
 |
 |
 |
 |
 |
Artigo
|
|
| Elucidação da quiralidade induzida na molécula dansilglicina na complexação com a proteína albumina do soro humano (HSA) Elucidation of the induced chirality of dansylglycine by its interaction with human serum albumin |
|
Aguinaldo Robinson de SouzaI; Izabelle Amorim Ferreira BozaI,*; Valdecir Farias XimenesI; Maurício Ikeda YoguinI; María-José Dávila-RodriguezIII,IV; Nelson Henrique MorgonII; Ignez CaracelliIII
I. Departamento de Química, Faculdade de Ciências, Universidade Estadual Paulista, 17033-360 Bauru - SP, Brasil Recebido em: 29/06/2018 *e-mail: izabelle.tif@gmail.com Human serum albumin (HSA) plays an important role in the transport of a wide variety of substances, including compounds with pharmacological properties. The dansylglycine (DanG) is a fluorescent amino acid derivative specific for the site II of HSA. This work aimed to elucidate the induction of chirality in the DanG due to its bonding to the HSA. Theoretical electronic circular dichroism spectra (ECDs) were simulated using the Density Functional Theory (DFT) and the implicit Solvation Model based on Density (SMD). The DanG-HSA complexation resulted in the appearance of a positive ECD spectrum centered at 346 nm. Focusing on the dihedral angles between the -N(CH3)2 group bounded to the naphthalene ring, the potential energy surface (PES) of the DanG was obtained. Analysis of the various conformations obtained revealed that the calculated dihedral angle (150º) is in agreed with the experimental ECD spectrum. In addition, we observed that the nitrogen atom of the -N(CH3)2 group presented the greatest contribution to the HOMO-LUMO transition that gives rise to the n→π* electronic transition involved in the generation of the ECD signal. Molecular docking analysis of the complexation between DanG and HSA revealed a conformation with a dihedral angle similar to that obtained by DFT. INTRODUÇAO A Albumina do Soro Humano (HSA) confere um importante papel no transporte de substâncias com propriedades farmacológicas devido à sua alta concentraçao no plasma e especificidade, colocando-a como a proteína fundamental responsável na implicaçao farmacocinética dos medicamentos.1 A proteína HSA é uma das mais abundantes no plasma humano, encontrada predominantemente nos fluídos corporais; possui importantes funçoes fisiológicas devido a sua alta capacidade como portadora e como reservatório para moléculas endógenas e exógenas. Apresenta-se como um monômero de 67 kDa; possui três domínios globulares, divididos em dois subdomínios: IA, IIA, IB, IIB, IIIA e IIIB. Os principais sítios de ligaçao para fármacos, referidos como sítio I e sítio II, estao localizados nas cavidades hidrofóbicas dos subdomínios IIA e IIIA, respectivamente,2-8 denominados de Sudlow I e II. A relevância em entendermos as bases estruturais da especificidade da interaçao entre a proteína HSA e moléculas consideradas como fármacos é devido ao fato de buscarmos e aperfeiçoarmos um melhor entendimento da interaçao, tanto ao nível atômico como molecular, que poderá definir entao as possíveis abordagens terapêuticas para o tratamento de enfermidades e no alívio de sintomas advindos de doenças ou síndromes.9-12 A Simulaçao Computacional utilizada neste trabalho tem como propósito a busca de um maior entendimento das interaçoes entre a proteína HSA e o aminoácido Dansilglicina (DanG), buscando subsídios teóricos para uma possível comparaçao com os resultados experimentais. Outra característica importante da simulaçao computacional, adotada neste trabalho, é o seu caráter preditivo das propriedades químicas e físicas de sistemas de interesse como, por exemplo, na prediçao da conformaçao adotada pela DanG no sítio de interaçao da HSA.13,14 A caracterizaçao de como um fármaco pode se ligar na proteína HSA torna-se de suma importância na busca do entendimento de sua distribuiçao pelo corpo humano, na sua taxa de metabolismo e a sua excreçao. Sendo assim, a caracterizaçao, tanto do ponto de vista estrutural como de variaçoes de energia de ligaçao, conduz a parâmetros que auxiliam na determinaçao da afinidade entre as espécies, da constante de ligaçao e do número de sítios de interaçao. Com este intuito, utilizamos o aminoácido DanG no estudo e caracterizaçao dos sítios de ligaçoes à proteína HSA, buscando uma explicaçao para a origem da sua quiralidade quando da formaçao do complexo entre ambos. Aminoácidos dansilados apresentam um alto rendimento quântico de fluorescência; esse rendimento aumenta consideravelmente quando o aminoácido interage com os sítios da HSA. Observamos um deslocamento nos picos de emissao para comprimentos de onda menores do espectro eletromagnético devido à ocorrência de um fenômeno típico de transferência, do aminoácido, do meio hidrofílico para o meio hidrofóbico no interior da proteína. Com a diminuiçao da fluorescência do complexo HSA-DanG, provocado pela adiçao de um fármaco específico, podemos obter um parâmetro analítico para a detecçao do seu sítio de ligaçao.15-20 Outra abordagem experimental que pode fornecer informaçoes fundamentais sobre a interaçao entre as biomoléculas e os fármacos sao os métodos espectrais de dicroísmo circular eletrônico, (ECD, Electronic Circular Dichroism). Além dos métodos experimentais de análise foram utilizados os métodos de simulaçao computacional baseados na Teoria do Funcional da Densidade (DFT) e no Docking Molecular na busca da interpretaçao dos dados obtidos experimentalmente e na elucidaçao da quiralidade induzida à molécula de DanG quando complexada à proteína HSA.21-27
PARTE EXPERIMENTAL Produtos químicos Dansilglicina (DanG - C14H16N2O4S), Albumina do Soro Humano, livre de ácidos graxos e de globulinas, 99% (HSA), Fosfato dibásico de sódio (Na2HPO4), Fosfato monobásico de sódio 99% (NaH2PO4), Acetonitrila 99,8% (C2H3N), Tetrahidrofurano 99,5% (THF, C4H8O), Metanol 99,9% (CH3OH), foram fornecidos pela Sigma Aldrich Chemical Co. (St. Louis, MO, EUA). Alcool etílico 99,66% (C2H6O) fornecido pela J. T Baker Chemical Company (Phillipsburg, New Jersey). A soluçao estoque de HSA 1 mmol L-1 foi preparada dissolvendo-se a massa de 0,066 g de HSA em 1,0 mL de tampao fosfato de sódio 0,05 mol L-1, pH 7,0. 60,0 µL dessa soluçao foram diluídas para 2,0 mL, gerando uma concentraçao final de 30,0 µmol L-1, a qual foi submetida às análises. Para o preparo de 1,0 mL da dansilglicina (DanG) 10 mmol L-1 (Massa Molar = 308,35 g mol-1), foi pesado 0,00323 g de DanG e dissolvido em 1 mL de álcool etílico. A partir desta fizeram-se as diluiçoes necessárias em água, metanol, etanol, acetonitrila e THF. A soluçao estoque foi mantida na geladeira e a soluçao de trabalho foi descartada após a mediçao. A concentraçao da soluçao estoque de HSA (1 mmol L-1), preparada em tampao fosfato 50 mmol L-1, pH 7,0, foi determinada por sua absorbância na regiao do UV-Vis (HSA, ε280 nm = 35.219 mol-1 L cm-1). Espectroscopia UV-Vis As medidas foram realizadas num espectrofotômetro UV-Vis Lambda 35 (Perkin Elmer, Shelton, CT, USA). O equipamento foi ajustado para determinar o espectro de absorbância no intervalo de 250-450 nm. O volume da soluçao, do aminoácido DanG, (100 µmol L-1) foi mantido fixo num valor de 10,0 µL adicionado em cada solvente num volume fixo de 990,0 µL, apresentando um volume final igual a 1.000,0 µL. Antes de cada análise as amostras e a placa transparente com as amostras foram incubadas a temperatura ambiente por 5 minutos. Elipticidade : dicroísmo circular eletrônico (ECD) Os estudos de ECD foram realizados em um espectropolarímetro Jasco J-815 (Jasco, Japan) equipado com controle termostatizado e agitaçao magnética no compartimento da amostra. Todas as medidas foram corrigidas para a absorçao do tampao fosfato 0,05 mol L-1, pH 7.0. Para os estudos na regiao do Ultra-Violeta próximo, foi utilizado um espectropolarímetro Jasco J-815 (Jasco, Japan). As medidas foram realizadas em uma cubeta de quartzo de 3 mL com 10 mm de caminho óptico, numa concentraçao de 30 µmol L-1 da proteína HSA e 30 µmol L-1 do aminoácido DG. Os estudos foram feitos em tampao fosfato 0,05 M, pH 7.0, à temperatura de 25 °C. Os espectros foram obtidos na faixa de comprimento de onda entre 250-500 nm, com uma largura de banda de 1 nm, com um tempo de resposta 1 s, e tempo de varredura de 50 nm/min. Metodologia computacional Teoria do Funcional da Densidade As conformaçoes do aminoácido DanG em fase gasosa e nos solventes água, etanol, metanol, acetonitrila e THF foram calculadas ao nível DFT utilizando os funcionais híbridos B3LYP e CAM-B3LYP com o conjunto de funçoes de base 6-311++G(2d,p), usando o programa Gaussian0928 e as facilidades computacionais do GridUNESP. No cálculo dos espectros teóricos de ECD e de absorçao UV-Vis foram utilizados os seguintes parâmetros no programa Gaussian 09: TD(FULL,SINGLET,NSTATES=15) SCRF(SMD, SOLVENT). As estruturas otimizadas nao apresentaram frequências imaginárias, nao sendo, portanto, estados intermediários; foi utilizado: CAM B3LYP/6 311++G(2d,p) Opt Freq. A visualizaçao e interpretaçao dos resultados foram feitas através do software GAUSSVIEWS 5.0.29 Docking molecular O docking foi realizado utilizando as seguintes estruturas cristalográficas obtidas do banco de dados "Protein Data Bank" (PDB): 2xvu30 e 2xw130 com resoluçao de 2,6 e 2,5 Å, respectivamente. Estas estruturas sao complexos cristalográficos: (a) 2xvu é uma HSA livre de ácidos graxos complexada com dansil-L-asparagina (DanN) em dois sítios, onde estao presentes os resíduos de aminoácidos W214 (sítio I) e Y411 (sítio II); (b) 2xvu é uma HSA livre de ácidos graxos complexada com dansil-L-norvalina (DanNV) em um único sítio, onde está presente o resíduo de aminoácido Y411 (sítio II). Os cálculos de docking molecular foram realizados utilizando o software computacional baseado em algoritmos genéticos GOLD (CCDC)31-33 v.5.6.2 e foi utilizada a funçao de ajuste GoldScore apresenta a seguir: 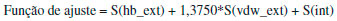 em que S(hb_ext) é a energia das ligaçoes de hidrogênio proteína-ligante, S(vdw_ext) é a energia das interaçoes de van der Waals proteína-ligante, e S(int) é uma penalidade para nao incentivar as interaçoes internas do ligante. Os cálculos de docking foram realizados de maneira similar nos dois casos:
Os ligantes para os cálculos de docking foram os seguintes:
Para os cálculos de docking somente os ligantes foram considerados flexíveis, ou seja, sem restriçoes aos ângulos de torçao. A seleçao dos compostos resultantes dos cálculos de docking foi feita em programas de visualizaçao gráfica (Accelrys DS Visualizer v3.5, Biovia Discovery Studio 2017 R2)34 considerando-se o padrao de poses (posiçao, conformaçao e orientaçao) do ligante e para aqueles dentro do padrao, com maior score. Após a seleçao da melhor pose, foram consideradas as interaçoes nao covalentes dos ligantes, dentro do sítio e analisadas quanto ao tipo e característica da interaçao, utilizando programas de visualizaçao gráfica.34,35 Também foram realizados cálculos para avaliar o gradiente reduzido da densidade eletrônica (RDG)36-39 a partir dos quais foi possível visualizar as isosuperfícies RDG das interaçoes nao covalentes (NCI).
RESULTADOS E DISCUSSAO Estudo do espectro de absorbância da DanG em diversos solventes Na Figura 3(a) apresentamos os espectros de absorçao na regiao do UV-Vis do aminoácido DanG nos diversos solventes considerados onde nao foi observada uma variaçao expressiva nos máximos de absorçao. Uma exceçao foi com o solvente água, na qual o espectro apresentou-se menos intenso e com uma menor definiçao entre as bandas de absorçao no valor de 324 nm. Os outros solventes apresentaram um máximo de absorçao (Tabela 1) identificada como uma transiçao n → π*, e no caso do solvente metanol esse valor foi igual a 338 nm, e de 338 nm para o etanol, de 343 nm para acetonitrila e de 340 nm para o solvente THF. Os diversos solventes foram utilizados pelo fato de terem caracteríticas próticas e apróticas, como constatado por seus valores de constante dielétrica (Tabela 1), onde buscamos identificar uma possível modificaçao no espectro na regiçao do UV-Vis e relacionar com uma possível interpretaçao para o fato de o aminoácido DanG ser transferido de um meio hidrofílico para um meio hidrofóbico quando da interaçao com a proteína HSA. No solvente água observamos um deslocamento hipsocrômico (para comprimentos de onda menores do espectro eletromagnético) junto com um efeito hipocrômico (uma diminuiçao da absorbância) na regiao de 250 nm, diferentemente dos demais solventes utilizados onde nao observamos modificaçoes tanto nas posiçoes como nas intensidades do espectro de absorçao. Na Figura 3(b) observamos uma pequena diminuiçao no valor da absorbância para os solventes: água, etanol e acetonitrila. No entanto, nao houve qualquer deslocamento no máximo de absorçao do aminoácido DanG quando comparado com os resultados experimentais. Este comportamento deverá ser mais bem analisado, pois no modelo teórico consideramos o solvente de maneira implícita, levando em consideraçao somente o valor da constante dielétrica dos mesmos.
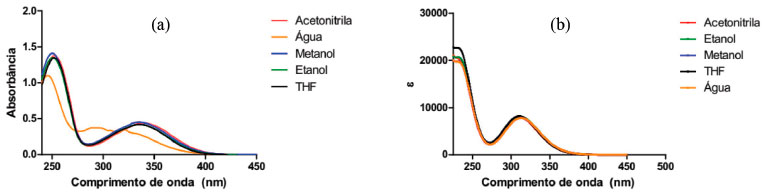 Figura 3. (a) Espectros de absorçao experimental da DanG, 100 µmol L-1 em diversos solventes: água; metanol; etanol; acetonitrila e THF (b) Espectros de absorçao teórico da DanG nos mesmos solventes
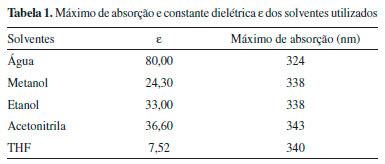
Para a determinaçao dos espectros de absorçao do aminoácido DanG na regiao do UV-Vis e ECD utilizamos o funcional CAM-B3LYP e a funçao de base 6-311++G(2d, p), nos mesmos solventes.40-42 Na Figura 3(b) apresentamos os espectros obtidos para o aminoácido DanG nos diversos solventes. Podemos observar uma boa concordância entre os valores obtidos na simulaçao computacional e aqueles obtidos experimentalmente, ressaltando o caso do solvente água que apresentou um deslocamento em relaçao, ao espectro obtido teóricamente, para a regiao do azul do espectro eletromagnético. Na Tabela 1 apresentamos o valor da constante dielétrica para os solventes estudados bem como o valor para o máximo de absorçao no espectro de UV-Vis. Interaçao do aminoácido DanG com a proteína HSA: dicroísmo circular eletrônico (ECD) De posse do conhecimento de que o aminoácido DanG liga-se ao sítio II da proteína HSA,26 utilizamos a técnica de ECD para descobrirmos em qual configuraçao, R ou S, a mesma se apresenta quando complexada com a HSA. Observamos que a molécula de DanG, quando diluída em tampao no pH 7,0, nao apresentou sinal de dicroísmo circular, o que já era previsto, pois nao se trata de uma molécula opticamente ativa, isto é, nao possui átomos de carbono quirais. Na Figura 4(a) o espectro de ECD foi medido na faixa de 250 nm a 500 nm e observamos que a complexaçao entre DanG e proteína HSA resultou no aparecimento de um sinal de ICD centrado em 346 nm. Esta induçao de quiralidade na molécula de DanG é devido à sua interaçao com a proteína, e pode ser explicada pela ligaçao ao ambiente assimétrico que caracteriza os sítios de ligaçao nas proteínas.
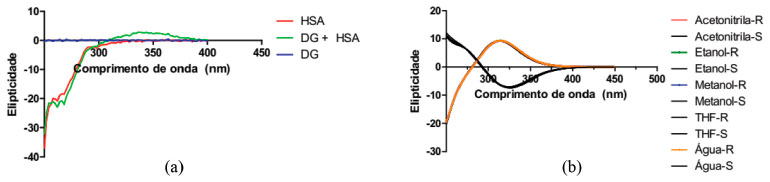 Figura 4. (a) Complexaçao da DanG-HSA (b) ECD da DanG em diferentes solventes e suas configuraçoes R e S
Foi utilizado o funcional híbrido B3LYP e o conjunto de base 6-311++G(2d, p) para o estudo teório do ECD,43,44 da DanG nas configuraçoes R e S. Na Figura 4(b) observamos que, na presença dos diferentes solventes, a molécula de DanG apresentou configuraçao R com sinal positivo, e na configuraçao S um sinal negativo no sinal de ECD. A comparaçao entre o espectro de ECD experimental com aqueles simulados em diferentes solventes nos leva a concluir que a conformaçao adquirida pelo aminoácido DanG ao se ligar à proteína deve apresentar uma configuraçao R. Estudo da superfície de energia potencial (SEP) e ECD da DanG Na Figura 5 apresentamos a estrutura da molécula de DanG com a numeraçao utilizada para a identificaçao dos átomos a serem considerados nos cálculos da SEP e de ECD referentes aos ângulos diedros.
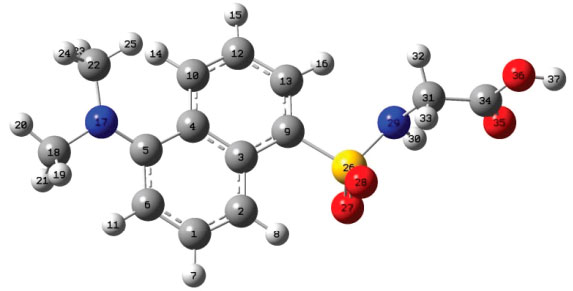 Figura 5. Estrutura da DanG com os átomos numerados
A SEP da DanG foi calculada a partir do funcional híbrido B3LYP e a funçao de base 6-31G(2d, p) no vácuo. Na Figura 6 apresentamos a SEP (com intervalos de 10° entre cada ponto da curva desde 0° até 360°), na qual podemos verificar a presença de mínimos e máximos de energia como uma funçao do ângulo diedro representado pelos átomos: C22-N17-C5-C4. A SEP foi calculada de maneira nao rígida, isso é, para cada valor do ângulo diedro a estrutura da molécula inteira foi otimizada.
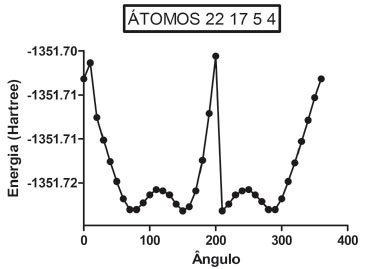 Figura 6. SEP do aminoácido DanG
Apresentamos na Figura 7 (a) o espectro da DanG no solvente água para os ângulos diedros de 0°, 10°, 120°, 130°, 140° e 150° onde podemos verificar que o sinal de ECD obtido foi positivo para todos os ângulos diedros considerados. Para o ângulo de 0° o máximo no espectro de ECD encontra-se em 350 nm, e à medida que aumentamos o ângulo diedro este máximo sofre um descolamento para comprimentos de onda menores: para o ângulo de 130° o valor máximo é igual a 310 nm e à medida que aumentamos o diedro o ângulo para 140° e 150° o valor máximo passa a ser igual 315 e 320 nm, respectivamente. Na Figura 7(b) apresentamos o espectro da DanG no solvente água para os ângulos diedros de 20°, 30°, 40°, 50°, 60°, 70° e 80°, a partir da qual podemos verificar que o sinal de ECD obtido foi negativo para todos os ângulos diedros considerados. A medida que aumentamos o valor do ângulo diedro no intervalo de 20° a 60°, o sinal de ECD nao sofre mudanças significativas situando-se próximo a 350 nm. A medida que aumentos o valor do ângulo diedro para 70° e 80°, foi observado um aumento na intensidade do sinal e um deslocamento hipsocrômico, com máximos de elipcitidade iguais a 326 e 317 nm, respectivamente.
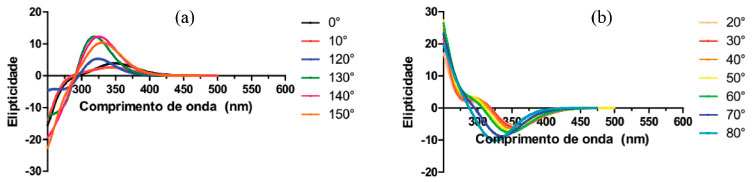 Figura 7. Espectros de dicroismo circular (a) positivo e (b) negativo da DanG no solvente água em funçao dos ângulos diedros
Nos cálculos da SEP da DanG, considerando os ângulos diedros formados pela ligaçao do grupo -N(CH3)2 do anel naftalenico, mostraram que a partir das várias conformaçoes possíveis e seus respectivos ECDs teóricos o valor do ângulo diedro igual a 150° (ECD positivo centrado em 320 nm) apreentaram uma boa concordância com o espectro obtido experimental. Por outro lado, o diedro de 80° apresentou um sinal do espectro de ECD oposto ao observado experimentalmente. Com o objetivo de obter e confirmar os resultados de ECD obtidos tanto teórica como experimentalmente, foi realizado cálculos de docking molecular. Esperávamos que os resultados de docking molecular para a estrutura da molécula de DanG, no sítio de interaçao, apresentasse um valor de ângulo diedro próximo àquele obtido pela técnica de DFT e confirmasse o sinal de ECD obtido experimentalmente. Os resultados obtidos para os cálculos de docking sao apresentados na Tabela 2, na qual estao inseridos os valores de energia para a melhor pose (posiçao, orientaçao e conformaçao do ligante) na cavidade da proteína HSA em cada cálculo realizado. Foram realizadas três simulaçoes: (a) uma para ligantes no sítio receptor de 2xw1; nesse sítio está presente o resíduo de aminoácido Tyr411; (b) uma para ligantes no sítio receptor de 2xvu; nesse sítio está presente o resíduo de aminoácido Trp214; (c) uma para ligantes no sítio receptor de 2xvu; nesse sítio está presente o resíduo de aminoácido Tyr411.
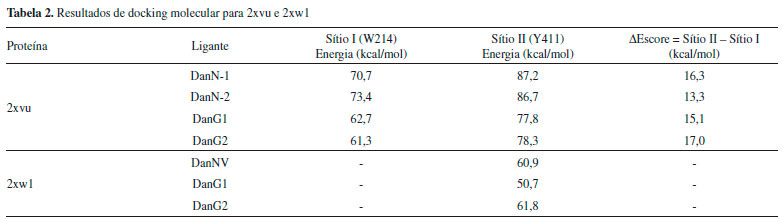
Os dados apresentados na Tabela 2 mostram que, para o caso em que o receptor é a proteína 2xw1, em que apenas o sítio II está ocupado pelo ligante, o escore do redocking (reconstruçao do complexo cristalográfico) e os de DanG sao muito próximos, indicando que o ligante DanG pode se acomodar neste sítio assim como o ligante cristalográfico. Para o receptor de 2xvu, oberva-se que os cálculos de redocking indicam uma preferência pelo sítio II, sugerindo que este sítio é o preferencial. Os ângulos de torsao das moléculas sao apresentados na Tabela 3.
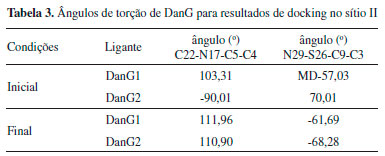
De acordo com os resultados apresentados na Tabela 3, podemos observar que apesar das moléculas ligantes DanG terem condiçoes iniciais diferentes, a condiçao final, obtida com o docking molecular á praticamente a mesma. Na Figura 8 apresentamos o ligante DanG no sítio II da proteína HSA em uma representaçao de superfície para a proteína, a partir da qual podemos observar que o grupo naftil ocupa o bolso interno da proteína e o grupo carboxílico está voltado para o solvente.
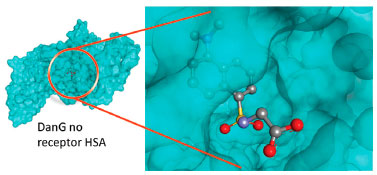 Figura 8. Molécula de DanG no sítio II da proteína HSA
A análise das interaçoes realizada com o programa de visualizaçao gráfica36 indica que o grupo naftil da molécula se estabiliza pelas interaçoes do tipo p entre os aminoácidos Asn391 e Leu453, conforme pode ser obervado na Figura 9. O grupo carboxílico da DanG faz ligaçoes de hidrogênio com Arg410 e o grupo sulfona faz interaçoes com a Tyr411.
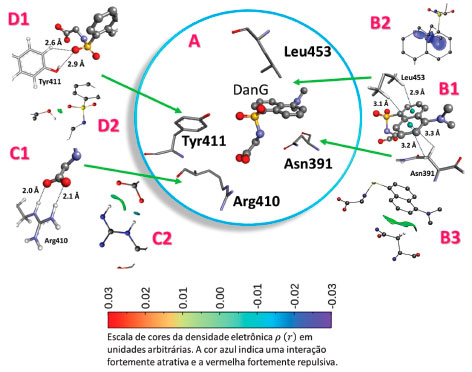 Figura 9. Interaçoes entre DanG no sítio II da HSA
A Figura 9 mostra que:
A partir da configuraçao obtida para a molécula de DanG nos cálculos de docking molecular, obtivemos os espectros de UV-Vis e de ECD da molécula de DanG a partir da simulaçao computacional utilizando o funcional CAM-B3LYP e a funçao de base 6-311++G(3df,2p). Na Figura 10 apresentamos os especros de UV-Vis (a) e ECD (b) representados como a força do oscilador e o poder rotatório, respectivamente. Podemos observar que ocorre uma boa concordancia entre os resultados do docking molecular e aqueles obtidos através da técnica DFT e dos resultados experimentais.
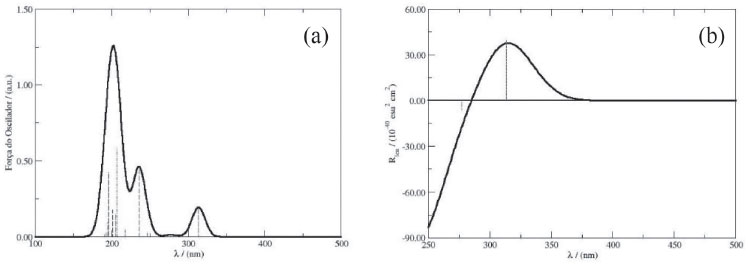 Figura 10. Espectros eletrônicos de (a) UV-Vis e (b) Dicroísmo Circular da DanG obtidos em etanol
Estudo dos orbitais moleculares HOMO-LUMO da DanG Na Figura 11 (a) apresentamos os orbitais moleculares HOMO e LUMO para a molécula de DanG para o ângulo diedro, formado entre os átomos C22-N17-C5-C4, igual a 80º. O orbital HOMO (de número 81) e o LUMO (de número 82) sao os responsáveis pela transiçao eletrônica observada no espectro de ECD, obtida no nível de teoria DFT com o funcional híbrido CAM-B3LYP e a funçao de base 6-311++G(2d, p) no vácuo. A transiçao HOMO-LUMO se refere à passagem de um elétron localizado no par de elétrons n do átomo de Nitrogênio N17 para um orbital p localizado no átomo de Carbono C5. Essa transiçao está relacionada ao sinal negativo no espectro de ECD apresentado na Figura 7(b) localizado em 317 nm.
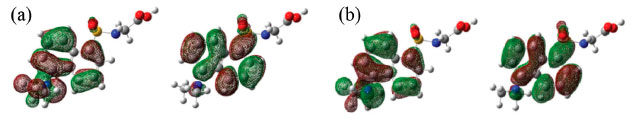 Figura 11. Principais orbitais moleculares (a) HOMO e LUMO da DanG para o diedro de 80º (b) HOMO e LUMO da DG para o diedro de 150º
Na Figura 11(b) apresentamos os orbitais moleculares HOMO e LUMO para a molécula de DanG para o ângulo diedro C22-N17-C5-C4 igual a 150º. O orbital HOMO (de número 81) e o LUMO (de número 82) sao os responsáveis pela transiçao eletrônica observada no espectro de ECD, obtida no nível de teoria DFT com o funcional híbrido CAM-B3LYP e a funçao de base 6-311++G(2d, p) no vácuo. A transiçao HOMO-LUMO se refere à passagem de um elétron localizado no par de elétrons n do átomo N17 para um orbital p localizado no átomo C5. Esta transiçao está relacionada ao sinal positivo no espectro de ECD apresentado na Figura 5 (a) localizado em 333 nm. As contribuiçoes dos orbitais atômicos da DanG para os orbitais moleculares HOMO e LUMO em funçao do ângulo diedro foram obtidas a partir da análise dos orbitais moleculares e foram escolhidos aqueles que sao predominantes para a formaçao do orbital molecular que descrevem as transiçoes eletrônicas. Podemos verificar que para o ângulo diedro igual a 80º a maior contribuiçao para o orbital molecular HOMO está relacionada ao átomo C5 com o orbital atômico 5s e para o LUMO a maior contribuiçao está relacionada ao átomo de Carbono (4) com o orbital atômico 5s. Para o ângulo diedro igual a 150º, a contribuiçao para o HOMO e LUMO está associada aos átomos de N17 com o orbital 4pz e o átomo de C5 com o orbital 5pz.
CONCLUSOES Os resultados obtidos mostraram que a DanG nos solventes: água, metanol, etanol, acetonitrila e THF, apresenta um comprimento de onda máximo de absorçao em aproximadamente em 340 nm, indicando uentificada como uma transiçao n→π*. Além disso, o uso desses dados experimentais foi útil para relacionarmos com os espectros teóricos, em que observamos que os espectros com as configuraçoes R e S nao apresentaram mudanças significativas em relaçao ao comprimento de onda máximo. Com os dados de ECD, pudemos demonstrar que a complexaçao de HSA com DanG leva ao aparecimento de uma banda ECD positiva centrada em 346 nm. Esse fenômeno pode ser explicado pelo aparecimento da quiralidade na DanG quando induzida pelo meio, no presente caso o sítio II da proteína HSA. A comparaçao entre o espectro de ECD experimental com aqueles obtidos por DFT, em diferentes solventes, nos leva a concluir que a conformaçao adquirida pela DanG ao se ligar à proteína se assemelha a configuraçao R. Cálculos da superfície de energia potencial (SEP) da DanG nos mostram que os grupos envolvidos no fenômeno do aparecimento do sinal de ECD está relacionado ao grupo -N(CH3)2 do anel naftalenico. A análise das várias conformaçoes obtidas e seus respectivos ECDs teóricos revelaram que o diedro de 150° (ECD positivo centrado em 320 nm) apresentou excelente similaridade com o espectro experimental. Por outro lado, o diedro de 80° apresentou um sinal do espectro de ECD oposto ao observado experimentalmente. Além disso, observamos que o átomo de nitrogênio do grupo -N(CH3)2 apresentou a maior contribuiçao para a transiçao HOMO-LUMO que dá origem à transiçao eletrônica n→π*envolvida na geraçao do sinal de ECD. Os resultados dos cálculos de docking realizados em proteína livre de ácidos graxos com compostos dansilados no sítio 2 (onde está o aminoácido Tyr411) mostram que o composto DanG tem possibilidade similar de se ligar a esse sítio da mesma forma que o ligante cristalográfico DanNV, para a estrutura de código pdb 2xw1. Os estudos de docking foram realizados em duas situaçoes no caso do receptor de código pdb 2xvu e o ligante DanG poderia ocupar o sítio I e/ou o sítio II do receptor. Observou-se que DanG liga-se preferencialmente ao sítio II com uma diferença de escore de mais de 15 kcal mol-1 em favor deste. As análises de docking molecular mostraram que no sítio II o grupo naftil está estabilizado, dos dois lados, por interaçoes do tipo CH...p entre os aminoácidos Leu453 e Asn391, ficando praticamente imobilizado por estas interaçoes. O grupo carboxílico e o átomo de oxigênio do grupo sulfona do ligante DanG fica estabilizado por interaçoes com Arg410 e Tyr411, respectivamente. Os cálculos de isosuperfície de densidade eletrônica mostram que estas interaçoes sao atrativas. As análises de docking molecular mostraram também que a complexaçao entre DanG e a proteína HSA gerou uma conformaçao com um diedro igual a 100°, muito similar àquele obtido teoricamente (DFT) e cujo ECD está em acordo com o resultado experimental. Em conclusao, o estudo das possíveis conformaçoes para a dansilglicina e o cálculo do seu ECD teórico permitiram identificar a origem do sinal de ECD obtido pela complexaçao entre DanG e HSA.
AGRADECIMENTOS Este trabalho foi financiado pela Fundaçao de Amparo à Pesquisa do Estado de Sao Paulo (FAPESP, auxílios: 2016/20549-5, 2015/22338-9, 308480/2016-3 e INCT.Bio.Nat 2014/50926-0), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq: 302793/2016-0, 306975/2013-0, IC:440503/2014-0, IC:308480/2016-3 e 305541/2017-0) e CAPES. Esta pesquisa tornou-se possível graças aos recursos computacionais disponibilizados pelo Núcleo de Computaçao Científica (NCC/GridUNESP) da Universidade Estadual Paulista (UNESP).
REFERENCIAS 1. Lindup, W. E.; Orme, M. C.; Br. Med. J. 1981, 282, 212. 2. Fanali G.; Di Masi A.; Trezza, V.; Marino, M.; Fasano, M.; Mol. Aspects Med. 2012, 33, 209. 3. Sudlow, G.; Birkett, D. J.; Wade, D. N.; Mol. Pharmacol. 1976, 12, 1052. 4. Sudlow, G.; Birkett, D. J.; Wade, D. N.; Mol. Pharmacol. 1975, 11, 824. 5. Kragh-Hansen, U.; Chuang, V. T. G.; Otagiri, M.; Biol. Pharm. Bull. 2002, 25, 695. 6. Gundry, R. L.; Proteomics: Clin. Appl. 2007, 1, 73. 7. Peters, T.; All about Albumin: Biochemistry, Genetics, and Medical Applications, 1st ed., Academic Press, Inc: San Diego, 1996. 8. Graham, A.; Li, G.; Chenm, Y.; Morgan, J.; Oseroff, A.; Dougherty, T. J.; Pandey, R. K. Photochem. Photobiol. 2003, 77, 561. 9. Chaves, O.; Echevarria, A.; Esteves-Souza, A.; Maciel, M.; Netto-Ferreira, J. C.; J. Braz. Chem. Soc. 2018, 29, 1786. 10. Chaves, O. A.; Santos, M. R. L.; Santánna, C. M.; Echevarria, A.; Ferreira, A. B. B.; Netto-Ferreira, J. C.; Mediterr . J. Chem. 2018, 7, 8. 11. Lakowicz, J. R.; Principles of Fluorescence Spectroscopy. 3rd ed., Springer: Boston, 2006. 12. Ximenes, V. F.; Morgon, N. H.; Souza, A. R.; Chirality 2018, 31, 1. 13. Souza, A.R.; Morgon, N. H.; Rev. Virtual Quim. 2016, 8, 417. 14. Petitpas, I.; Bhattacharya, A. A.; Twine, S.; East, M.; Curry, S.; J. Biol. Chem. 2001, 276, 22804. 15. Bertucci, C.; Domenici, E. C.; Med. Chem. 2002, 9, 1463. 16. Bhattacharya, A. A.; Curry, S.; Franks, N. P.; J. Biol. Chem. 2000, 275, 38731. 17. Ryan, A. J.; Ghuman, J.; Zunszain, P. A.; Chung, C. W.; Curry, S.; J. Struct. Biol 2011, 174, 84. 18. Simard, J. R.; Zunszain, P. A.; Hamilton, J. A.; Curry, S.; J. Mol. Biol. 2006, 361, 336. 19. Bertucci, C.; Pistolozzi, M.; Simone, A. D.; Anal. Bioanal. Chem. 2010, 398, 155. 20. Ali J. R.; Ghuman, J.; Zunszain, P. A.; Chung, C. W.; Curry, S.; J. Struct. Biol. 2011, 174, 84. 21. Chatterjee, T.; Pal, A.; Dey, S.; Chatterjee, B. K.; Chakrabarti, P.; PLoS One 2012, 7, e37468. 22. Bertucci, C.; Pistolozzi, M.; Chirality 2008, 20, 552. 23. Koch, W.; Holthausen, M. C.; A Chemist's Guide to Density Functional Theory, 2nd ed., Wiley-VCH: Weinheim, 2001. 24. Allenmark, S.; Chirality 2003, 15, 409. 25. Junior, N. N.; Tese de Doutorado, Universidade de Sao Paulo, Brasil, 2013. 26. Graciani, F. S.; Ximenes, V. F.; PLoS One 2013, 8, 1. 27. Arruda, P. M.; Dissertaçao de Mestrado, Universidade Federal do Espírito Santo, Brasil, 2009. 28. Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Petersson, G. A.; Nakatsuji, H.; Li, X.; Caricato, M.; Marenich, A. V.; Bloino, J.; Janesko, B. G.; Gomperts, R.; Mennucci, B.; Hratchian, H. P.; Ortiz, J. V.; Izmaylov, A. F.; Sonnenberg, J. L.; Williams-Young, D.; Ding, F.; Lipparini, F.; Egidi, F.; Goings, J.; Peng, B.; Petrone, A.; Henderson, T.; Ranasinghe, D.; Zakrzewski, V. G.; Gao, J.; Rega, N.; Zheng, G.; Liang, W.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Throssell, K.; Montgomery, J. A., Jr.; Peralta, J. E.; Ogliaro, F.; Bearpark, M. J.; Heyd, J. J.; Brothers, E. N.; Kudin, K. N.; Staroverov, V. N.; Keith, T. A.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A. P.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Millam, J. M.; Klene, M.; Adamo, C.; Cammi, R.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Farkas, O.; Foresman, J. B.; Fox, D. J.; Gaussian 09,Revision D.01; Gaussian Inc., Wallingford CT, 2013. 29. Foresman, J. B.; Frisch, E.; Exploring chemistry with electronic structure methods, 2nd ed., Gaussian Inc., Pittsburg, 1996. 30. Chaves, O. A.; Tavares, M. T.; Cunha, M. R.; Parise, R. F.; Santánna, C. M.; Netto-Ferreira, J. C.; Biomolecules 2018, 8, 78. 31. Chaves, O. A.; Mathew, B.; Cesarin, D. S.; Lakshminarayanan, B.; Joy, M.; Mathew, G. E.; Suresh, J.; Netto-Ferreira, J. C.; J. Mol. Liq. 2017, 242, 1018. 32. Ryan, A. J.; Ghuman, J.; Zunszain, P. A.; Chung, C. W.; Curry, S.; J. Struct. Biol. 2011, 84, 174. 33. Jones, G.; Willet, P.; Glen, R. C.; Leach, A. R.; Taylor, R.; J. Mol. Biol. 1997, 267, 727. 34. Jones, G.; Willett, P.; Glen, R. C.; J. Mol. Biol. 1995, 245, 43. 35. http://www.ccdc.cam.ac.uk/products/life_sciences/gold/, acessada em janeiro de 2019. 36. http://accelrys.com, acessada em janeiro de 2019. 37. Humphrey, W.; Dalke, A.; Schulten, K.; J. Mol. Graphics. 1996, 14, 33. 38. Johnson, E.R.; Keinan, S.; Mori, P. S.; Contreras, J. G.; Cohen, A. J.; Yang, W. J. Am. Chem Soc. 2010, 132, 6498. 39. Contreras-Garcia, J.; Johnson, E. R.; Keinan, S.; Chaudret, R; Piquemal, J. P.; Beratan, D. N.; Yang, W.; J. Chem. Theory Comput. 2011, 7, 625. 40. Marques M. A. L.; Botti, S.; Gaz. Fis. 2006, 29, 10. 41. Morgon, N. H.; Custodio, R.; Quim. Nova 1994, 18, 44. 42. http://www.chemkeys.com, acessada em janeiro de 2019. 43. Jensen, F.; Introduction to Computational Chemistry. 3rd ed., Wiley: Chichester, 2017. 44. de Souza, A. R.; Ximenes, V. F.; Morgon, N. H.; Rev. Virtual Quim. 2016, 8, 525. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access