence other properties such as solubility and wettability. Cordeiro,25 for example, produced chitosan biofilms at a fixed glycerol concentration (20%) to compare with biofilms produced with chitosan/gelatine blends in different proportions. In its turn, Rico26 prepared chitosan biofilms with a fixed glycerol amount (2g) and added microcapsules of carvacrol and grape seed extract to evaluate their influence on this biofilm's properties. Cerqueira et al.27 investigated glycerol influence over chitosan biofilm physicochemical properties by adding glycerol at 0.5, 1.25 and 2% (m/v) to the film-forming solution. Authors reported that biofilm's solubility increased and tensile strength decreased by increasing glycerol concentration; the latter effect is an indicator of sample flexibility improvement. Chagas22 investigated the effects of glycerol addition to chitosan biofilm over its proprieties by adding this plasticizer to chitosan powder at 10% and 25% proportions (m / m), obtaining biofilms by casting technique. The author observed that 25% glycerol biofilm showed to be more flexible and with a higher hydrophilic nature. However, this study was carried out through the traction test and swelling index values, which have low precision when compared to other more specific techniques. In this scenario, to evaluate more accurately the influence of glycerol over chitosan biofilm properties, this work was performed using the nanoindentation technique, which quantifies materials mechanical properties more accurately than the tensile test. In addition, to evaluate the wettability, the contact angle was measured with a specific goniometer apparatus. A programming language was used to obtain quantitative morphological data from AFM images in order to complement the wettability assessment, as these parameters can significantly influence biofilm's surface properties.
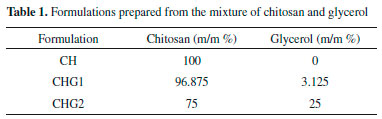
EXPERIMENTAL Materials The following reagents were used to obtain the chitosan biofilms: chitosan - low molecular weight (Sigma-Aldrich®, Brazil) with 95% of deacetylation degree, glycerol (Alphatec Química Fina®, Brazil), and glacial acetic acid (Cromaline Química Fina Ltda®, Brazil). All the other reagents were analytical grade without prior treatment. Figure 1 shows the chemical structure of the main reagents used. Biofilm preparation Chitosan biofilms were prepared by casting technique; the concentrations were evaluated as described in Table 1. Chitosan solutions at 2% (m/v) were prepared from the dissolution of this polymer in an aqueous solution of acetic acid at 3% (v/v), kept under magnetic stirring at room temperature (25 ± 2 °C) for 1 hour. Then, 40 mL aliquots of this solution were placed in a 9 cm diameter Petri dishes kept at room temperature for 5 days, until complete drying of the solvent.
A spatula and sodium hydroxide solution at 2 mol L-1 were used to neutralize remaining acid on the biofilm surface. Then, the samples were washed in distilled water and kept at room temperature for more 5 days for the final drying, at this time covered in absorbent paper. For glycerol containing biofilms, this same methodology was used but adding the plasticizer to polymer in acetic acid aqueous solution for subsequent stirring. Biofilms characterization Scanning Electron Microscopy (SEM) Morphologic profile and thickness of the biofilms were analyzed with a Scanning Electron Microscope model Tabletop Microscope TM3030 plus (Hitachi) in Standard mode. Biofilm samples (1.0 cm2) were fixed using double-sided adhesive tape. The images were assessed at 50x, 180x, 400x and 600x magnifications. Atomic Force Microscopy (AFM) The topographical analysis was performed using an Atomic Force Microscope (AFM) Park NX-10 model (Park Systems) in Tapping mode. The probe used for scanning was made of monocrystalline silicon, conical shaped, with 10° of opening angle and 6 nm of apex radius. The cantilever was made of the same material, with an elastic constant of about 5.1 N.m-1 and 150 kHz of resonance frequency. The study was performed at ambient conditions of relative air humidity (45%) and temperature (23 ± 2 °C). Biofilms samples (1.0 cm2) were fixed to the device's sample holder using a double-sided adhesive tape, then, for each biofilm six randomly chosen area (2.5 µm2) were assessed. The scanning was performed using the Tapping mode with 30.39 nm of range, setpoint of 21.05 nm and 2 seconds of scanning per line corresponding to 256 pixels per line, totalizing 256 lines. This process provided a matrix of data with 65536 pixels, where each pixel represents a value of height registered. After obtaining the topographical images by AFM, they were analyzed using the software Gwyddion. It was verified the presence of nanopores on the surface of the biofilms, and 100 diameter measurement of nanopores were assessed from 40 profiles obtained from random image areas, as described by Assis and Silva21 and Wang and Clapham.28 This topographical data were analyzed with the software WSxM to calculate the roughness RMS (standard deviation of the heights) and to obtain the matrices of heights. After obtaining the matrices of the heights, they were further assessed using two programs in R language to calculate Topographical Entropy (TE) and Surface Pore Percentage (SPP). The first algorithm was described by Matos et al.,29 it provides a quantitative measurement of roughness distribution, by giving entropy values between 0 and 1, where the closer to 1 is the Topographical Entropy value, the more uniform is the surface. The second algorithm was created exclusively for this present study and provides values for the SPP. This program finds the average of the heights and defines it as the cut-off value, heights above or equal to this cut-off value receive 1, whereas heights under the cut-off value receive 0, resulting in a binary image of 1 and 0 points, with pixels accessible to water called 0 and pixels inaccessible by water called 1. After obtaining the binary matrix, the program counts number ni of pixels accessible to water per scanned line and calculate the SPP value through the equation (1): 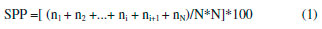 where N is the number of pixels per scanned line; AFM scans squares of N*N pixels area. Sorption and solubility tests In order to evaluate the sorption capacity and water solubility of biofilms, five pieces (1.0 cm2 area) from each produced biofilm were cut, then kept in a greenhouse at 40 °C for 48 h to remove remaining water from the preparation process. After weighing, the pieces were distributed in bottles containing 15 mL of distilled water and kept in this conditions for 4 days. Then, the excess of water was removed with absorbent paper and pieces were weighed again to evaluate the water sorption capacity of each film sample through the equation (2): 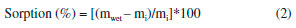 where mwet is the mass of the wet film, and mi is the mass of the dry film after the first weighing. Right after the second weighing, the pieces were retaken to the greenhouse at 70 °C until weight becomes constant. Then, they were weighed again to determinate the amount remaining material at the solution, and to calculate the solubility through the equation (3): 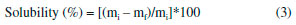 where mi is the same initial mass of the equation 1, and mf is the final mass after the last weighing. Contact Angle Measurement To measure the contact angle (Figure 1) between a drop of deionized water and the surface of the biofilms was used a Goniometer model 590 F3 (Ramé-Hart Instrument Co). The study was performed in ambient conditions of relative humidity (45%) and temperature (23 ± 2 °C). Biofilm samples (2.0 cm2 area) were placed in the sample holder, then a drop of 8 µL of deionized water was dropped on their surface using the equipment's built-in microsyringe. Contact angle measurements were performed soon after the contact between drops and surface, using a sequence of a hundred photos taken by the device's camera (0.0001 seconds per photo). Simultaneously to photo shooting, the equipment's software measured the contact angle (lateral mean), providing 100 measurements for each sample. Nanoindentation tests Nanoindentation tests were performed using a Nanoindentator TI 700 Ubi-1 (Hysitron Incorporated), with a Bekovich-type penetrator (a three faces pyramid where each one of them form an angle of about 65.3° with the plane perpendicular to the pyramid's base) made of diamond with elasticity modulus (Ei) equal to 1141 GPa, Poisson coefficient (νi) equal to 0.07 and apex radius between 50-100 nm. The device was used in the semi-static nanoindentation mode, with load limit between 100-1000 µN. During each indentation, 1024 depth measurements were registered with the precision of 0.011836 seconds per measurement, with 12.120064 seconds per indentation. For each test, was performed 12 indentations of biofilm samples (1.0 cm2 area). This way, device's software provided the values of rigidity (S), residual depth (hf) (plastic deformation), hardness (H) and reduced elastic modulus (Er), which is used to calculate elasticity modulus (E) of the sample through the equation (4): 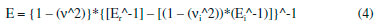 where ν is the Poisson ratio of polymer samples, which according to García et al.30 has an approximate value of 0.35. Fourier transformation infrared spectroscopy (FTIR) Biofilms spectra were assessed using a Spectrometer FTIR-ATR Shimadzu model IRAffinity-1, over the range 400-4000 cm-1, with a resolution of 4 cm-1 and 12 accumulated scans. X-Ray Diffractometry (XRD) Diffractograms were obtained using an X-Ray Diffractometer model D2 Phaser (Brunker), operating in mode Continuous PSD Fast, with CuKα radiation (λ = 1,5406 Å) generated by an electric current of 10 mA and voltage of 30 kV. The scan speed used was of 3° per minute in a scan range of 5-50°. For each scan, a square-shaped piece of biofilm (3 cm2 area) was placed at the device's sample holder. Statistical Analysis The results obtained were expressed as a mean ± standard deviation. Significant differences between average values were assessed through analysis of variance (ANOVA) using the software Statistica version 7 (StatSoft, Oklahoma, EUA), followed by the Tukey's Test, considering 5% as the significance level.31
RESULTS AND DISCUSSION Biofilms characterization Scanning Electron Microscopy (SEM) and Atomic Force Microscopy (AFM) The images acquired by SEM show that all obtained biofilms were very stable, with few apparent failures, similarly to what was observed by Assis and Silva,21 who did not use a plasticizer, and Rotta17 who used sorbitol at 10% as a plasticizer in his biofilms. Compared to other samples, biofilm CHG2 was the one with fewer irregularities on its surface, proving to be the most homogeneous. Transversal section images, which were used to measure the thicknesses, as shown in Figure 2, reveal an internal structure with a fibrous and plate-shaped appearance for all the chitosan biofilms. This was already expected since most of the films produced by casting have an irregular thickness, and are characterized by a very heterogeneous fibrous structure.21
 Figure 2. SEM images; surface (left) and transversal (right) sections of biofilms: (a) CH, (b) CHG1 e (c) CHG2
The increase of thickness occurred proportionally to the increase of glycerol concentrations, as shown in Table 2, proving the plasticizer incorporation into the structure of chitosan biofilm, once this molecule becomes positioned between the polymeric chains of the biomaterial, weakening the hydrogen bonds and hence increasing the system's volume.32-34 According to Pires and Moraes,35 skin substitutes are usually thinner than the human dermis, whose thickness ranges from 0.5 mm to 2 mm, depending on the age, the gender and body area. From this aspect, it is possible to say that biofilms prepared in this study have great potential to be used, for example, as cutaneous dressings.

By using AFM, it was found that even the most homogeneous areas of biofilms were not as regular as they seemed to be. In fact, at nano-scale (Figure 3), there is a common characteristic to all biofilms: an irregular and nanoporous topography, such as that obtained in the work of Assis and Silva,21 where the average pore diameter was close to 12.85 nm. The right side of Figure 3 shows the topographic profile indicated by the blue line in the enlarged images of AFM, providing a measurement of the pore diameter through the horizontal axis.
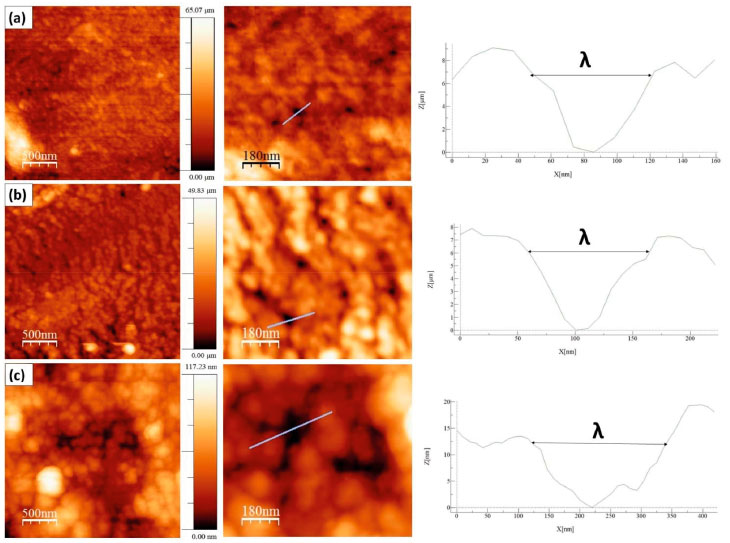 Figure 3. Biofilms topography: (a) CH, (b) CHG1 e (c) CHG2
Porosities on biofilms surface explain their high permeability to water and the difficulty in measuring contact angles more accurately. Table 2 show results from SEM and AFM. All the biofilms presented randomness for RMS values (the standard deviation was too high, implying in false results of Analysis of the Variance). However, this was already expected, since the roughness suffers high variations due to scale transformations (temporal, dimensional, among others). It is precisely due to this randomness that surface characterization of biofilms requires other parameters besides the roughness.36 In fact, it is noticed that roughness of the chitosan biofilm decreases with higher glycerol concentration, similarly what was reported by Chagas,22 whose biofilm with glycerol at 25% presented less roughness compared to one with glycerol at 10%. The sample with glycerol at 25% (CHG2) presented molecular agglomerates in its surface (partially round nuclei) distributed over the biofilm, which produces a distinct morphology with hierarchical structures, minimizing the standard deviation of heights, and hence explaining the low RMS value of this sample. These agglomerates are possibly formed by glycerol molecules due to the sample saturation, as Assis e Silva21 suggests. Moreover, the agglomerates explain the increase of surface interaction between water and surface of biofilm CHG2, since the accumulation of polar molecules in water-solid interface tends to increase the drop-surface adhesion, as seen in measurements of contact angle (Table 3).
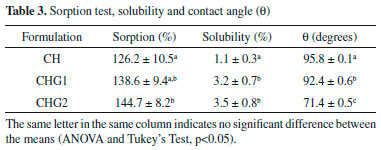
Regarding the porosities, biofilms containing glycerol had higher pores diameter compared to the standard sample (CH). However, the Surface Pore Percentage did not present the same behavior by the addition of plasticizer, showing similar values for all samples. This shows that glycerol did not influence the amount of water access points in biofilms surface. On the other hand, biofilms CHG1 and CHG2 still had pores with the same diameter values, showing that the porosity was not a determining factor for the difference of wettability (Table 3) and flexibility (Table 4) of samples.

Concerning topographical uniformity was noticed that the roughness, although its randomness, had a uniform distribution on the areas scanned by AFM for all the samples since the entropy values became close to 1. In fact, even if the presence of glycerol did not influence entropy, considering the biofilm application as a cutaneous dressing, the uniform surface helps to prevent the formation of air bubbles under the biomaterial.29 This effect further intensifies the hydrophilic nature of samples, since preventing air bubble formation increases the drop-surface contact area.37 Sorption test, solubility and contact angle The assessment of sorption capacity and water solubility shows how the addition of glycerol to biofilm can influence these properties, which is essential since the application of the biofilms as biomaterial involves contact with the aqueous media and biological fluids. Indeed, all the samples presented high water sorption capacity, retaining nearly 1.5 times its own dry mass of water, as shown in Table 3. This behavior is in accordance with the sorption values for chitosan biofilms found in the literature. Assis and Silva,21 for example, reported values close to 90%, for chitosan biofilms without a plasticiser, while Cordeiro25 obtained values around 150% using the glycerol as a plasticiser. In respect to water solubility, even though all the biofilms present a low percentage, it was noticed that the addition of glycerol to this biomaterial improved this property. This was expected since the presence of a polar molecule like glycerol would increase the interaction with water. Garcia, Pinotti and Zaritzkya,38 for example, reported that the addition of glycerol to chitosan/starch biofilms increased its solubility by 56%. In contrast, from the values of contact angle, it is noticed that biofilm CH (standard sample) had low interaction with water, revealing specific hydrophobic nature, this is probably due to protonated amine groups (NH3+) disposed at the chitosan structure, appearing when the polymer is diluted in acid medium forming a cationic polymer.7-10,39 These positive charges scattered over the biofilm structure possibly repel themselves by electrostatic repulsion, consequently migrating to sample surface, culminating in a higher affinity for anionic molecules by the chitosan biofilm,40 and hence weakening its adhesion with water, whose molecules are polar. It is clear that glycerol addition influenced the hydrophilicity of the biofilms since its addition reduced the contact angle values proportionally to its concentration. This is probably due to the high affinity between molecules of glycerol and water considering that both of them are polar. Indeed, when samples are saturated with glycerol, these molecules dispose themselves between the polymeric chains and between the biofilm's most external molecules, increasing water-surface adhesion, as Chagas22 suggests. A correlation test followed by linear regression was performed to prove the correlation between glycerol concentration and the contact angle. It was observed that the variables had a high negative linear correlation (r = -0.92) and the glycerol concentration variable describes nearly 85% of contact angle variation, as shown in Figure 4.
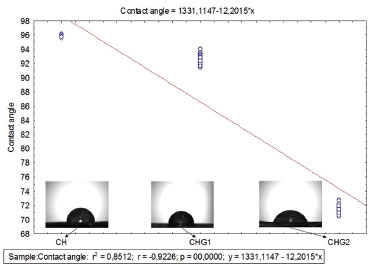 Figure 4. Correlation and linear regression test for measurement of contact angle in function of glycerol concentration
The behavior shown in the graph of Figure 4 is in agreement with the results of Chagas,22 who observed through values of the swelling index that among biofilms of chitosan containing glycerol at 0%, 10% and 25%, that the ones with higher glycerol concentration had a higher hydrophilic nature. Its also in agreement with results of Leceta et al.,41 who observed through contact angle measurements that among chitosan biofilms with glycerol at 0%, 15% and 30%, the sample with highest glycerol content had the most hydrophilic nature. Furthermore, the drops photos on samples surface (Figure 4) indicate a better scattering by biofilm CHG2 (glycerol at 25%), confirming that glycerol significantly improves the hydrophilicity of chitosan biofilms. The increase of hydrophilicity also favored water absorption, as shown in Table 3. Nanoindentation tests Through a nanoindentator were obtained curves of charge and discharge to evaluate the stiffness and elasticity modulus of samples quantitatively. From these curves were obtained the results shown in Table 4. The values of stiffness and residual depth show no significative difference between samples. However, the fact that material presented residual depth after the mechanical test indicates an elasto-plastic behavior of these films, which means that a part of the deformation is elastic and the other part is plastic. Materials with this behavior are subjected to cold work, in other words, they are subjected to either hardening or softening following the plastic deformation.42,43 Biofilms CHG1 and CHG2 had statically similar values of hardness compared to CH, showing that addition of glycerol did not change the biomaterial resistance mechanical stress deformations. However, regarding the elasticity module, both biofilms had significative differences compared to the standard sample (CH), with a decrease of about 8.6% for CHG1 and about 12.9% for CHG2, evidencing an improvement in biofilm flexibility with higher plasticizer concentration, in agreement with the literature. Chagas,22 Cerqueira et al.27 and Leceta et al.,41 for example, adding different glycerol concentrations to chitosan biofilm, reported that biofilms tensile strength decreased by the addition of this plasticizer, which indicates the increased flexibility. In fact, when incorporated to the biofilm, glycerol tends to decrease its resistance to stress deformation, since by placing itself among polymer's chains, it makes them more distant from each other, resulting in weaker intermolecular forces of the biomaterial.24,25 The weakening of intermolecular forces makes it easier to suffer distortions in bonds between polymer chains due to mechanical stress, increasing elastic properties of the biofilm. Fourier Transformation Infrared Spectroscopy (FTIR) In the chitosan biofilm spectra, frequency bands 3650-3600 cm-1 and 3400-3200 cm-1, corresponding to free hydroxyls or amine, and ones associated to hydrogen bonds,44 respectively, possibly suffered a displacement to lower frequencies due to the effect of secondary hydrogen bonds by free amine groups (-NH2) of chitosan molecule. Nitrogen is less electronegative than oxygen, and hence amine forms weaker hydrogen bonds, explaining the low solubility of chitosan in water. Besides that, weaker chemical bonds have a lower force constant and vibrate in lower frequencies.45,46 In this sigh, in order to make this vibration appear at the infrared spectrum, the molecule should undergo a variation of its dipole moment due to this vibration. Molecular vibrations without dipole moment variation do not generate absorption bands at the infrared spectrum.44 Therefore, secondary hydrogen bond formed by amines nitrogen, possibly softened the effects of intermolecular vibration over chitosan molecule dipole moment, explaining the displacement of frequency bands 3650-3600 cm-1 and 3400-3200 cm-1 for lower frequencies, as shown in Figure 5.
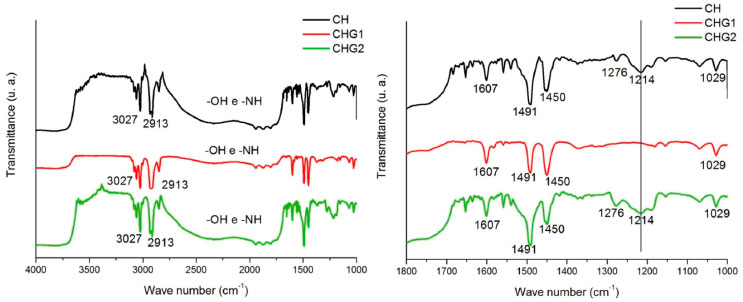 Figure 5. FTIR Spectra of biofilms
The peaks at 3027 cm-1 and 2913 cm-1 can correspond to the axial deformation of chemical bonds O-H and N-H from free hydroxyls and amines which did not form hydrogen bonds. While bands in the range 2830-1669 cm-1 possibly are a stretch superposition between -OH and -NH of hydroxyls and amines forming the hydrogen bonds among chitosan polymer. It is noticed a peak at 1607 cm-1, which according to Horn,47 corresponds to chitosan protonated amines (-NH3+), confirming the presence of positive charges in the biopolymer structure. Furthermore, the peak at 1491 cm-1 corresponds to angular deformation vibration by -NH group from amines or secondary amides.44 The peak at 1450 cm-1 is caused by the angular deformation out of the -CH2 bonding plane. Peaks at 1214 cm-1 and 1029 cm-1 correspond to the axial deformation C-O from saturated esters due to the reaction between the acetic acid carboxyl and the hydroxyls from chitosan or glycerol. Overall, the biofilms spectra did not present remarkable differences. However, biofilm CHG1 spectrum did not show the peak 1214 cm-1. This was not expected since glycerol is an alcohol, then esters formation was supposed to be bigger. Probably, glycerol concentration of this biofilm was not sufficient to intensify the presence of esters. X-Ray Diffractometry (XRD) Biofilms diffractograms show no remarkable differences of chitosan crystalline phase since all of them had diffraction peaks near to 2θ = 20°, and a diffraction halo at 2θ = 10°. According to Uragami and Tokura,16 strong intramolecular and intermolecular interactions caused by the hydrogen bonds between amines, alcohol and amide groups of chitosan, causes this biomaterial to show some crystallinity, as shown in Figure 6.
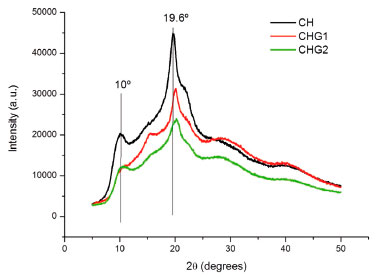 Figure 6. Biofilms diffractograms
It is possible to notice that material crystallinity decreases with higher glycerol concentration since biofilms CHG1 and CHG2 had lower peaks compared to the standard sample (CH), similar to that observed by Dallan48 who also used glycerol as plasticizer in chitosan biofilms. This explains why sample CHG2 had higher flexibility, once its elasticity module was the lowest among the analyzed samples.
CONCLUSION In this study was produced chitosan biofilms; without glycerol (CH), with glycerol at 10% (CHG1), and at 25% (CHG2), with good appearance and homogeneity, despite the morphological irregularities at the nanoscale. However, the chitosan biofilm, unlike expected, did not show remarkable hydrophilic nature, since the standard sample (CH) had a contact angle of 95.8°. Chitosan provides a biomaterial with high sorption capacity and a nanoporous structure. For these reasons, it was plausible to suppose that biofilm's hydrophilicity was evident, but results obtained showed the wettability of the biofilms were affected by the possible presence of positive charges on their surface, these charges were generated by protonated amines present in chitosan structure (which was confirmed by the FTIR spectra). In this context, using glycerol as a plasticizer on biofilms production was decisive for improving their wettability and flexibility, since values of contact angle and elasticity modulus decreased with higher glycerol content added to the film-forming solution, confirming the initial hypothesis of this study. Besides the improvement of physical properties, the chemical structure of chitosan biofilm was not significantly influenced by the presence of glycerol, ensuring that therapeutical and toxicological properties of chitosan were not affected, which is highly attractive for future applications on the biomedical field. Thus, biofilm containing glycerol at 25% (CHG2) was the one with the best results from the analysis, showing that this type of biomaterial has a high potential for pharmaceutical applications.
ACKNOWLEDGMENTS To Capes (financial assistance), to Laboratory of Quality Control, Bromatology and Microbiology (LCBM) of the Federal University of Amapá (UNIFAP), to the Laboratory Van De Graaff (LVDG) of the Pontifical Catholic University of Rio de Janeiro (PUC-RJ), to Laboratory of Research in Drugs (LPFar) of UNIFAP and the Laboratory of Instrumental Analysis (LAI) of the State University of Amapá (UEAP).
REFERENCES 1. Silva, S. S.; Luna, S. M.; Gomes, M. E.; Macromol. Biosci. 2008, 8, 568. 2. Dash, M.; Chiellini, F.; Ottenbrite, R. M.; Chiellini, E.; Prog. Polym. Sci. 2011, 36, 981. 3. España-Sánchez, B. L.; Cruz-Soto, M. E.; Elizalde-Peña, E. A.; Sabasflores-Benítez, S.; Roca-Aranda, A.; Esquivel-Escalante, K.; Luna-Bárcenas, G.; IntechOpen (2018), doi: 10.5772/intechopen.75401. 4. Laranjeira, M. C. M.; De Fávere, V. T.; Quim. Nova 2009, 32, 672. 5. Darmadji, P.; Izumimoto, M.; Meat Sci. 1994, 38, 243. 6. Kumar, R.; React. Funct. Polym. 2000, 96, 1. 7. Li, Y.; Guo, X.; Lin, P.; Fan, C.; Song, Y.; Carbohydr. Polym. 2010, 81, 484. 8. Kim, I-Y.; Seo, S-J.; Moon, H-S.; Biotechnol. Adv. 2008, 4, 1. 9. Cissé, M.; Montet, D.; Tapia, M. S.; Loiseau, G.; Ducampcollin, M. N.; Food Hydrocolloids 2012, 28, 361. 10. Wang, S. F.; Shen, L.; Tong, Y. J.; Chen, L.; Phang, I. Y.; Lim, P. Q.; Liu, T. X.; Polym. Degrad. Stab. 2005, 90, 123. 11. Shi, C.; Zhu, Y.; Ran, X.; J. Surg. Res. 2006, 133, 185. 12. Silva, S. S.; Luna, S. M.; Gomes, M. E.; Macromol. Biosci. 2008, 8, 568. 13. Chatelet, C.; Damour, O.; Domard, A.; Biomaterials 2001, 22, 261. 14. Santos, T. C.; Marques, A. P.; Silva, S. S.; J. Biotechnol. 2007,132, 218. 15. Escárcega-Galaz, A. A.; Cruz-Mercado, J. L. D.; López-Cervantes, J.; Sánchez-Machado, D. I.; Brito-Zurita, O. R.; Ornelas-Aguirre, J. M.; Saudi J. Biol. Sci. 2018, 25, 130. 16. Uragami, T.; Tokura, S.; Material Science of Chitin and Chitosan, Kodansha: Japan, 2006. 17. Rotta, J.; Dissertaçao de Mestrado, Universidade Federal de Santa Catarina, Brasil, 2008. 18. Hsu, S. H.; Whu, S. W.; Tsai, C.; J. Polym. Res. 2004, 11, 141. 19. Hamilton, V.; Yan, Y.; Rigney, D. A.; J. Mater. Sci.: Mater. Med. 2006, 17, 1373. 20. Tsai, H. S.; Wang, Y. Z.; Polym. Bull. 2008, 60, 103. 21. Assis, O. B. G.; Silva, V. L.; Polim.: Cienc. Tecnol. 2003, 13, 223. 22. Chagas, J. A. O.; Dissertaçao de Mestrado, Universidade Federal do Rio Grande do Norte, Brasil, 2017. 23. Khajuria, D. K.; Patil, O. N.; Karasik, D.; Razdan, R.; Arch. Oral Biol. 2018, 85, 120. 24. Pérez-Córdoba, L. J.; Norton, I. T.; Batchelor, H. K.; Gkatzionis, K.; Spyropoulos, F.; Sobral, P. J. A.; Food Hydrocolloids 2018, 79, 544. 25. Cordeiro, C. M. S.; Dissertaçao de Mestrado, Universidade Técnica de Lisboa, Portugal, 2010. 26. Rico, B. P. M.; Dissertaçao de Mestrado, Universidade de Algarve, Portugal, 2015. 27. Cerqueira, M. A.; Souza, B. W. S.; Teixeira, J. A.; Vicente, A. A.; Food Hydrocolloids 2012, 27, 175. 28. Wang, H.; Clapham, D. E.; Biophys. J. 1999, 77, 241. 29. Matos, R. S.; Lopes, G. C. L.; Ferreira, N. S.; Pinto, E. P.; Carvalho, J. C. T.; Figueiredo, S. S.; Oliveira, A.F.; Zamora, R. M.; Arabian J. Sci. Eng. 2018, 43, 3371. 30. García, M. E.; Domíngues, C. G.; Pérez, J. J. C.; Rebollo, R. R. F.; Adame, J. A.; Vázquez, I. A.; Mendez, J. V. M.; Ruiz, L. A. M.; Int. J. Biol. Macromol. 2013, 61, 106. 31. Martins, G. A; Domingues, O. A.; Estatística Geral e Aplicada, Atlas S. A.: Sao Paulo, 2011. 32. Mchugh, T. H; Krochta, J. M.; J. Agric. Food Chem. 1994, 42, 841. 33. Lima, A. M. F.; Andreani, L.; Soldi, V.; Quim. Nova 2007, 30, 832. 34. Rahmat, N.; Abdullah, A. Z.; Mohamed, A. R.; Renewable Sustainable Energy Rev. 2010, 14, 987. 35. Pires, A. L. R., Moraes, A. M.; 7º Congresso Latino Americano de Biomateriais e Orgaos Artificiais, Natal, Brasil, 2012. 36. Salcedo, M.; Dissertaçao de Mestrado, Universidade Federal do Amapá, Brasil, 2016. 37. Oliveira, L. R.; Dissertaçao de Mestrado, Universidade Federal de Santa Maria, Brasil, 2010. 38. Garcia, M. A.; Pinotti, A.; Zaritzkya, N. E.; Starch/Stärke 2006, 58, 453. 39. Azevedo, V. V. C.; Chaves, S. A.; Bezerra, D. C.; Lia Fook, M. V.; Costa, A. C. F. M.; Revista Eletrônica de Materiais e Processos 2007, 23, 27. 40. Mathur, N. K.; Narang, C. K.; J. Chem. Educ. 1990, 67, 938. 41. Leceta, I.; Guerrero, P.; Caba, K.; Carbohydr. Polym. 2013, 93, 339. 42. Souza Neto, E.A.; Peric, D.; Owen, D.R J.; Computacional methods for plasticity: theory and applications, 1st ed., Wiley: New York, 2008. 43. Callister, W. D.; Rethwisch, D. G.; Ciência e Engenharia de Materiais: uma introduçao, 9th ed., LTC: Rio de Janeiro, 2016. 44. Pavia, D. L.; Lampman, G. M.; Kriz, G. S.; Vyvyan, J. R.; Introduçao à Espectroscopia, trans. 4th North American edition, Cengage Learning: Sao Paulo, 2010. 45. Nussenzveig, H.M.; Curso de Física Básica: fluídos, oscilaçoes, ondas e calor, Blucher: Rio de Janeiro, 2003. 46. Hollauer, E.; Quimica Quântica, LTC: Rio de Janeiro, 2007. 47. Horn, M. M.; Tese de Doutorado, Universidade de Sao Paulo, Brasil, 2012. 48. Dallan, P. R. M.; Tese de Doutorado, Universidade Estadual de Campinas, Brasil, 2005.
#Grupo de Pesquisa Materiais Amazônicos §Grupo de Biocatálise e Síntese Orgânica Aplicada do Departamento de Química |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access