
 |
 |
 |
 |
 |
Artigo
| Ring opening polymerization of L-lactide with two different zinc(II) phenoxy-imine complexes as initiators |
|
Alana L. C. OliveiraI; Leonardo C. FerreiraI; Marcos L. DiasI,*; Rodrigo S. BitzerII; Marco A. C. NascimentoII
I. Instituto de Macromoléculas Professora Eloisa Mano, Universidade Federal do Rio de Janeiro, 21941-598 Rio de Janeiro - RJ, Brasil Recebido em 18/02/2019 *e-mail: mldias@ima.ufrj.br This article reports the synthesis of two zinc(II) complexes containing phenoxy-imine ligands and their application as initiators of bulk ring opening polymerization (ROP) of L-lactide (LLA). The phenoxy-imine ligands (L1 and L2) were obtained by condensation reactions of salicylaldehyde derivatives with 2,6-diisopropylaniline. The reactions of L1 and L2 with ZnEt2 in the presence of n-BuOH afforded [Zn(L1)(OBu)] (C1) and [Zn(L2)(OBu)] (C2), which were characterized by elemental analysis, FTIR and 1H NMR spectroscopy. In addition, the geometries of both complexes were investigated by DFT (B3LYP/LACV3P**) calculations. C1 and C2 were tested as initiators of ROP of LLA at 180 °C using different LLA/Zn molar ratios, namely 500, 1000 and 2500. Both complexes showed good activity, resulting in conversions up to 90-92% in 2 h. The poly(L-lactide) obtained displayed Mn ranging from 40.000-109.000, relatively low polydispersity (Mw/Mn = 1.2-2.1) and high stereoregularity with Tm = 176 °C. INTRODUCTION Linear aliphatic polyesters, such as those derived from lactides and their copolymers, have attracted considerable attention due to their biodegradability and biocompatibility. An effective way of producing polylactides (PLAs) involves ring-opening polymerization (ROP) of lactides using metal complexes as initiators.1,2 In effect, metal-initiated ROP of lactides offers large degree of reaction and polymer tacticity controls as well as generates high molecular weight polymers.2-4 ROP initiators generally affect the properties of the final polymers in different ways.5,6 When metal complexes are used as initiators, the structure and properties of the final polymers are dictated by the type of the metal and the stereochemical attributes of the ancillary ligands. Moreover, as catalyst residues are likely to be present in the material at the end of polymerization, the use of biocompatible initiators has been largely recommended when producing polymers with potential biomedical applications.7-11 Over the past fifteen years, the use of organocatalysts or metal complexes as ROP catalysts has been exploited in an attempt of controlling polymer micro and macrostructures as well as limiting transesterification and other uncontrolled chain transfer processes.12,13 In this context, complexes with oxophilic and Lewis acidic metals, such as alkaline metals, AlIII, SnII, ZnII, MgII, CaII, LnIII, TiIV, and ZrIV, are efficient ROP catalysts and account for chain length control and possible stereocontrol.14 Mono and polynuclear zinc(II) complexes containing different types of ligands, such as phenolates, biphenolates, diiminates, diimines and phenoxy-imines, have been widely employed as initiators for ROP of several lactones.10-21 The presence of zinc(II) in these reactions seems to stimulate transesterification processes as well as a decrease in chain epimerization during PLA formation. In addition, ROP reactions catalyzed by zinc(II) complexes are controlled processes that give rise to high molecular weight polyesters.15 Accordingly, the suitable choice of ligands constitutes an important step toward improving the performance of the initiators. In a previous article, we showed that bimetallic zinc(II) complexes containing phenoxy-imine ligands are active initiators for lactide polymerization, generating poly(L-lactic acid) (PLLA) with low degree of isomerization.22 In this work, we have undertaken the preparation of two mononuclear zinc(II) complexes with phenoxy-imine ligands with different degree of steric hindrance and the study of their performance toward bulk ROP of L-lactide as well as their possible influence on the stereoregularity of the formed PLLA. In addition, the thermal properties of the PLLA obtained from these initiators are discussed.
EXPERIMENTAL Materials The preparation of the zinc(II) complexes and the polymerization reactions were carried out under inert atmosphere using Schlenk-type glassware and glove bags. Toluene was dried over Na-benzophenone ketyl under N2. Salicylaldehyde, 3-tert-butyl-2-hydroxybenzaldehyde, 2,6-diisopropylaniline (Aldrich) and diethylzinc (ZnEt2) (15 wt% solution in hexane) (Akzo Nobel) were purchased from commercial sources and used as received. Butyl alcohol (Aldrich) was dried over molecular sieve. Polymerizations were performed using L-lactide (Purac) recrystallized from dry toluene. Synthesis of 2-((2,6-diisopropylphenylimine)methyl)phenol (L1) L1 was prepared according to a procedure described in the literature with some modifications.23-25 An ethanolic solution (15 mL) of salicylaldehyde (0.86 mL, 8.19 mmol) was added dropwise to a solution of 2,6-diisopropylaniline (1.56 mL, 8.19 mmol) in the same solvent (15 mL). The reaction mixture was refluxed for 3 hours and kept under stirring overnight. Then, the resulting yellow solution was cooled, leading to the formation of a yellow precipitate, which was filtered off and washed with hexane. Yield: 62%. Melting point (M.p.): 61-62 °C. FTIR (ATR, ±4 cm-1): 3500-3300 cm-1 (νO-H); 3100-2800 cm-1 (νC-H); 1626 cm-1 (νC=N, imine); 1572 and 1462 cm-1 (νC=C, aromatic); 1277 cm-1 (νC-N); 1173 cm-1 (νC-O, phenol). 1H NMR (400 MHz, CDCl3, 25 °C): δ 10.15 (s, 1H, OH phenol); δ 8.29 (s, 1H, CH=N); δ 7.50-7.0 (m, 7H, ArH); δ 3.01 (m, 2H, CH); δ 1.5-1.0 (m, 12H, CH3). Synthesis of 2-tert-butyl-6-((2,6-diisopropylphenylimine) methyl)phenol (L2) L2 was prepared using a procedure similar to that described for L1. An ethanolic solution (15 mL) of 3-tert-butyl-2-hydroxybenzaldehyde (1.00 g, 5.61 mmol) was added dropwise to a solution of 2,6-diisopropylaniline (0.8 mL, 5.61 mmol) in the same solvent (15 mL). The reaction mixture was refluxed for 3 hours and kept under stirring overnight. Thereafter, the resulting yellow solution was cooled, leading to the formation of a yellow solid, which was filtered off and washed with hexane. Yield: 60%. M.p.: 95-97 °C. FTIR (ATR, ±4 cm-1): 3500-3300 cm-1 (νO-H); 3100-2800 cm-1 (νC-H); 1623 cm-1 (νC=N, imine); 1585 and 1465 cm-1 (νC=C, aromatic); 1250 cm-1 (νC-N); 1166 cm-1 (νC-O, phenol). 1H NMR (400 MHz, CDCl3, 25 °C): δ 10.25 (s, 1H, OH phenol); δ 8.30 (s, 1H, CH=N); δ 7.5-7.0 (m, 5H, ArH); δ 3.05 (m, 2H, CH); δ 1.5-1.0 (m, H, CH3). Synthesis of [Zn(L1)(OBu)] (C1) Complex C1 was prepared according to a procedure described in the literature with some modifications.26-31 In a 25 mL Schlenk tube, ZnEt2 (15 wt% solution in hexane) (1.46 mL, 1.78 mmol) was slowly added to a solution of L1 (0.50 g, 1.78 mmol) in dry toluene (15 mL). The reaction mixture was stirred overnight at room temperature. After this period, 0.16 mL (1.78 mmol) of n-BuOH was added, and the reaction mixture was kept under stirring for additional 3 hours. The resulting greenish-yellow solid was filtered off, washed three times with dry toluene, and dried under vacuum. Yield: 80%. M.p.: >300 °C. Elemental analysis for [Zn(C19H22NO) (O(CH2)3CH3)]: (Calcd. (Found)) C, 65,99 (66.20); H, 7.40 (7.20); N, 3,36 (3,45). 1H NMR (400 MHz, CDCl3, 25 °C): δ 8.29 (s, 1H, CH=N); δ 7.50-6.96 (m, 7H, ArH); δ 3.31 (m, 2H, OCH2); δ 2.05 (m, 2H, CH); δ 3.31-3.05 (m, 2H, (CH2) butyl alcohol); δ 2.05 (m, 2H, (CH2) butyl alcohol); δ 1.28-1.20 (m, 12H, Pr(CH3)); δ 1.14 (m, 3H, (CH3) butyl alcohol). Synthesis of [Zn(L2)(OBu)] (C2) Complex C2 was prepared using a procedure similar to that described for C1.26-31 In a 25 mL Schlenk tube, ZnEt2 (15 wt% solution in hexane) (1.22 mL, 1.48 mmol) was slowly added to a solution of L2 (0.50 g, 1.48 mmol)) in dry toluene (15 mL). The reaction mixture was stirred overnight at room temperature. After this period, 0.14 mL (1.48 mmol) of n-BuOH was added, and the reaction mixture was kept under stirring for additional 3 hours. The resulting greenish-yellow solid was filtered off, washed three times with dry toluene, and dried under vacuum. Yield: 85%. M.p.: >300 °C. Elemental analysis for [Zn(C23H30NO)(O(CH2)3CH3)]: (Calcd. (Found)) C, 68.32 (69.18); H, 8.22 (8.30); N, 2.95 (3.11). 1H NMR (400 MHz, CDCl3, 25 °C): δ 8.30 (s, 1H, CH=N); δ 7.58-7.18 (m, 6H, ArH); δ 3.05 (m, 2H, 2xCH); δ 3.30-2.86 (m, 2H, OCH2); δ 1.34 (m, 2H, (CH2) butyl alcohol); δ 1.52 (s, 9H, tBu(CH3)3); δ 1.29 (s, 12H, 4xiPr(CH3)); δ 1.22 (m, 2H, (CH2) butyl alcohol); δ 1.15 (m, 3H, (CH3) butyl alcohol). Characterization of ligands and complexes 1H NMR spectra were recorded at 25 °C on a Varian Mercury VX spectrometer using CDCl3 as internal reference. FTIR spectra were recorded on a Frontier FTIR/FIR using the Attenuated Total Reflectance (ATR) technique. Melting points were determined using a Quimis equipment (model 0340513) with controlled heating. Elemental analyses were performed on a HANAU Elementar vario MICRO cube. L-Lactide polymerization The activity of C1 and C2 toward bulk ROP of L-lactide (LLA) was investigated using the following monomer/initiator (LLA/Zn) molar ratios: 500, 1000 and 2500. Each mixture containing the desired amount of initiator and L-lactide (about 2 g) was prepared in a Schlenk flask under N2. Each polymerization reaction was conducted at 180 °C for 2 h.3,4 After this period, the reaction was quenched by cooling to room temperature. Then, the solid residue was dissolved in chloroform, and the polymer was precipitated from ethanol. The isolated polymers were dried under vacuum at 50 °C. Polymers characterization 1H and 13C NMR spectra were recorded at 25 °C using a Varian Mercury VX-300 NMR spectrometer and CDCl3 as internal reference. Gel permeation chromatography (GPC) was carried out on a Shimadzu LC 20 instrument equipped with a set of two Phenogel columns and RID-20A differential index detector. GPC analyses were performed at 25 °C using chloroform as eluent at a 1 mL min-1 rate. A calibration curve using ten polystyrene (PS) standards was used for molecular weight determination by the Shimadzu software. Thermal properties were determined by DSC using a TA Q1000 calorimeter with a heating and cooling rate of 10 °C min-1 from -10 to 200 °C under N2 flow. Glass transition (Tg), crystallization temperature on heating (cold crystallization, Tcc) and melting temperature (Tm) were obtained from a second heating after a quenching run. The crystallization temperature on cooling (Tc) was recorded in a cooling run after the second heating run. The degree of crystallinity (Xc) was determined using the following equation: Xc = ΔHm/ΔHmo, where ΔHm is the melting enthalpy and ΔHmo is the standard melting enthalpy for 100% crystalline PLA (106 J g-1).32 Thermogravimetric analyses (TGA) were performed on a TA TGA Q500 thermoanalyzer. Measurements were carried out under N2 at a heating rate of 20 °C min-1 up to 700 °C, with a gas flow rate of 20 mL min-1. DFT calculations The molecular structures of C1 and C2 were studied at the density functional theory (DFT) level. All calculations were carried out using JAGUAR 7.9 with an energy convergence criterion of 1.00 x 10-8 hartree.33 Gas-phase geometry optimizations were performed without constraints using the B3LYP hybrid functional along with the LACV3P** (6-311G** for non-metallic elements and Los Alamos ab initio ECP, which incorporates the mass-velocity and Darwin relativistic effects, plus valence triple-zeta basis set for Zn) basis set.34,35 The vibrational frequencies of each optimized geometry were evaluated at the same level of calculation and verified to be real. An excellent agreement between experimental and calculated frequencies was obtained.
RESULTS AND DISCUSSION Ligands and complexes The condensation reaction of salicylaldehyde with anilines constitutes an ordinary method for the preparation of phenoxy-imines. L1 and L2 were synthesized in high yields.23-25 Complexes C1 and C2 were also obtained in high yields (Figure 1).
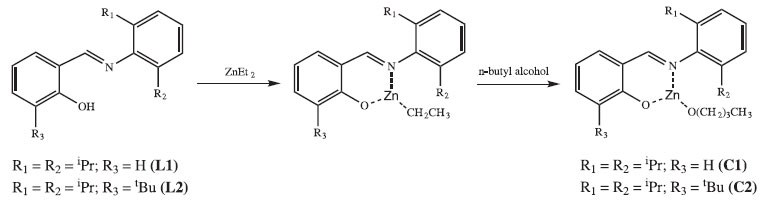 Figure 1. Synthesis of C1 and C2
According to our preliminary characterization (M.p. and microanalysis), ligands and complexes were isolated as analytically pure solids. The elemental analyses indicate that C1 and C2 are monomeric and exhibit only one phenoxy-imine group. In the FTIR spectrum of L1, the C=N stretching band appears at 1626 cm-1, which is consistent with the literature.36,37 Also, it is possible to observe a broad band in the range 3500-3300 cm-1, ascribed to the phenolic ν(OH) vibration. Comparing the FTIR spectra of L1 and C1, it is possible to observe changes associated with the bidentate coordination of the ligand. For C1, the imine ν(CN) band lies at 1605 cm-1, indicating coordination of the imine group. Moreover, the disappearance of bands in the 3500-3300 cm-1 region suggests deprotonation and coordination of the phenolic O atom. In the FTIR spectrum of L2, a strong band at 1623 cm-1 is assigned to the ν(CN) mode, whereas in the spectrum of C2 this band is shifted to a lower wavenumber (1610 cm-1). In addition, the absence of phenolic ν(OH) bands in C2 also indicates bidentate coordination of L2. The 1H NMR spectra of C1 and C2 (Figures 2 and 3) are also consistent with the proposed structures. As expected, the appearance of resonances for the -(CH2)3CH3 group between 1.5-1.0 ppm confirms the presence of coordinated butoxide. Also, it is possible to observe the disappearance of the downfield signal for the phenol group present in the ligands (~10 ppm), which also corroborates to the binding of the O atom.9
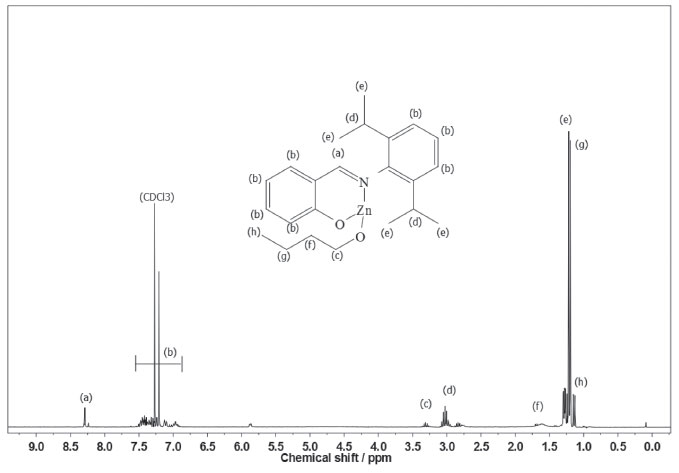 Figure 2. 1H NMR spectrum of C1 in CDCl3
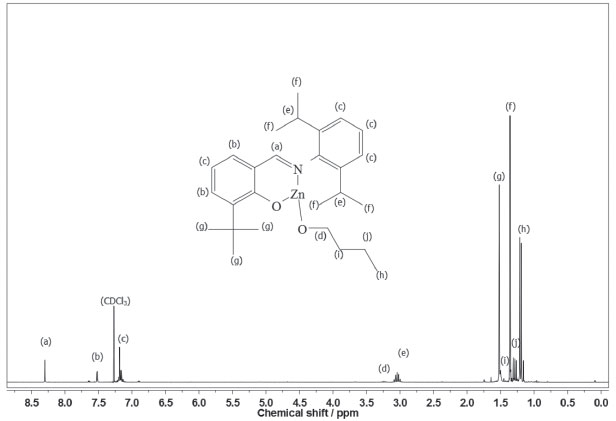 Figure 3. 1H NMR spectrum of C2 in CDCl3
DFT calculations at the B3LYP/LACV3P** level were used to elucidate the molecular structures of C1 and C2. The geometries optimized for C1 and C2 are depicted in Figure 4. The geometric parameters obtained for both complexes are very similar. In both alkoxides, the metal center is three-coordinated and displays a trigonal planar coordination geometry, which is essential for the activity in ROP of cyclic esters.9 In C1, the Zn-N, Zn-OPhO and Zn-OBuO distances amount to 2.070, 1.942 and 1.858 Å, respectively. In C2, the Zn-N, Zn-OPhO and Zn-OBuO distances are equal to 2.057, 1.939 and 1.857 Å, respectively. Regardless of the complex, the Zn-OBuO distance is more than 0.08 Å shorter than the Zn-OPhO distance. The N-Zn-OPhO bite angle for C1 is 93.7o, whereas for C2 it amounts to 92.9o. Examples of other three-coordinate zinc(II) phenoxides supported by bulky Schiff base ligands are available in the literature.38-42 Their geometries are similar to those calculated for C1 and C2.
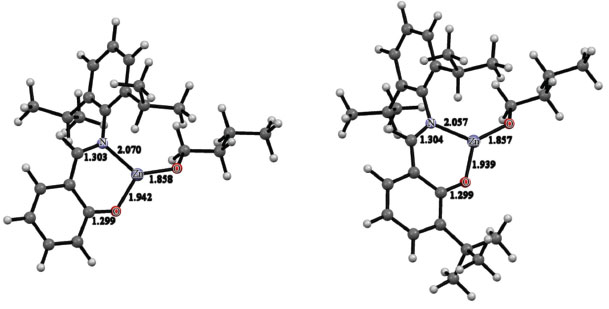 Figure 4. Molecular structures optimized for C1 (left) and C2 (right) at the B3LYP/LACV3P** level of calculation
Complexes C1 and C2 are soluble in L-lactide above 100 °C and catalyze the ring-opening polymerization of this lactone to produce PLLA. Herein, we assume that they exist as mononuclear entities in solution before coordinating to L-lactide in the beginning of the catalytic cycle. Ring opening polymerization of L-Lactide Bulk polymerization of L-lactide using C1 and C2 as initiators (Figure 5) were investigated in order to gain some insight into their performance in producing poly(L-lactic acid) (PLLA). The reactions were carried out at 180 °C for 2 h using different lactide/initiator molar ratios (LLA/Zn = 500, 1000 and 2500). Both complexes show good activity. Here, we observe that the smallest LLA/Zn ratio favors the highest monomer-polymer conversion at the condition used in this work (Table 1).
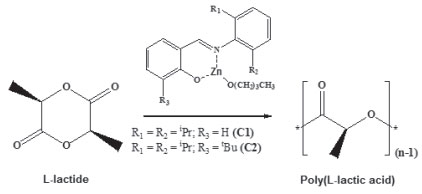 Figure 5. Polymerization of L-lactide using C1 and C2 as catalysts
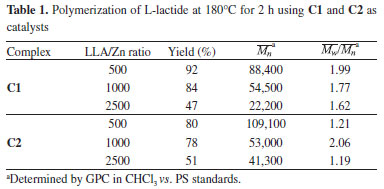
Alcohol is generally added in some ROP reactions in order to activate the catalytic process through alkoxide formation.9 No additional alcohol was needed in this work, since both initiators contain an alkoxide group in their structures, displaying a metal-alkoxide bond necessary to initiate the catalytic coordination-insertion process.9 The results in Table 1 show that for both initiators the reactions produced polymers with the highest molecular weights at the smallest monomer/initiator molar ratio (LLA/Zn = 500) in 2 h polymerization. The PLLA exhibiting the highest molecular weight was obtained with C2, suggesting that steric hindrance due to the tert-butyl group does not play an important role in the polymerization mechanism. On the other hand, depending on the position of the tert-butyl group in the aromatic ring, the ligand-to-metal inductive effect should favor the activation of the metal center, which can explain the highest molecular weight when the LLA/Zn(C2) = 500 molar ratio is used.43 According to Table 1, both polymer yield and number average molecular weight (Mn) increase as the LLA/Zn ratio decreases. The results of yield and Mn attained in this work are in the same order of magnitude reported for tin octoate, which is considered one of the best initiator for ROP of lactides.14 The evolution of lactide conversion to PLLA with time in reactions catalyzed by C1 and C2 was examined from bulk polymerization at 180 °C and LLA/Zn = 1000. We have chosen an intermediate value for the LLA/Zn molar ratio to study the reaction evolution. Although it has not provided the best results in terms of molar mass and yield, it is a representative condition. Conversion as a function of time was determined by taking aliquots of the reaction medium along the polymerization, analyzing the monomer-polymer mixture by 1H NMR (Figure 6).44,45 The curves show that the polymerization with C1 exhibits a higher reaction rate when compared with the reaction catalyzed by C2. The curve for C1 shows the expected behavior for this type of polymerization, with a fast increase in the conversion and tendency to attain a constant value (equilibrium).
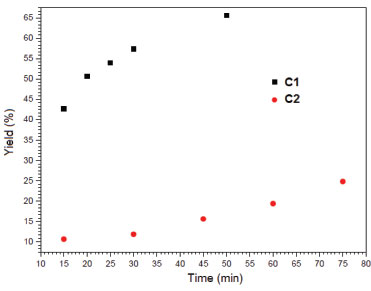 Figure 6. Yield vs. time plot for the polymerization of LLA with C1 and C2. Conditions: bulk polymerization, LLA/Zn = 1000, 180 °C
The polymerization reactions initiated by C1 and C2 display different evolution behavior, which can be accounted for by the structure of the complexes. The main structural difference concerns the presence of a tert-butyl group in C2. This feature may explain our results if ligand-to-metal inductive effect is taking into account. The presence of a tert-butyl group in the aromatic ring should increase the basicity of the ligand, leading to a more stable ZnII-Ophenolate bond. In this condition, the coordination of the monomer to the metal is less effective but enough for insertion in the polymeric chain. Complex C1 has a more acidic metal, thereby generating a more effective coordination of the monomer to the metal. Microstructure of PLLA Highly isotactic PLLA is produced by the ROP of L-lactide if no side reactions take place during polymerization. Racemization is one of these reactions and it can be induced by metals.3 Thus, the zinc(II) phenoxy-imine complexes could cause racemization reactions. Racemization produces inversion of the CH3 configuration of the repeating units, with the introduction of D-lactyl units in the polymer chain. The microstructures of the PLLA obtained by ROP with C1 and C2 were analyzed by 13C NMR. Figures 7 and 8 show the PLLA regions in the 13C NMR spectra. The spectra show three signals at 16, 68.5 and 169 ppm, attributed to the methyl, methine and carbonyl groups, respectively.44-47 D-lactyl units in the polymer chain is manifested by the appearance of lateral side signals in the methine and carbonyl signals.4 The figures show the carbonyl and the methine regions of the 13C NMR spectra of the polymers obtained with LLA/Zn = 2500 and 500. In these spectra, tetrad stereosequences were considered for evaluation of polymer stereoregularity, where, i and s are isotactic (two L-lactyl units) and syndiotactic (L-lactyl and D-lactyl units) diads, respectively. For methine carbons, tetrad iii, which indicates pure isotactic sequence (68.97 ppm) may appear overlapped with iis, sii, sis tetrads, which contain syndiotactic diads.
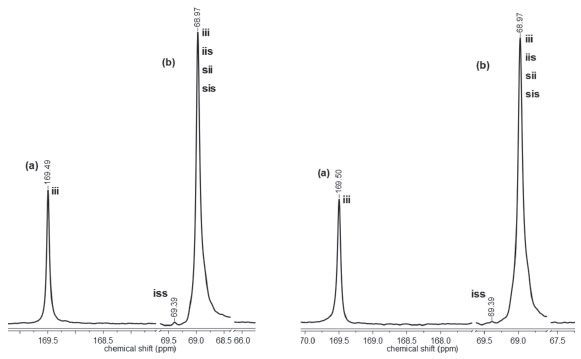 Figure 7. Carbonyl (a) and methine (b) regions of the 13C NMR spectra of PLLA obtained with C1 at LLA/Zn = 2500 (left) and LLA/Zn = 500 (right)
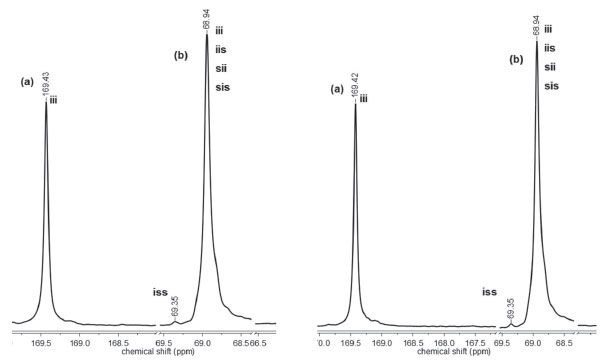 Figure 8. Carbonyl (a) and methine (b) regions of the 13C NMR spectra of PLLA obtained with C2 at LLA/Zn = 2500 (left) and LLA/Zn = 500 (right)
The region corresponding to the carbonyl groups shows low intensity lateral signals, which indicate the presence of very small amounts of other tetrad sequences different from iii. These results suggests that the polymers exhibit high stereoregularity. The methine region (68.97 ppm) seems to be more sensitive to the small difference in the stereoregularity of the samples, showing a split at 69.35 ppm (tetrad iss). The evaluation of stereoregularity based on NMR tetrad analysis for PLLA prepared at two LLA/Zn molar ratios (500 and 2500) is presented in Table 2. The tetrad percentage was calculated from the integration of the corresponding 13C peaks of iss and iii, iis, sii, sis tetrads.48
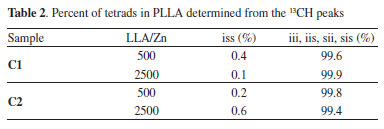
According to Table 2, all polymers have a high stereoregularity with percentage of isotactic rich tetrads (iii, iis, sii) superior to 99%. It was not possible to observe any influence of the ligand structure or LLA/Zn molar ratio on the stereoregularity of these polymers, which means that the microstructure of the polymers generated from C1 and C2 is controlled by the nature of the metal center. Polymer thermal properties DSC curves for PLLA synthesized using C1 and C2 were obtained in order to access the main transition temperatures. The values for the transition temperatures are shown in Table 3. Glass transition (Tg), crystallization temperature on heating (cold crystallization) (Tcc) and melting temperature (Tm) were obtained from a second heating run at 10 °C min-1 after a fast cooling run. Crystallization temperature on cooling (Tc) was accessed from a cooling run at 10 °C min-1. The Figures 9 and 10 show the DSC curves for the polymers obtained at different LLA/Zn molar ratios with C1 and C2, respectively.
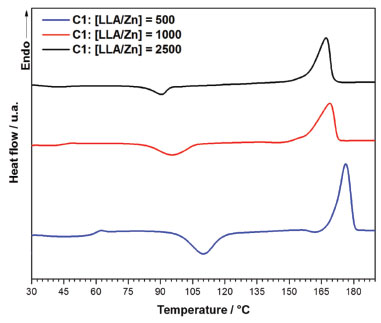 Figure 9. DSC curves for the polymers obtained with C1
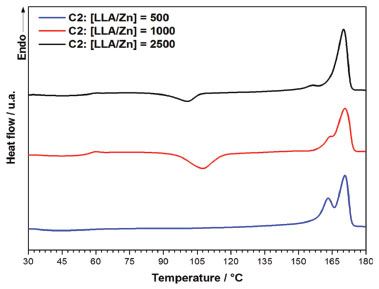 Figure 10. DSC curves for the polymers obtained with C2
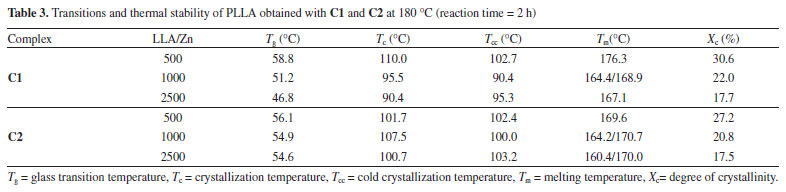
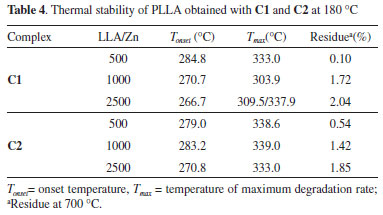
In general, the polymers show Tg in the expected range for high molecular weight PLLA (Tg = 50-60 °C) and high Tm (> 175 °C), confirming that they have high degree of stereoregularity. The PLLA obtained with C1 at LLA/Zn = 500 show the highest Tm (176.3 °C) and degree of crystallinity (Xc= 30.8%). While for PLLA prepared with C1 the increase in LLA/Zn decreases the Tm, for the PLLA prepared with C2 practically no difference was noted in the Tm when the LLA/Zn ratio is changed. It is important to pay the attention to the presence of two melting endotherms for the polymers obtained at LLA/Zn = 100 and 2500, which suggests two different types of crystals in these polymers. Table 4 summarizes the thermal stability data with the degradation temperatures (onset and maximum degradation rate) and the residue percentage at 700 °C of PLLA obtained using C1 and C2. The PLLA show a weight loss profile with only one degradation step with the onset temperature (Tonset) and temperature of maximum degradation rate (Tmax) around 270 °C and 330 °C, respectively. Tonset and Tmax obtained for these polymers are below those reported in the literature for commercial PLLA obtained with tin octoate initiator, which are about 350 °C and 370-390 °C, respectively.49 As these lower values of Tonset and Tmax are also observed for the PLLA produced at LLA/Zn = 500, with high molecular weight (Mn = 109,100 g/mol), these lower degradation temperatures do not seem to be related to molecular weight, but to the nature of metal residue. Residual zinc is likely to accelerate thermal chain degradation in these polymers.
TGA data (Figure 11) show that the content of final residues obtained at 700 °C is very small (0.1 to 2.0 wt%) and increases as the LLA/Zn ratio increases. This is probably due to a decrease in the polymer yield and increase in the residual initiator/polymer ratio.
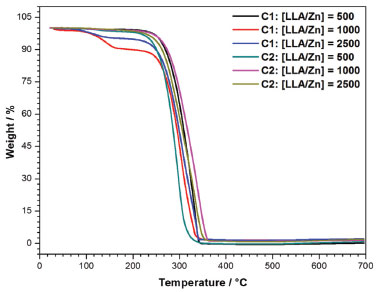 Figure 11. TG curves for the polymers obtained at different LLA/Zn molar ratios
CONCLUSIONS Two phenoxy-imine ligands and their zinc(II) complexes were synthesized and characterized by different spectroscopic techniques. Elemental analyses suggest that the complexes are monomeric, and the FTIR results indicate that the ligands behave as N^O donors. Their geometries were investigated by DFT calculations, which showed that the bite angles amount to ~93o for both complexes. The ligands and the coordination environment around the metal were chosen in a way that the complexes display suitable stereochemical attributes for ROP of lactides, which was confirmed by the successful synthesis of high molecular weight poly(L-lactide) (PLLA) under various reaction conditions. The PLLA produced using these complexes as initiators exhibit high stereoregularity, showing Tm up to 176 °C and high degree of crystallinity. Nevertheless, they showed lower thermal stability than those PLLA obtained with classical tin(II) initiators.
ACKNOWLEGDEMENTS The authors are grateful to the following Brazilian agencies, CAPES, CNPq (Grant 310917/2014-0) and FAPERJ (Grant E-26/201.304/2014), for financial support.
REFERENCES 1. Cabaret, O. D., Vaca, B. M.; Bourissou, D.; Chem. Rev. 2004, 104, 6147. 2. Idage, B. B.; Idage, S. B.; Kasegaonkar, A. S.; Jadhav, R. V.; Mater. Sci. Eng. B 2010, 168, 193. 3. Dias, M. L.; Palermo, L. C.; Silvino, A. C.; Macromol. Symp. 2011, 299/300, 156. 4. Silvino, A. C.; Correa, P. S.; Dias, M. L.; J. Appl. Polym. Sci. 2014, 131, 40771. 5. Sauer, A.; Kapelski, A.; Fliedel, C.; Dagorne, S.; Kol, M.; Okuda, J.; Dalton Trans. 2013, 42, 9007. 6. Hormnirun, P.; Marshall, E. L.; Gibson, V. C.; Pugh, R. I.; White, A. J. P.; Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 15343. 7. Chisholm, M. H.; Eilerts, N. W.; Huffman, J. C.; Iyer, S. S.; Pacold, M.; Phomphrai, K.; J. Am. Chem. Soc. 2000, 122, 11845. 8. Agatemor, C.; Arnold, A. E.; Cross, E. D.; Decken, A.; Shaver, M. P.; J. Organometal. Chem. 2013, 745, 335. 9. Chen, C.; Hung, C.; Chang, Y.; Peng, K.; Chen, M.; J. Organometal. Chem. 2013, 738, 1. 10. Appavoo, D.; Omondi, B.; Guzei, I. A.; van Wyk, J. L.; Zinyemba, O.; Darkwa, J.; Polyhedron 2014, 69, 55. 11. Duan, R.; Gao, B.; Li, X.; Panga, X.; Wang, X.; Shao, H.; Chen, X.; Polymer 2015, 71, 1. 12. Kamber, N. E.; Jeong, W.; Waymouth, R. M.; Pratt, R. C., Lohmeijer, B. G. G.; Hedrick, J. L.; Chem. Rev. 2007, 107, 5813. 13. Kapelski, A.; Okuda, J.; J. Polym. Sci., Part A: Polym. Chem. 2013, 51, 4983. 14. Dechy-Cabaret, O.; Martin-Vaca, B.; Bourissou, D.; Chem. Rev. 2004, 104, 6147. 15. Thomas, C. M.; Chem. Soc. Rev. 2010, 39, 165. 16. Saunders, L. N.; Dawe, L. N.; Kozak, C. M.; J. Organomet. Chem. 2014, 749, 34. 17. Zhang, C.; Wang, Z.; J. Organomet. Chem. 2008, 693, 3151. 18. Di Lulio, C.; Middleton, M.; Kociok-Kohn, G.; Jones M. D.; Johnson, A. L.; Eur. J. Inorg. Chem. 2013, 9, 1541. 19. Saha, T. K.; Mandal, M.; Thunga, M.; Chakraborty, D.; Ramkumar, V.; Dalton Trans. 2013, 42, 10304. 20. Klatt, T.; Markiewicz, J. T.; Sämann, C.; Knochel, P.; J. Org. Chem. 21. Pappalardo, D.; Annunziata, L.; Pellecchia, C.; Macromolecules 2009, 42, 6056. 22. Oliveira, A. L. C.; Ferreira, L. C.; Dias, M. L.; Bitzer, R. S.; Nascimento, M. A. C.; Marques, M. F. V.; Crociani, L.; J. Braz. Chem. Soc. 2017, 29, 217. 23. Ghosh, S.; Chakraborty, D.; Varghese, B.; Eur. Polym. J. 2015, 62, 51. 24. Bhunora, S.; Mugo, J.; Luximon, A. B.; Mapolie, S.; Van Wyk, J.; Darkwa, J.; Nordlander, E.; Appl. Organomet. Chem. 2011, 25, 133. 25. Jones, M. D.; Davidson, M. G.; Keir, C. G.; Hughes, L. M.; Mahon, M. F.; Apperley, C.; Eur. J. Inorg. Chem. 2009, 5, 635. 26. Ghosh, S.; Chakraborty, D.; Ramkumar, V.; Polym. Chem. 2015, 53, 1474. 27. Honrado, M.; Otero, A.; Fernández-Baeza, J.; Sánchez-Barba, L. F.; Garcés, A.; Lara-Sánchez, A.; Martinez-Ferrer, J.; Sobrino, S.; Rodriguez, A. M.; Organometallics 2015, 34, 3196. 28. Fliedel, C.; Mameri, S.; Dagorne, S.; Aviles, T.; Appl. Organomet. Chem. 2014, 28, 504. 29. Kong, W. L.; Chai, Z. Y.; Wang, Z. X.; Dalton Trans. 2014, 43, 14470. 30. Miller, L. Z.; Shatruk, M.; McQuade, D. T.; Chem. Commun. 2014, 50, 8937. 31. Wei, Y.; Yingzhao, X.; Tiantian, L.; Aihong, G.; Yanfeng, W.; Chem. Res. Chin. Univ. 2014, 30, 87. 32. Sarasua, J. R.; Rodriguez, N. L.; Arraiza, A. L.; Meaurio, E.; Macromolecules. 2005, 38, 8362. 33. JAGUAR (Version 7.9), 2011, Schrodinger, LLC, New York, NY. 34. Becke, A.D.; J. Chem. Phys. 1993, 98, 5648. 35. Hay, P. J.; Wadt, W. R.; J. Chem. Phys. 1985, 82, 270. 36. Garcia-Valle, F. M.; Estivill, R.; Gallegos, C.; Cuenca, T.; Mosquera, M. E. G.; Tabernero, V.; Cano, J.; Organometallics 2015, 34, 477. 37. Saha, T. K.; Mandal, M.; Thunga, M.; Chakraborty, D.; Ramkumar, V.; Dalton Trans. 2013, 42, 10304. 38. Cheshmedzhieva, D.; Angelova, I.; Ilieva, S.; Georgiev, G. S.; Galabov, B.; Comput. Theor. Chem. 2012, 995, 8. 39. Cheng, M.; Attygalle, A. B.; Lobkovsky, E. B.; Coates, G. W.; J. Am. Chem. Soc. 1999, 121, 11583. 40. Chisholm, M. H.; Gallucci, J. C.; Zhen, H. S.; Inorg. Chem. 2001, 40, 5051. 41. Chamberlain, B. M.; Cheng, M.; Moore, D. R.; Ovitt, T. M.; Lobkovsky, E. B.; Coates, G. W.; J. Am. Chem. Soc. 2001, 123, 3229. 42. Huang, B. H.; Hsueh, M. L.; Lai, S. L.; Lin, C. C.; Polymer 2005, 46, 9784. 43. Golcu, A.; Tumer, M.; Demirelli, H.; Wheatley, R. A.; Inorg. Chim. Acta 2005, 358, 1785. 44. Silvino, A. C.; Martins, D. B. A. T.; Rodrigues, A. C.; Dias, M. L.; J. Polym. Environ. 2013, 21, 1002. 45. Wang, X.; Liao, K.; Quan, D.; Wu, Q.; Macromolecules 2005, 38, 4611. 46. Suganuma, K.; Horiuchi, K.; Matsuda, H.; Cheng, H. N.; Aoki, A.; Asakura, T.; Macromolecules 2011, 44, 9247. 47. Suganuma, K.; Horiuchi, K.; Matsuda, H.; Cheng, H. N.; Aoki, A.; Asakura, T.; Polym. J. 2012, 44, 838. 48. Suganuma, K.; Matsuda, H.; Cheng, H. N.; Iwai, M.; Nonokawa, R.; Asakura, T.; Polym. Test. 2014, 38, 35. 49. Souza, D. H. S.; Borges, S. V.; Dias, M. L.; Andrade, C. T.; Polym. Compos. 2012, 33, 555. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access