
 |
 |
 |
 |
 |
Artigo
| Isolation of antifungal quinoid derivatives from leaves of Pentacalia desiderabilis (Vell.) Cuatre. (Asteraceae) using ionic liquid in the microwave assisted extraction |
|
Kaio de S. GomesI; Cinthia I. TamayoseII; Marcelo José P. FerreiraII; Cynthia MurakamiIII; Maria Claudia M. YoungIII; Guilherme M. AntarII; Fernanda F. CamiloIV; Patricia SartorelliIV; Joao Henrique G. LagoI,*
I. Centro de Ciências Naturais e Humanas, Universidade Federal do ABC, 09210-580 Santo André - SP, Brasil Recebido em: 06/08/2018 *e-mail: joao.lago@ufabc.edu.br In the present work, leaves of Pentacalia desiderabilis (Asteraceae) were subjected to extraction using aqueous 1-butyl-3-methylimidazolium bromide (BMImBr) in the microwave assisted extraction (MAE). The obtained extract was partitioned using CH2Cl2 and antifungal activity against Cladosporium cladosporioides and C. sphaerospermum of the organic phase was evaluated. Sequentially, this material was subjected to several chromatographic procedures to afford four quinoid derivatives identified as jacaranone (1), methyl-jacaranone (2), quinolacetic acid (3), and methyl 1-hydroxy-4-oxo-cyclohexaneacetate (4), being the first occurrence of compounds 2 - 4 in P. desiderabilis. Isolated quinoid derivatives were submitted to bioautography assay and the obtained results indicated that compounds 1 - 3 are active against Cladosporium cladosporioides (0.78, 12.5 and 12.5 µg, respectively) and C. sphaerospermum (3.13, 12.5 and 12.5 µg, respectively). INTRODUCTION Despite the high diversity found in the family Asteraceae and its genera, only two species of the genus Pentacalia occur in Brazil: P. tropicalis, found in Espírito Santo and Rio de Janeiro states, and P. desiderabilis, located in the South and Southeast regions, especially in Sao Paulo, Paraná and Santa Catarina states.1 Known as "catiao-trepador", P. desiderabilis is a shrub with yellow-crowned flowers.1 Phytochemical studies with this species have reported the sesquiterpene germacrene D2 and the quinoid jacaranone, which displays antiparasitic,3 antifungal4 and antitumor5 activities in vitro. However, no other studies were reported in the literature concerning phytochemistry or pharmacological aspects of P. desiderabilis. Ionic liquids have been used as substitutes of organic solvents in several extraction procedures due to their reduced vapor pressure, high solubilization capacity, chemical stabilities, and possibility of reuse.6 Due to these characteristics, ionic liquids have been recognized as "green solvents".7 Furthermore, since ionic liquids are excellent microwave absorbers, the use of this method during the extraction process reduced the extraction time and increases the process efficiency. Recently, this method has been successfully used in our group in order to obtain different metabolites such as terpenoids from Schinus terebinthifolius8 and lignoids from Saururus cernuus.9 Based on these innovative aspects, this work has two main objectives - conduct the extraction of metabolites from leaves of P. desiderabilis using aqueous 1-butyl-3-methylimidazolium bromide (BMImBr) under microwave and evaluate the antifungal activity of isolated compounds. After chromatographic separation procedures, quinoid derivatives were identified by NMR and MS techniques. Their antifungal potential was tested against Cladosporium cladosporioides and C. sphaerospermum.
RESULTS AND DISCUSSION After extraction of dried leaves of P. desiderabilis using aqueous 1-butyl-3-methylimidazolium bromide (BMImBr) under MAE during 10 min at 60 ºC, the obtained material was filtered and extracted using CH2Cl2. This organic phase displayed activity against Cladosporium sp. and was subjected to chromatographic fractionation to afford four related quinoid derivatives: jacaranone (1), methyl-jacaranone (2), quinolacetic acid (3) and methyl 1-hydroxy-4-oxo-cyclohexaneacetate (4), as showed in Figure 1.
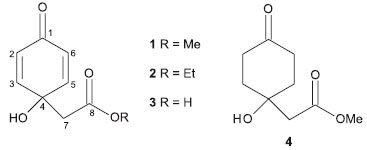 Figure 1. Structures of compounds 1 - 4 isolated from P. desiderabilis
The 1H NMR spectra of 1 - 4 showed signals at δ 6.96/6.96/6.94 (d, J = 10.0 Hz, H-3/H-5) and at δ 6.28/6.20/5.97 (d, J = 10.0 Hz, H-2/H-6) characteristic of a quinoid system.3,5,10 Besides those signals, a singlet at δ 2.71/2.69/2.54 (2H) was attributed to hydrogen H-7. Additionally, the 1H NMR spectrum of compound 1 displayed a singlet at δ 3.77 (3H), consistent with a methoxy group at C-8. This analysis allowed the identification of compound 1 as jacaranone, after comparison of obtained data with those reported in the literature.11 For compound 2, a triplet was observed at δ 1.29 (J = 7.0 Hz, 3H) and a quartet at δ 4.22 (J = 7.0 Hz, 2H), suggesting the presence of an ethoxyl group at C-8. By comparison with literature data, compound2 was identified as methyl-jacaranone.10 On the other hand, in the 1H NMR spectrum of compound 3, no signals attributed to methoxy or ethoxy groups were observed, suggesting that this component corresponds to quinolacetic acid, the carboxylic acid precursor of compounds 1 and 2. The 13C NMR spectra of compounds 1-3 showed carbons C-2/C-6, C-3/C-5 and C-1 at δ 128.4/128.5/125.4, 148.7/149.1/154.2 and 184.4/185.2/185.1 respectively, confirming the structural similarity of these compounds. Furthermore, the 13C NMR spectra of 2 and 3 showed the signals of C-8, C-7 and C-4 at δ 171.1/174.5, 43.7/43.8 and 67.2/67.5, respectively, similar of those reported in the literature to methyl-jacaranone4 and quinolacetic acid.11) 1H NMR spectrum of compound 4 showed, among other signals, singlets at δ 2.57 (2H, H-7) and at δ 3.75 (3H, OCH3), consistent with a structure related to compound 1. The absence of signals for olefinic hydrogens indicated the presence of a cyclohexanone moiety, as well as signals at δ 2.80 (ddd, J = 14.1, 14.0 and 6.3 Hz) and at δ 1.76 (ddd, J = 14.1, 14.0 and 4.8 Hz) assigned to axial hydrogens H-2/H-6 and H-3/H-5, respectively. Furthermore, signals at δ 2.24 (ddd, J = 14.0, 4.8 and 2.5 Hz) and at δ 2.11 (ddd, J = 14.0, 6.3 and 2.5 Hz) were attributed to equatorial hydrogens H-2/H-6 and H-3/H-5, respectively. DEPT 135° NMR spectrum confirms the proposed structure due to signals at δ 211.4 (C, C-1), 36.9 (CH2, C-2/C-6), 36.6 (CH2, C-3/C-5), 68.4 (C, C-4), 44.5 (CH2, C-7), 173.2 (C, C-8) and d 51.9 (OCH3). Finally, LR-MS analyses of compounds 1 - 4 were consistent with the molecular formulae C9H10O4, C10H12O4, C8H8O4, and C8H12O4, respectively. The comparison of obtained data with those reported in the literature12-14 allowed the identification of compound 4 as methyl 1-hydroxy-4-oxo-cyclohexaneacetate. Although already identified in other plant species,2,4,15 this is the first occurrence of compounds 2 - 4 in P. desiderabilis. The bioautography assay (Table 1) showed that compound1 was the most active in inhibiting the growth of Cladosporium cladosporioides and C. sphaerospermum. As reported in the literature,4 compounds 1 and 2 possess antifungal activity against Fusarium oxysporum and Botrytis cinerea. However, the isolation of compounds 1 - 4 allowed the recognition of some structural features associated to antifungal activity. As observed in compound 1, the presence of a methyl ester moiety enhances the activity against Cladosporium cladosporioides and C. sphaerospermum when compared to the corresponding ethyl ester and free carboxylic acid (compounds 2 and 3), which display lower bioactivity. Moreover, replacing the unsaturated quinoid ring by a saturated cyclohexanone ring renders compound 4 completely inactive. These results corroborate the importance of the quinoid ring (α,β-unsaturated system) and methyl esterification at C-7 to the antifungal activity of this class of secondary metabolites.
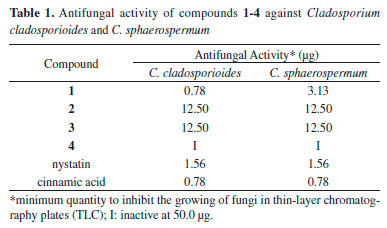
CONCLUSIONS In the present work, the leaves of P. desiderabilis were subjected to a microwave-assisted extraction (MAE) procedure using aqueous solution of ionic liquid BMImBr (1-butyl-3-methylimidazolium bromide). After partition with CH2Cl2, the bioactive organic phase was subjected to chromatographic fractionation to afford three antifungal (1-3) and one inactive (4) quinoid derivatives. Isolated compounds were identified by NMR and MS techniques and comparison with spectral data reported in the literature. Compound 1 displayed higher antifungal potential against both tested fungi while its hydrogenated derivative (compound 4) was inactive. These results indicate that the presence of a conjugated system in the six member ring is crucial to antifungal activity of these related compounds. Furthermore, the use of 1-butyl-3-methylimidazolium bromide (BMImBr) in MAE system consists in an efficient and selective method of extraction of antifungal quinoid derivatives from P. desiderabilis. Therefore, the obtained results contribute to future uses of several other ionic liquids using MAE system in plant extraction, aiming at the selective extraction of certain metabolites, especially those which display bioactivity.
EXPERIMENTAL SECTION General experimental procedures Silica gel (Merck, 230-400 mesh) was used for column chromatographic separation, while silica gel 60 PF254 (Merck) was used for analytical (0.25 mm) TLC. 1H NMR spectra were recorded at 300 MHz and 13C NMR at 75 MHz on a Bruker Avance 300 spectrometer using CDCl3 and DMSO-d6 as solvents. LRESIMS (negative mode) and LREIMS (70 eV) spectra were recorded, respectively, using a Platform II-Micromass (quadrupole) and INCOS 50 Finnigan-Mat (quadrupole) mass spectrometers. Microwave assisted extraction (MAE) experiments were performed with a MAS-I microwave oven (2450 MHz, Sineo Microwave Chemistry Technology Company, Shanghai, China) with a maximum delivered power of 1000 W. The temperature was monitored by an infrared probe inside the microwave oven. Plant material Leaves of P. desiderabilis were collected in Campos do Jordao, Sao Paulo State in May, 2015. The botanical identification was made by MSc. Guilherme M. Antar (University of Sao Paulo/SP) and the voucher specimen was deposited in the Herbarium of Biosciences Institute - USP/SP under number SPF220668. Extraction and isolation 1-butyl-3-methylimidazolium bromide (BMImBr) was prepared as previously described in the literature.8,9 Dried and powdered leaves (10 g) of P. desiderabilis were extracted by microwave-assisted extraction (MAE) with 20 mL of mixture containing H2O:BMImBr 1:1 (v/v) during 10 min at 60 ºC. After this procedure, the solution was filtered, extracted using CH2Cl2 (3 X 20 mL) and dried over Na2SO4. After distillation of the solvent under reduced pressure, were obtained 720 mg of CH2Cl2 phase. Part of this material (500 mg) was chromatographed over SiO2 using n-hexane with increasing amounts of EtOAc as eluent. This procedure afforded eight groups (A - H). Groups A - F showed to be composed by waxy material. Group G (150 mg), eluted with n-hexane:EtOAc 8:2, was chromatographed over Sephadex LH-20 (4 x 50 cm) eluted with MeOH resulting in seven groups (G1 - G7). Group G5 (92 mg) was purified over SiO2 column chromatography eluted with n-hexane:EtOAc 8:2 to give 13 mg of 1 and 14 mg of 2. Group H (150 mg), eluted with n-hexane:EtOAc 7:3 was chromatographed over Sephadex LH-20 (4 x 50 cm) eluted with MeOH resulting in seven groups (H1 - H7). Groups H5 (40 mg) and H3 (6 mg) were constituted by 3 and 4, respectively. All isolated compounds displayed purity higher than 97% (HPLC analysis). Jacaranone (1). White amorphous solid. 1H NMR (δ, 300 MHz, CDCl3): 6.97 (d, J = 10.0 Hz, H-3/H-5), 6.28 (d, J = 10.0 Hz, H-2/H-6), 2.71 (s, H-7), 3.77 (s, OCH3). 13C NMR (δ, 75 MHz, CDCl3): 184.4 (C-1), 148.7 (C-3/C-5), 128.4 (C-2/C-6), 67.4 (C-4), 43.2 (C-7), 171.7 (C-8), 53.2 (OCH3). LREIMS (70 eV) m/z (rel. int.): 182 (5) [M]+, 166 (10), 150 (30), 122 (30), 109 (100), 106 (40), 94 (10), 81 (40), 74 (90), 53 (30), 43 (40). Methyl-jacaranone (2). White amorphous solid. 1H NMR (δ, 300 MHz, CDCl3): 6.96 (d, J = 10.0 Hz, H-3/H-5), 6.19 (d, J = 10.0 Hz, H-2/H-6), 4.22 (q, J = 7.0 Hz, OCH2CH3), 2.69 (s, H-7),1.29 (t, J = 7.0 Hz, OCH2CH3). 13C NMR (δ, 75 MHz, CDCl3): 185.2 (C-1), 171.1 (C-8), 149.1 (C-3/C-5), 128.5 (C-2/C-6), 67.2 (C-4), 61.7 (OCH2CH3), 43.7 (C-7), 14.3 (OCH2CH3). LRESIMS (positive mode) m/z 197 [M+H]+ Quinolacetic acid (3). White amorphous solid. 1H NMR (δ, 300 MHz, DMSO-d6): 6.94 (d, J = 10.0 Hz, H-3/H-5), 5.97 (d, J = 10.0 Hz, H-2/H-6), 2.54 (s, H-7). 13C NMR (δ, 75 MHz, DMSO-d6): 185.1 (C-1), 154.2 (C-3/C-5), 125.4 (C-2/C-6), 67.5 (C-4), 43.8 (C-7), 174.5 (C-8). LRESIMS (negative mode) m/z 167 [M-H]- Methyl 1-hydroxy-4-oxo-ciclohexaneacetate (4). White amorphous solid. 1H NMR (δ, 300 MHz, CDCl3): 2.80 (ddd, J = 14.1, 14.0 and 6.3 Hz, H-2ax/H-6ax), 2.57 (s, H-7), 3.75 (s, OCH3), 2.24 (ddd, J = 14.0, 4.8 and 2.5 Hz, H-2eq/H-6eq), 2.11 (ddd, J = 14.0, 6.3 and 2.5 Hz, H-3eq/H-5eq), 1.76 (ddd, J = 14.1, 14.0 and 4.8 Hz, H-3ax/H-5ax). 13C NMR (δ, 75 MHz, CDCl3): 211.4 (C-1), 173.2 (C-8), 68.4 (C-4), 51.9 (OCH3), 44.5 (C-7), 36.9 (C-2/C-6), 36.6 (C-3/C-5). LREIMS (70 eV) m/z (rel. int.): 186 (5) [M]+, 168 (80), 155 (10), 154 (20), 140 (40), 137 (20), 129 (80), 116 (20), 112 (70), 99 (20), 98 (90), 84 (50), 81 (50), 74 (90), 65 (10), 55 (80), 45 (10), 43 (100) Antifungal assay Bioautography assays were conducted using the microorganisms Cladosporium cladosporioides Fresen (SPC 140) and C. sphaerospermum Perzig (SPC 491), which have been maintained at the Instituto de Botânica, Sao Paulo, Brazil. For the antifungal assay, 10.0 µL of solutions corresponding to 100.0 µg of crude extracts or semi-purified fractions were applied to precoated SiO2 TLC plates, developed with hexane-EtOAc (7:3), and dried for complete removal of solvents. For pure compounds 1 - 4, 10.0 µL of solutions corresponding to 50.0, 25.0, 12.5, 6.25, 3.12, 1.56 and 0.78 µg were applied to precoated SiO2 TLC plates. The chromatograms were sprayed with a spore suspension of C. cladosporioides or C. sphaerospermum in glucose and salt solution (5x107 spore/mL) and incubated for 48 h in darkness in a moistened chamber at 27 °C, following the previously reported procedure.16 Fungal growth inhibition appeared as clear zones against a dark background, indicating the minimum amount of pure compounds 1 - 4 required for it. Nystatin and cinnamic acid were used as positive controls.
ACKNOWLEDGMENTS To FAPESP, CNPq and CAPES for the financial suppor.
REFERENCES 1. Teles, A. M.; Stehmann, J. R.; Check List: Journal of Species Lists and Distribution 2008, 4, 62. 2. Deuschle, R. A.; Camargo, T.; Alves, S. H.; Mallmann, C. A.; Heizmann, B. M.; Rev. Bras. Farmacogn. 2007, 17, 220. 3. Morais, T. R.; Romoff, P.; Fávero, O. A.; Reimao, J. Q.; Lourenço, W. C.; Tempone, A. G., Hristov, A. D.; Di Santi, S. M.; Lago, J. H. G.; Sartorelli, P.; Ferreira, M. J. P.; Parasitol. Res. 2012, 110, 95. 4. Pedrozo, J. A.; Torrenegra, R. D.; Téllez, N. A.; Granados, A.; Rev. Bras. Farmacogn. 2006, 16, 591. 5. Massaoka, M. H.; Matsuo, A. L.; Figueiredo, C. R.; Farias, C. F.; Girola, N.; Arruda, D. C.; Scutti, J. A. B.; Romoff, P.; Fávero, O. A.; Ferreira, M. J. P.; Lago, J. H. G.; Travassos, L. R.; PLoS One 2012, 7, e38698. 6. Daí, Y.; Spronsen, J. V.; Witkamp, G. J.; Veepoorte, R.; Choi, Y. H.; J. Nat. Prod. 2013, 76, 2162. 7. Rogers, R. D.; Seddon, K. R.; Ionic Liquids: industrial applications for green chemistry, 1st ed., American Chemical Society: New York, 2002. 8. Morais, T. R.; Coutinho, A. P. R.; Camilo, F. F.; Martins, T. S.; Sartorelli, P.; Massaoka, M. H.; Figueiredo, C. R.; Lago, J. H. G.; J. Braz. Chem. Soc. 2017, 28, 492. 9. Brito, J. R.; Camilo, F. F.; Figueiredo, C. R.; Azevedo, R. A.; Romoff, P.; Buturi, F. O. S; Fávero, O. A.; Lago, J. H. G.; Ferreira, E. A.; Quím. Nova 2018, 41, 778. 10. Lajide, L.; Escoubas, P.; Mizutani, J.; Experientia 1996, 52, 259. 11. Brownlee, J. M.; Heinz, B.; Bates, J.; Moran, G. R.; Biochemistry 2010, 49, 7218. 12. Mericli, A. H.; Mericlim, F.; Jakupovic, J.; Bohlmann, F.; Dominguez, X. A.; Vega, H. S.; Phytochemistry 1989, 28, 1149. 13. Tian, Y. Q.; Niu, Y. F.; Shen, T.; Weng, C. W.; Xie, W. D.; Row, K. H.; J. Chem. Res. 2010, 9, 25. 14. Ma, H.; Yang, L.; Zhang, M.; Wang, C. H.; Wang, Z. T.; Acta Pharm. Sin. 2008, 43, 626. 15. Wu, C.; Zhang, L.; Zhou, P.; Sun, M.; Gao, K.; Phytochem. Lett. 2015, 14, 245. 16. Homans, A. L.; Fuchs, A.; J. Chromatogr. 1970, 51, 327. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access