
 |
 |
 |
 |
 |
Artigo
|
|
| Pré-concentração baseada na coprecipitação usando cromato de prata como carreador para determinação de cobre por FAAS Preconcentration based on coprecipitation using silver chromate as a carrier for copper determination by FAAS |
|
Ivero P. Sá; Luana N. Santos; Erik G. P. da Silva; Daniel de C. Lima; Fábio Alan C. Amorim*
Departamento de Ciências Exatas e Tecnológicas, Universidade Estadual de Santa Cruz, 45662-900 Ilhéus - BA, Brasil Recebido em: 09/05/2018 *e-mail: facamorim@uesc.br In the present paper, a preconcentration procedure based in the coprecipitation of copper by silver dichromate (Ag2CrO4) and consecutive determination by flame atomic absorption spectrometry (FAAS) was proposed. The experimental factors were optimized from application of the Doehlert matrix and the conditions set: chromate concentration 5.8 x 10-4 mol L-1, silver concentration 1.1 x 10-3 mol L-1, pH 7.3 and disperser concentration HNO3 0.38 mol L-1. Under these conditions, analytical parameters of the procedure were obtained, with the quantification limits of 0.76 µg L-1, precision (based in the relative standard deviation, RSD %) with values lower than 2.7%, and preconcentration factor of 28. The accuracy of the procedure was given by addition/recovery test in water samples groundwater and mineral, and by comparison with results obtained by ICP-MS. The procedure developed presented be simple, sensitive and low cost, thus can be applied in the preconcentration and determination of copper in several water samples. INTRODUÇAO O cobre é um elemento de importância considerável nos sistemas ambientais e na fisiologia animal.1 A essencialidade do cobre é firmemente estabelecida na dieta nutricional humana, e este apresenta um papel fundamental na síntese de diversas enzimas responsáveis pela produçao de proteínas.1,2 Apesar de sua relevância, a toxicidade do Cu2+ desempenha um papel importante no desenvolvimento da doença de Alzheimer, mostrando que até pequenas quantidades deste analito em água potável causa a rápida perda da cogniçao.1-5 Por isso, é de extrema necessidade o monitoramento desse analito em amostras ambientais, alimentos e de água. Técnicas espectroanalíticas convencionais como a espectrometria de absorçao atômica com chama (FAAS) tem sido eficiente na detecçao de metais traços em vários tipos de amostras devido a sua alta seletividade, rapidez, fácil manuseio e baixo custo. No entanto, FAAS apresenta algumas limitaçoes como a impossibilidade de determinar analitos em baixas concentraçoes e/ou alta influência de sais dissolvidos em amostras de águas, causando interferência no sinal dos analitos.6-9 Para solucionar essas limitaçoes, vários métodos de separaçao/pré-concentraçao têm sido empregados, como, por exemplo, a extraçao em fase sólida,10 extraçao em ponto nuvem,11 extraçao por solventes,12 flotaçao,13 adsorçao,14 coprecpitaçao,15 e membrana filtrante.16,17 A coprecipitaçao é um importante método de separaçao/pré-concentraçao para metais traços devido a sua simplicidade, baixo consumo de solvente e alto fator de pré-concentraçao. Além disso, diversos analitos podem ser separados e pré-concentrados em uma única etapa da matriz utilizando diferentes coprecipitantes, orgânicos ou inorgânicos.18-21 Hidróxidos metálicos como érbio,22 cério,23 túlio,7 gadolínio,24 zircônio15 têm sido empregados como coprecipitantes inorgânicos. Coprecipitantes orgânicos que tendem a formar quelatos neutros com espécies metálicas como 8-hidroxiquinolina, ditiocarbamato e ácido violúrico têm sido usados na pré-concentraçao de metais traços em várias matrizes.25,26 A coprecipitaçao envolve a incorporaçao de traços de impurezas de uma fase líquida solúvel para dentro do precipitado. A habilidade dos precipitados de capturar impurezas pode ser utilizada para concentrar elementos traço.27 Os mecanismos de coprecipitaçao incluem a adsorçao superficial, a troca iônica, precipitaçao superficial e a oclusao.28 O cromato de prata (Ag2CrO4), composto bastante conhecido e empregado na química analítica clássica com indicador para quantificaçao de íons cloreto pelo método de Mohr. O coprecipitante cromato de prata (Ag2CrO4) é praticamente insolúvel em água, com Kps = 1,12x10-12 e apresenta um interessante equilíbrio químico dependente do pH com a espécie dicromato, que é solúvel.29 O mecanismo de coprecipitaçao predominante é baseado no fenômeno de adsorçao superficial, em que após a precipitaçao do cromato de prata forma-se a primeira camada de adsorçao na qual sao retidos os analitos, os quais sao contrabalanceados com a camada de adsorçao secundária. Ainda que seja um composto popular e comumente utilizado como indicador em métodos titulométricos de precipitaçao, a aplicaçao do Ag2CrO4 como carreador para separaçao e pré-concentraçao por coprecipitaçao nao foi avaliada. Diante do exposto, o objetivo deste trabalho foi desenvolver um método de pré-concentraçao com o emprego do carreador cromato de prata para coprecipitaçao de quantidades traço de Cu2+ em amostras de águas para determinaçao por FAAS.
PARTE EXPERIMENTAL Instrumentos Um espectrômetro de absorçao atômica com chama, modelo SpectrAA 240FS (VARIAN, Mulgrave, Austrália) equipado com chama ar-acetileno foi utilizado para detecçao do analito. As condiçoes instrumentais foram: comprimento de onda do cobre 324,8 nm, corrente da lâmpada 10 mA, largura da fenda 0,5 nm, altura do queimador 13,5, mm, vazao de acetileno 2,00 L min-1 e vazao de ar 13,5 L min-1. Um espectrômetro de massas com plasma indutivamente acoplado (ICP-MS) modelo NexION 300X (Perkin-Elmer, Shelton, CT) equipado com a tecnologia de célula universal (Universal Cell TechnologyTM - UCT) foi utilizado para determinaçoes diretas de cobre (63Cu) nas amostras. As condiçoes intrumentais foram: potência RF 1600W, vazao de gás no plasma 18 L min-1, vazao de gás auxiliar 1,2 L min-1, vazao de gás no nebulizador 1,01 L min-1. As medidas dos pH foram realizadas em pHmetro HANNA modelo pH 21, uma centrífuga Solab modelo L-700 foi utilizada para acelerar o processo de decantaçao do precipitado, uma balança analítica GEHAKA foi utilizada para medir as massas de reagentes e amostras. Uma centrífuga Eppendorf modelo 5804 foi utilizada para decantaçao do precipitado. Reagentes e soluçoes Para o preparo de todas as soluçoes foi utilizado água desionizada obtida por osmose reversa (18,2 MΩ cm, GEHAKA modelo OS10 LX). Todos reagentes utilizados foram de grau analítico. Toda vidraria utilizada foi descontaminada em soluçao HNO3 10% (v/v) por 24 horas e lavada duas vezes com água desionizada. As soluçoes padrao de Cu(II) foram preparadas a partir da diluiçao do padrao estoque 1000 mg L-1(Specsol). Soluçoes de cromato de potássio (K2CrO4, Merck) e nitrato de prata (AgNO3, Vetec) foram preparadas a 0,055 mol L-1. Tris (Sigma-Aldrich) 0,1 mol L-1foi preparado e o pH ajustado de acordo ao experimento. Acido nítrico 65 % m/m (Merck) foi utilizado para preparaçao das soluçoes de ácido nítrico utilizadas para solubilizaçao do precipitado. Método de pré-concentraçao O método de coprecipitaçao do cromato de prata foi otimizado em soluçoes padroes. Em tubos de ensaio contendo 10 mL de soluçao-amostra contendo Cu2+ a 20 µg L-1 foram adicionados 5,8 x 10-4 mol L-1 de cromato e 1,1 x 10-3 mol L-1de prata. O pH da soluçao foi ajustado para 7,3 utilizando soluçao tampao tris 0,1 mol L-1. O tubo foi agitado manualmente por alguns segundos e em seguida foi centrifugado em 1780 G durante 10 min. O sobrenadante foi removido e o precipitado permaneceu aderido ao tubo, e este foi solubilizado com 200 µL de HNO3 0,38 mol L-1. O cobre foi determinado por espectrometria de absorçao atômica com chama, e como resultado analítico foi utilizado absorvância relativa a área do pico do analito em funçao do tempo (10 s) para 200 µL de amostra pré-concentrada. Estratégia de otimizaçao O procedimento de coprecipitaçao foi otimizado através da análise dos quatro fatores: (1) concentraçao de cromato; (2) concentraçao de prata; (3) pH; (4) concentraçao do dispersor HNO3. Os níveis foram determinados a partir da revisao de trabalhos publicados e experimentos preliminares.30-34 A matriz Doehlert foi empregada para a otimizaçao dos fatores, sendo os experimentos realizados em triplicata. Para análise dos dados foi utilizado o programa STATISTICA tendo nível de confiança fixo em 95% e a Tabela 1 mostra os fatores e níveis estudados neste procedimento.
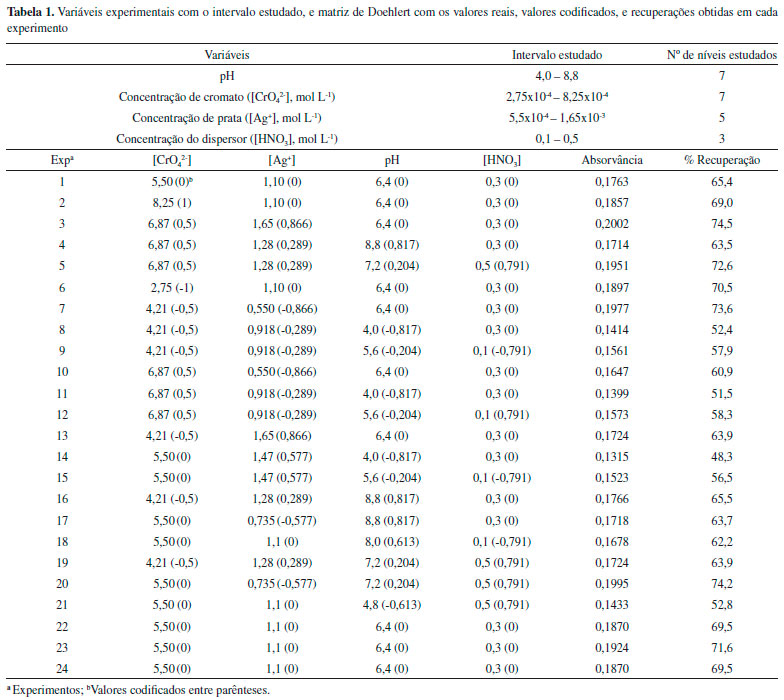
Aplicaçao em amostras reais As amostras de água mineral engarrafada foram coletadas na cidade de Ilhéus, BA, Brasil e as amostras de água subterrânea coletadas em vários pontos no bairro do Banco da Vitória e em bairros da regiao norte da cidade de Ilhéus-Ba, Brasil. As amostras foram acondicionadas em frascos de polipropileno e mantidas refrigeradas em aproximadamente 4 °C e transportadas até o laboratório. No laboratório, antes da aplicaçao do procedimento de pré-concentraçao, as amostras foram filtradas utilizando membrana de acetato de celulose com poros de 0,45 µm, acidificadas com HNO3 a pH 1 e armazenadas sob refrigeraçao entre 2 e 4 °C. O pH das amostras foi ajustado para pH 7,3 com soluçao tampao Tris. Posteriormente, o procedimento de pré-concentraçao descrito anteriormente foi aplicado às amostras, sempre em triplicata.
RESULTADOS E DISCUSSAO Otimizaçao das condiçoes experimentais O emprego de ferramentas quimiométricas é uma interessante opçao na otimizaçao de processos de pré-concentraçao. A maior vantagem na aplicaçao dessas ferramentas está na análise da interaçao das variáveis estudadas. Dentre as ferramentas utilizadas têm-se a matriz Doehlert, o planejamento Box-Behnker e o planejamento de composto central que têm sido amplamente descritos na literatura.35-39 Para execuçao da otimizaçao do procedimento de coprecipitaçao utilizou-se a matriz Doehlert, e foram necessários 24 experimentos expresso em uma matriz com 4 fatores, sendo 21 experimentos e mais 3 experimentos no ponto central, a qual está demonstrada na Tabela 1. Os resultados foram analisados de acordo ao percentual de recuperaçao, e estes foram calculados a partir do fator de pré-concentraçao teórico do procedimento. Validaçao do modelo estatístico e experimental A avaliaçao da qualidade do modelo matemático é necessária para a obtençao de dados confiáveis, ou seja, se este é capaz de descrever o comportamento dos valores experimentais satisfatoriamente. A qualidade do modelo matemático pode ser testada por meio da Análise de Variância (ANOVA). Os modelos matemáticos, linear e quadrático foram avaliados a fim de obter a melhor descriçao da regiao experimental. Segundo Pimentel e Neto,40 para avaliar a qualidade do modelo, deve-se verificar o valor F da falta de ajuste. Se o modelo matemático estiver ajustado aos dados experimentais, a média quadrática da falta de ajuste (MQfaj) deve refletir apenas os erros aleatórios inerentes ao sistema. Além disso, a média quadrática do erro puro (MQep) também deve ser uma boa estimativa desses erros e presume-se que estes dois valores nao sao estatisticamente diferentes. Se a razao entre a MQfaj/MQep for inferior ao valor de F tabelado, o ajuste do modelo é considerado satisfatório. Com ANOVA apresentada na Tabela 2, observou-se que a falta de ajuste para o modelo quadrático nao é significativa, pois o valor da razao da MQfaj/MQep é (8,01) inferior ao valor de Ftabelado (8,78) para 10 e 3 graus de liberdade, respectivamente. Assim, o modelo quadrático está bem ajustado aos dados obtidos apresentando menores resíduos e desta forma boa capacidade de previsao.
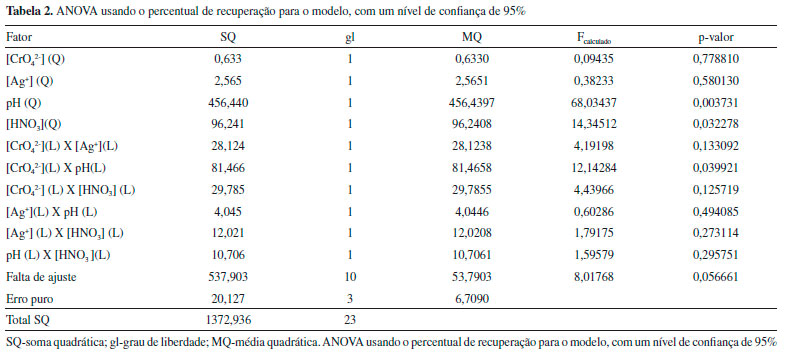
Além do teste da falta de ajuste, o modelo quadrático foi avaliado também pela análise do gráfico dos valores preditos em funçao dos valores observados, apresentado na Figura 1(a). A concordância entre os valores preditos pelo modelo e os valores experimentais (R2 = 0,93) confirma que o modelo quadrático está bem ajustado aos dados experimentais obtidos no procedimento, com percentagem de variaçao explicada em torno de 93%.
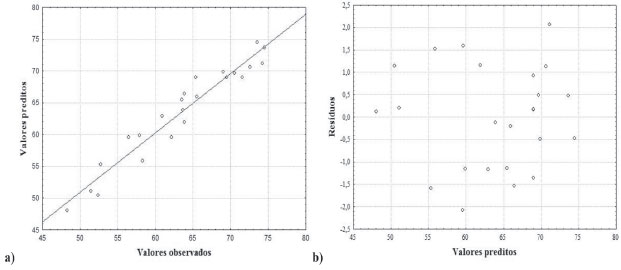 Figura 1. (a) Gráfico de valores preditos em funçao dos valores observados e (b) gráfico de resíduos em funçao dos valores preditos para o modelo quadrático usando o percentual de recuperaçao
O modelo quadrático também foi avaliado através do gráfico de valores preditos em funçao dos resíduos. Assim como a falta de ajuste apresenta importância na avaliaçao do modelo, é necessário que avalie os resíduos deixados pelo modelo. Para um modelo bem ajustado, esses resíduos nao devem apresentar nenhum indício de anormalidade. Um modelo que deixa resíduos muito grandes ou tendenciosos é inadequado para inferir precisamente sobre o comportamento destes dados no campo experimental em questao.40,41 A Figura 1(b) apresenta o gráfico de resíduos e, pode-se observar que os valores dos resíduos sao de magnitude na faixa de ±2,2% de recuperaçao e se distribuem aleatoriamente mostrando mais uma vez que o modelo quadrático está bem ajustado. A determinaçao das condiçoes ótimas para os dados modelados pode ser realizada a partir da equaçao da regressao apresentada na equaçao 1. É possível calcular as coordenadas do ponto crítico, ou seja, estabelece as condiçoes em que os fatores estudados fornecem a melhor resposta, através das primeiras derivadas desta funçao matemática que descreve a superfície de resposta, em relaçao a cada variável e igualando-as a zero.41 Atraves desses cálculos, foram obtidos os valores críticos para cada variável estudada, como sendo: [CrO42-] = 5,8x10-4, [Ag+] = 1,1x10-3, [HNO3] = 0,38 e pH = 7,3. 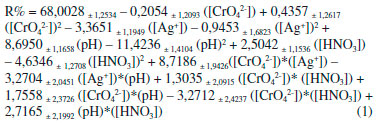 A inspeçao visual das superfícies de resposta é outra forma de extrair informaçoes do modelo referente as condiçoes que melhor beneficiam a recuperaçao de cobre para esse procedimento.42 Entretanto, só é possível projetar duas variáveis em cada projeçao gráfica e o terceiro eixo é dedicado a resposta analítica. Nas superfícies de resposta obtidas, a regiao entre os eixos onde a cor vermelha é mais intensa denota o máximo da resposta para os fatores projetados. Três, das seis, superfícies de resposta sao apresentadas na Figura 2, e todas elas apresentam pontos de máximo coerentes com os pontos críticos obtidos.
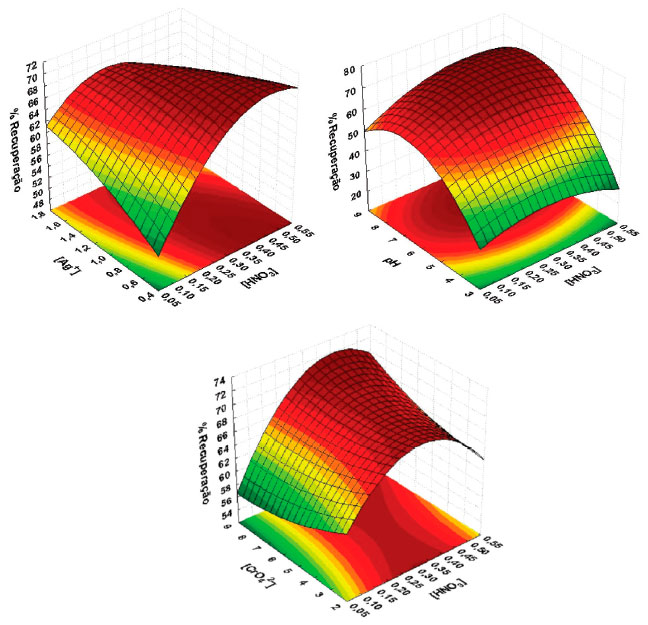 Figura 2. Superfícies de resposta obtidas a partir da matriz Doehlert para as variáveis
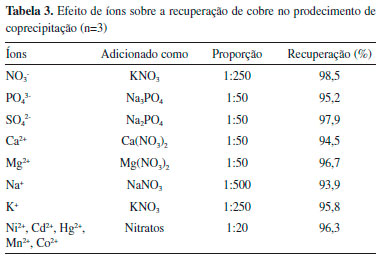
O pH influencia diretamente no equilíbrio químico de formaçao do coprecipitante, cromato de prata. Em pH mais ácido o equilíbrio é deslocado para formaçao do íon dicromato, equaçao 2, ao qual é solúvel e isto implica diretamente na recuperaçao de cobre.43 Em pH elevado a concentraçao de OH- interfere no equilíbrio favorecendo a formaçao do hidróxido de prata (equaçao 3). A precipitaçao da prata como hidróxido só ocorre em pH superior a 10 e por este motivo a faixa de trabalho nao excedeu ao pH 9.43 A condiçao que maximiza a precipitaçao de Ag2CrO4 para a extraçao de cobre em relaçao ao pH foi determinado pelos valores críticos do modelo e da superfície de resposta. 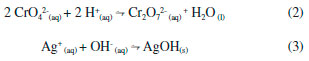 As concentraçoes de cromato e de prata sao de fundamental importância para o procedimento, pois nao há coprecipitaçao sem o precipitado cromato de prata. A concentraçao do agente dispersor HNO3 é um importante fator, necessário para solubilizaçao do precipitado liberando assim o cobre para a soluçao, a adiçao do ácido nítrico deslocou o equilíbio para a formaçao do dicromato o que favoreceu a solubilizaçao do precipitado disponibilizando o cobre para a determinaçao. Assim, as condiçoes críticas obtidas pelos cálculos ([CrO42-] = 5,8x10-4, [Ag+] = 1,1x10-3, [HNO3] = 0,38 e pH = 7,3) corroboram com a inspeçao visual dos pontos máximos de resposta, estando todos os valores críticos dentro do domínoi experimental estudado. Características analíticas Sob as condiçoes otimizadas foram estabelecidos os parâmetros analíticos do procedimento e os mesmos foram determinados de acordo com as recomendaçoes da Uniao Internacional de Química Pura e Aplicada (IUPAC).44 A curva de calibraçao foi construída variando a concentraçao de cobre de 2 até 100 µg L-1, com equaçao para a reta sendo Abs = 0,0028±0,0004 [Cu, µg L-1] - 0,0168±0,0075 e R2 = 0,9987. O fator de enriquecimento experimental foi calculado utilizando a relaçao entre os coeficientes angulares das curvas de calibraçao com e sem pré-concentraçao, e o valor calculado foi de 28. Considerando a razao entre os volumes da amostra (10 mL) e da fase rica (0,2 mL), o fator de enriquecimento teórico é de 50, resultando numa eficiência de transferencia de fases de 56%. O limite de detecçao (LD), como a concentraçao equivalente a 3 vezes o desvio padrao do branco, obtido por 10 determinaçoes distintas, dividido pela inclinaçao da curva analítica, foi de 0,23 µg L-1. O limite de quantificaçao (LQ), como a concentraçao equivalente a 10 vezes o desvio padrao do branco, obtido por 10 determinaçoes distintas, dividido pela inclinaçao da curva analítica, foi de 0,77 µg L-1. A precisao do procedimento foi avaliada como desvio padrao relativo (RSD %) de 10 determinaçoes distinas nas concentraçoes 5,0 µg L-1 e 100 µg L-1 e os valores encontrados foram 2,7% e 1,3%, respectivamente. Efeito de outros íons Um dos problemas que mais compromete uma análise em espectrometria de absorçao atômica na detecçao de metais traços é a interferência de matriz. Esses experimentos devem ser conduzidos para examinar o efeito de outros íons na coprecipitaçao, porque alguns desses metais traços estao presentes em amostras reais e pode interferir tanto na determinaçao do analito quanto no procedimento de coprecipitaçao desenvolvido. Neste estudo, diferentes quantidades de outros íons foram adicionadas a uma soluçao contendo cobre a 20 µg L-1 e a amostra tratada de acordo ao procedimento recomendado. Os resultados obtidos mostraram que as recuperaçoes foram quantitativas para o cobre na presença de outros elementos (Tabela 3). O limite de tolerância foi definido como a concentraçao de íons que conduz a um erro menor que 5% em relaçao a separaçao e detecçao do analito. Elevadas concentraçoes do íon cloreto (> 30‰) podem inviabilizar a aplicaçao do procedimento de coprecipitaçao, pois na presença deste íon a prata pode precipitar como cloreto de prata e nao como cromato de prata. A constante de solubilidade do cromato de prata é 1,12 x 10-12 e do cloreto de prata 1,77 x 10-10, apesar do Ag2CrO4 apresentar Kps menor, o AgCl é o precipitado formado, o que ocorre devido à solubilidade molar do cromato de prata ser maior que a do cloreto de prata. Considerando esse fator, o presente método nao é aplicado a análises de maostra de água salobra ou salgada, isso é, contendo íons cloreto em concentraçoes maiores que 5 ‰.
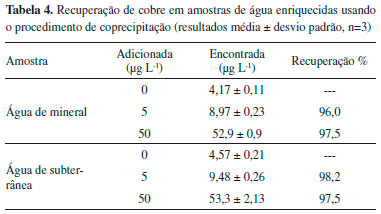
Exatidao e aplicaçao A exatidao de um procedimento é determinada pela proximidade do valor medido com o valor real da amostra. A acurácia pode ser medida de três formas, a primeira delas é a análise da amostra com concentraçao conhecida e comparaçao do valor medido com o verdadeiro, vários materiais de referência sao utilizados com essa finalidade. A segunda maneira é comparar os resultados do procedimento desenvolvido com os resultados de um método alternativo existente que é conhecido por ser preciso. A terceira abordagem é mais utilizada em estudos de recuperaçao, e é realizada por adiçao de um analito em amostras.45 Para avaliar a exatidao do procedimento, diferentes quantidades de analitos foram adicionadas em amostras de água mineral, água subterrânea. O procedimento de coprecipitaçao foi aplicado às amostras de águas e os resultados estao apresentados na Tabela 4. Obteve-se uma ótima concordância entre as quantidades de analitos adicionadas e encontradas. Os valores de recuperaçao para os analitos variaram entre 94 e 98%. Estes valores foram quantitativos e confirmam a exatidao do procedimento proposto, mostrando que o método de coprecipitaçao apresentado pode ser aplicado à separaçao e pré-concentraçao de cobre em amostras de água subterrânea e mineral.
Após verificar a exatidao do procedimento proposto, a coprecipitaçao de cobre foi aplicada para pré-concentraçao em outras 6 amostras de água subterrânea amostradas em diferentes localidades da regiao litorânea norte da cidade de Ilhéus, Bahia, Brasil. Além disso, foi realizado uma comparaçao entre os valores obtidos por análise em FAAS e por análise direta destas mesmas amostras por ICP-MS (Tabela 5).
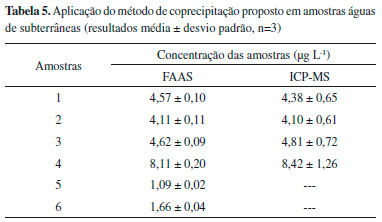
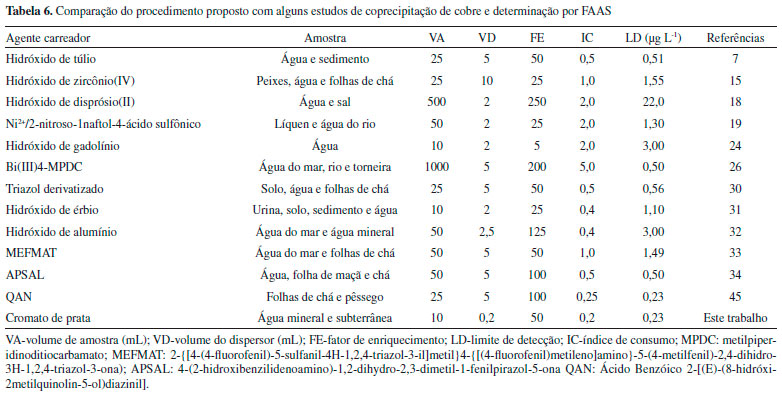
A avaliaçao estatística aplicando o teste-t pareado ao nível de confiança de 95% mostrou nao haver diferença estatística entre os valores obtidos pelo procedimento de coprecipitaçao e aqueles obtidos por análise direta no ICP-MS, considerando que o t calculado é de 0,09, enquanto que o t-crítico foi de 2,78. O Conselho Nacional de Meio Ambiente (CONAMA) preconiza que em águas de classe 1, tidas como própria para o consumo humano, os valores máximos permitidos para cobre nao devem ser superiores a 9 µg L-1. Nas amostras analisadas todas as concentraçoes de cobre obtidas estao abaixo do valor máximo permitido pelo CONAMA.46 O procedimento de coprecipitaçao desenvolvido apresentou LQ bem mais baixo que o limíte máximo permitido pela legislaçao brasileira, consumo de pequeno volume de amostra, e mostrou uma eficiência de extraçao superior a outros procedimentos descritos na literatura, evidenciado principalmente pelo parâmetro índice de consumo, expresso pela razao entre volume amostra e o fator de enriquecimento, conforme apresentado na Tabela 6.
CONCLUSAO Nesse trabalho foi proposto um procedimento de separaçao e pré-concentraçao baseado na coprecipitaçao de cobre usando como agente carreador o cromato de prata, e esse procedimento foi aplicado nas amostras de água subterrânea e água mineral. Com a matriz Doehlert pôde-se determinar as melhores condiçoes para os fatores estudados de forma mais rápida e com menos experimentos quando comparado com o método de otimizaçao univariada. O procedimento proposto apresentou RSD% sempre inferior a 5% e ótima exatidao, evidenciada pela análise adiçao e recuperaçao nas amostras de água, sendo que as recuperaçoes obtidas foram quantitativas na faixa de 94 a 98%, com baixos limites de detecçao e quantificaçao. Dentre as espécies químicas avalidadas no efeito de matriz, somente o íon cloreto pode interferir na coprecipitaçao do cobre, mostrando que o procedimento nao é aplicável a amostras consideradas salobras (salinidade compreendida entre 0,50‰ e 30‰). As principais vantagens do procedimento desenvolvido foram a sua simplicidade, baixo custo e bons parâmetros analíticos. Estes resultados indicam a viabilidade de aplicaçao deste procedimento de coprecipitaçao na separaçao/pré-concentraçao de cobre e determinaçao por FAAS.
REFERENCIAS 1. Brewer, G. J. Em Molecular, Genetic, and Nutritional Aspects of Major and Trace Minerals; Collins, J. F., ed.; Elsevier: Amsterda, 2017, cap. 7-10. 2. Subramanian, K. S.; Spectrochim. Acta, Part B 1996, 51, 291. 3. Mohammadi, S. Z.; Shamspur, T.; Baghelani, Y. M.; Arab. J. Chem. (2014), DOI: 10.1016/j.arabjc.2014.11.054. DOI: http://dx.doi.org/10.1016/j.arabjc.2014.11.054. 4. Marczenko, Z.; Balcerzak, M.; Separation, Preconcentration and Spectrophotometry in Inorganic Analysis, Elsevier B.V.: Amsterda, 2000. 5. Bishop, M. L.; Fody, E. P.; Schoeff, L. E. Química Clínica, 5ª. ed, Manole: Barueri, 2010. 6. Korn, M. D. G. A.; de Andrade, J. B.; de Jesus, D. S.; Lemos, V. A.; Bandeira, M. L. S. F.; dos Santos, W. N. L.; Bezerra, M. A.; Amorim, F. A. C.; Souza, A. S.; Ferreira, S. L. C.; Talanta 2006, 69, 16. 7. Soylak, M.; Aydin, A.; Food Chem. Toxicol. 2011, 49, 1242. 8. Evans, E. H.; Pisonero, J.; Smith, C. M. M.; Taylor, R. N. T.; J. Anal. At. Spectrom. 2013, 29, 779. 9. Hywel Evans, E.; Pisonero, J.; Smith, C. M. M.; Taylor, R. N.; J. Anal. At. Spectrom. 2015, 30, 1017. 10. Camel, V.; Spectrochim. Acta, Part B 2003, 58, 1177. 11. Bezerra, M. D. A.; Zezzi Arruda, M. A.; Costa Ferreira, S. L.; Appl. Spectrosc. Rev. 2007, 40, 269. 12. Jain, V. K.; Pillai, S. G.; Mandalia, H. C.; TrAC, Trends Anal. Chem. 2016, 85, 46. 13. Alexandrova, L.; Grigorov, L.; Int. J. Miner. Process. 1996, 48, 111. 14. Ilaiyaraja, P.; Singha Deb, A. K.; Sivasubramanian, K.; Ponraju, D.; Venkatraman, B.; J. Hazard. Mater. 2013, 250-251, 155. 15. Citak, D.; Tuzen, M.; Soylak, M.; Food Chem. Toxicol. 2009, 47, 2302. 16. Soylak, M.; Narin, I.; Divrikli, U.; Saracoglu, S.; Elci, L.; Dogan, M.; Anal. Lett. 2004, 37, 767. 17. Alothman, Z. A.; Unsal, Y. E.; Habila, M.; Tuzen, M.; Soylak, M.; Desalin. Water Treat. 2015, 53, 3457. 18. Peker, D. S. K.; Turkoglu, O.; Soylak, M.; J. Hazard. Mater. 2007, 143, 555. 19. Uluozlu, O. D.; Tuzen, M.; Mendil, D.; Soylak, M.; J. Hazard. Mater. 2010, 176, 1032. 20. Mendil, D.; Karatas, M.; Tuzen, M.; Food Chem. 2015, 177, 320. 21. Elçi, L.; Şahin, U.; Öztaş, S.; Talanta 1997, 44, 1017. 22. Soylak, M.; Saracoglu, S.; Divrikli, U.; Elci, L.; Talanta 2005, 66, 1098. 23. Divrikli, Ü.; Elçi, L.; Anal. Chim. Acta 2002, 452, 231. 24. Soylak, M.; Balgunes, H.; J. Hazard. Mater. 2008, 155, 595. 25. Feist, B.; Mikula, B.; Food Chem. 2014, 147, 225. 26. Efendioğlu, A.; Yağan, M.; Batı, B.; J. Hazard. Mater. 2007, 149, 160. 27. Alfasi, Z. B.; Wai, C. M.; Preconcentration Techniques for Trace Elements, CRC Press: Boca Raton, 1992. 28. Zhu, C.; Geochim. Cosmochim. Acta 2004, 68, 3327. 29. Barnes, J. D.; Thomas, M. J. K.; Denney, R. C.; Mendham, J.; Vogel - Análise Química Quantitativa, 5a. ed., LTC: Rio de Janeiro, 2002. 30. Bahadır, Z.; Bulut, V. N.; Ozdes, D.; Duran, C.; Bektas, H.; Soylak, M.; J. Ind. Eng. Chem. 2014, 20, 1030. 31. Saracoglu, S.; Soylak, M.; Elci, L.; Talanta 2003, 59, 287. 32. Doner, G.; Ege, A.; Anal. Chim. Acta 2005, 547, 14. 33. Duran, C.; Ozdes, D.; Sahin, D.; Bulut, V. N.; Gundogdu, A.; Soylak, M.; Microchem. J. 2011, 98, 317. 34. Gouda, A. A.; Talanta 2016, 146, 435. 35. Soylak, M.; Onal, G.; J. Hazard. Mater. 2006, 137, 1130. 36. Yıldız, E.; Saçmacı, Ş.; Kartal, Ş.; Saçmacı, M.; Food Chem. 2016, 194, 143. 37. Ferreira, S. L. C.; Dos Santos, W. N. L.; Quintella, C. M.; Neto, B. B.; Bosque-Sendra, J. M.; Talanta 2004, 63, 1061. 38. Bezerra, M. A.; Santelli, R. E.; Oliveira, E. P.; Villar, L. S.; Escaleira, L.; Talanta 2008, 75, 965. 39. Lundstedt, T.; Chemom. Intell. Lab. Syst. 1998, 42, 3. 40. Pimentel, M. F.; Barros Neto, B.; Quim. Nova 1996, 19, 268. 41. Novaes, C. G.; Yamaki, R. T.; de Paula, V. F.; do Nascimento Júnior, B. B.; Barreto, J. A.; Valasques, G. S.; Bezerra, M. A.; Rev. Virtual Quim. 2017, 9, 1184. 42. Passari, L. M. G. Z.; Soares, P. K.; Bruns, R. E.; Quim. Nova 2011, 34, 888. 43. Vogel, A. I.; Quimica Analitica Qualitativa, 1a. ed, Mestre Jou: Sao Paulo,1981. 44. Thompson, M.; Ellison, S. L. R.; Wood, R.; Pure Appl. Chem. 2002, 73, 835. 45. Green, J. M.; Anal. Chem 1996, 68, 305. 46. Rosini, F.; Matos, W. O.; Santos, M. C.; Nóbrega, J.; Analytica 2006, 22, 74. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access