
 |
 |
 |
 |
 |
Artigo
| Characterization of silicon-aluminum-zirconium oxide obtained by the sol-gel process |
|
Cristiano Nunes da SilvaI; José Márcio Siqueira JúniorII; Amanda Garcez VeigaI; Germildo Juvenal MuchaveIII; Donato Alexandre Gomes ArandaIII; Júlio Carlos AfonsoI; Emerson Schwingel RibeiroI,*
I. Instituto de Química, Universidade Federal do Rio de Janeiro, 21941-909 Rio de Janeiro - RJ, Brasil Recebido em 25/02/2019 *e-mail: emersonsr@iq.ufrj.br A new silicon-aluminum-zirconium ternary oxide (SiO2/Al2O3/ZrO2) was prepared by the sol-gel method followed by one-step washing with absolute ethanol or two-step washing with absolute ethanol followed by ultra-pure water. Both ternary oxides presented high surface area (over 340 m2 g-1). The binding energy values for Zr3d and Al2p levels showed the insertion of Zr and Al atoms in silica matrix, confirming the dispersion surface of these elements as shown by scanning electron microscope (SEM) and energy dispersive X-ray fluorescence spectroscopy (EDXRF) data. A weight loss around 40 wt.% was found for both samples submitted to thermogravimetric analysis (TGA). Crystallographic data showed the presence of monoclinic and tetragonal ZrO2 phases in different proportions according to the calcination temperature. Regarding the acid-base properties, temperature-programmed desorption of CO2 (CO2-TPD) profiles suggested that washing with water reduced the overall basicity of the samples and removed the strongest basic sites. However, according to temperature-programmed desorption of NH3 (NH3-TPD) profiles, washing significantly increased the amount of weak acid sites, in agreement with pyridine adsorption data. Bronsted acid sites (BAS) are ever dominant but washing with ethanol followed by water reduced the number of Lewis acid sites (LAS). INTRODUCTION The first studies on the sol-gel process are assigned to Ebelman and Graham, who underwent hydrolysis of tetraethyl orthosilicate (TEOS; Si(OC2H5)4) in acidic conditions. Silica was obtained as a material similar to glass.1,2 The products obtained by this method present high purity and homogeneity, besides being obtained at much lower processing temperatures than those used in other traditional procedures.3,4 In recent years, several ternary oxides (SiO2/MxOy/NxOy) have been synthesized by the sol-gel process.5-9 For example, they show potential applications as molecular adsorbents8, metal ion preconcentration systems,9 photocatalysis,10 electrocatalysis11 and dye adsorption from effluents.12 Oxides such as SiO2, Al2O3 and ZrO2 are important on an industrial scale. Their surface properties play a fundamental role for many applications.13 Silica (SiO2) is widely used in manufacture of glass,14 in the production of electronic components15 and also in civil construction, either as a grit or as a component of Portland cement.16 It may be present in various crystalline forms: α-quartz, β-quartz, α-tridymite, β-tridymite, α-cristobalite, β-cristobalite, keatite, moganite, coesite, stishovite, seifertite, melanophlogite and fibrous silica.17,18 Silica is suitable as a catalyst support in catalytic reforming19 because of its high tolerance to sulfur poisoning and its high surface area. Its surface acidity is due to a lower water content that is restricted to the inside pores of the material. However, its thermal stability is lower compared to alumina.19 Alumina (Al2O3) usually presents a high specific surface area and is thermally very stable. This oxide may exist in several crystalline forms, such as gibbsite (γ-Al(OH)3), bayerite (α-Al(OH)3), boehmite (γ-AlO(OH)), diaspore (α-AlO(OH)) and corundum (α-Al2O3).20 Gamma-alumina (γ-Al2O3) and delta-alumina (δ-Al2O3) are widely employed as catalyst supports for reforming reactions19 and hydrodesulfurization (HDS).21,22 The main use of aluminum oxide on an industrial scale is in the production of metallic aluminum through the Hall-Héroult process.23,24 Zirconia (ZrO2) presents high strength, fracture toughness, chemical compatibility and ionic conductivity.25 Three phases of zirconia are stable at atmospheric pressure: cubic, tetragonal and monoclinic.26 This is widely used commercially, from the manufacture of laser mirrors, broadband interference filters, ionic conductors, components coatings of architectural glass, the coating of some types of exterior service paints and for the manufacture of jewelry of low-cost.27,28 SiO2, Al2O3 and ZrO2 present distinct acid-base behaviors.29,30 Silica is generally considered acidic, zirconia is relatively basic30 while alumina is an amphoteric substance.31 Reactions between these oxides lead to the formation of compounds with intermediate basicities. It is of interest that the metal oxide dispersed in the silica matrix may exhibit different chemical and physical properties from those observed in the oxide alone.32 Accordingly, mixed oxides are catalytically interesting substrates because their species have acid sites of different types, especially those of Brønsted present on silica and those of Lewis on the other oxides.33 In this work, we prepared SiO2/Al2O3/ZrO2 samples by the sol-gel process and purified them using two distinct washing procedures. A set of structural, textural and chemical analyzes were performed to characterize in detail these samples in view of a future application as an absorbent of anions and metal ions from effluents and/or as a heterogeneous catalyst, for example.
EXPERIMENTAL Materials Tetraethylorthosilicate (TEOS, 98%), zirconium(IV) butoxide solution (80 wt. % in 1-butanol) and aluminum-tri-sec-butoxide (97%) were acquired from Sigma-Aldrich (Saint Louis, MO, USA). Absolute ethanol (99.8%), HCl (37% v/v) and HNO3 (95% v/v) were purchased from Vetec (Duque de Caxias, RJ, Brazil). Ultrapure water (resistivity >18.2 MΩ cm-1 at 298 K) was obtained using a Millipore© Milli-Q© purification system. Synthesis of the SiAlZr ternary oxides SiO2/Al2O3/ZrO2 samples were prepared according to the following procedure: 15.0 mL of 3.5 mol L-1 HCl were added to 230.0 mL of ethanol/TEOS (50% v/v). The mixture was stirred for 3 h at 343 K to initiate prehydrolysis. In the next step, 15.0 mL of aluminum tri-sec-butoxide were added dropwise to the mixture, followed by 3.5 mol L-1 HCl. The mixture was stirred for 2 h at 343 K. Then, 62.0 mL of zirconium(IV) butoxide solution were added dropwise, followed by 15.0 mL of 3.5 mol L-1 HCl. The reaction mixture was stirred for 20 h at 353 K. The solvent was slowly evaporated at 373 K until formation of a gel. This gel was subsequently heated in an oven at 383 K for 4 h, milled and dried under vacuum (10-5 Pa) at 363 K. Two different washing procedures were tested. In Procedure A, the solid (SiAlZr-A) was washed with absolute ethanol in a Soxhlet apparatus for 24 h. In Procedure B (SiAlZr-B), washing was performed in two steps: i) with absolute ethanol; ii) with ultra-pure water. Both steps were carried out in a Soxhlet apparatus for 24 h. In both routes the final solid was washed with 100.0 mL of 0.1 mol L-1 HNO3, (10 mL g-1), absolute ethanol, ultra-pure water and absolute ethanol again. The washed solids were dried under vacuum (10-5 Pa) for 6 h at 363 K and stored in a desiccator. Characterization The crystallinity of the thermally treated powders was analyzed by powder x-ray diffraction (XRD). Samples were previously heated at 473, 673, 873, 1073 or 1273 K for 8 h (283 K min-1). A Bruker AXS D8 Advance instrument (Cu Kα1, α2 radiation, 40 kV, 40 mA) operating in a Bragg-Brentano θ/θ configuration was used for this purpose. The diffraction patterns were collected in a flat geometry with steps of 0.02° and accumulation time of 1 s per step. The X-ray powder diffraction data were refined following the Rietveld method with the TOPAS Academic V.5®. In order to verify the stability of the oxides in the silica network, we studied the thermal behavior of the samples at various temperatures. The composition of ternary oxides was determined by energy dispersive X-ray fluorescence spectroscopy (EDXRF) on a Shimadzu EDX 800 HS. The objective was to evaluate the efficiency of proposed synthetic routes using the sol-gel process in this work. Samples were pressed using boric acid as the substrate and were analyzed under He with an X-ray source operating at 5-50 kV according to the measured region. The specific surface area (SBET) was determined on a Micromeritics Model TriStar II instrument using the BET (Brauner, Emmett and Teller) multipoint method. Samples were previously activated at 393 K under vacuum for 24 h. The BJH (Barrett-Joyner-Halenda) method was used to obtain the average pore size and volume in the mesoporous region.34 Thermogravimetric analysis (TGA) (293-1173 K, 5 K min-1) was performed on a Shimadzu DTG-60-instrument under argon flow (50 mL min-1). Scanning electron micrographs (SEM) were acquired using a Quanta 400 scanning electron microscope (FEI Company). The maximum operating voltage was 10 kV, according to the analyzed sample (magnification factor 550x). Samples were dispersed on a double-sided conductive tape. Electron dispersive spectroscopy (EDS) analyses were run on an INCA PentaFETx3 (Oxford Instruments) Energy Dispersive X-ray Spectrometer linked to the SEM equipment. X-ray photoelectron spectroscopy (XPS) spectra were carried out using a Thermo Scientific Escalab 250Xi spectrometer. The pressure inside the vacuum chamber was kept below 5 x 10-9 mbar. An Al Kα X-ray monochromatized radiation (hν = 1486.6 eV) was used as X-ray source and operated at 216 W. The pass energy of the spectrometer was 100 eV and 25 eV for XPS survey and core level spectra, respectively. C1s (284.8 eV) was used for binding energy calibration. The data acquisition and peak fitting was performed using the Thermo Avantage© software. The determination of the value of optical band gap of the thermally treated samples was performed through the Kubelka-Munk function (F(R)) of data interpretation from the diffuse reflectance spectroscopy (DRS) obtained in a Cary 5000 Varian UV-Vis-NIR Spectrophotometer, with wavelengths between 190-950 nm and barium sulfate as reference. Acid-base properties were measured through temperature-programmed desorption of CO2 (CO2-TPD) and NH3 (NH3-TPD). This technique allows the differentiation between the sites in relation to the basic/acidic strength. CO2-TPD and NH3-TPD were performed on a QMS-200 mass spectrometer (Balzers) using the m/z ratios 15 and 18 for quantification of carbon dioxide and ammonia, respectively. The sample pretreatment procedure consisted of heating at 423 K with a rate of 283 K min-1 and remaining for 30 min. After pretreatment, the assay procedure consisted of heating the samples to 1073 and 1273 K at 293 K min-1 for both desorption procedures. In both TPD procedures, the dissolved gases in the ternary oxides were quantified by integrating the area under the curve intensity versus time. Pyridine was also used as a probe molecule to detect Lewis and Brønsted acidic sites in the samples. This characterization allows identification of the types of acidic sites present in the samples (Brønsted, Lewis, hydrogen bonding and silanols).35 Drops of pyridine were added to the samples, after which they were dried under vacuum at two different temperatures (298.15 and 423.15 K). After drying, the samples were analyzed by FTIR spectroscopy using KBr pellets containing 12 wt.% sample.
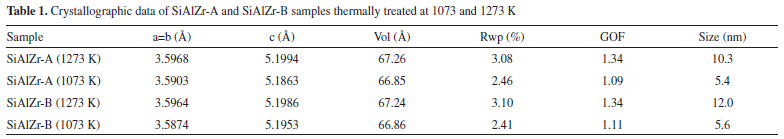
RESULTS AND DISCUSSION Chemical composition The theoretical weight percent of SiO2, Al2O3 and ZrO2 in both SiAlZr-A and SiAlZr-B materials are 60.0, 10.0 and 30.0 wt.%, respectively. However, EDXRF data are not in full agreement with the expected values. The SiAlZr-A material contains 34.9 wt.% of ZrO2 and 4.7 wt.% of Al2O3, while the SiAlZr-B material contains 33.2 wt.% of ZrO2 and 4.8 wt.% of Al2O3. Chlorine was also found in both samples: 9.6 wt.% on SiAlZr-A and 8.2 wt.% on SiAlZr-B. The content decreased ~15 wt.% after washing with ultra-pure water using a Soxhlet apparatus for 24 h. It is possible that some chlorine is inaccessible to ethanol or water during washing under our experimental conditions. The presence of chlorine in ternary oxides is attributed to hydrochloric acid, employed as catalyst during their preparation. Regarding the synthesis, the products obtained showed a significantly lower incorporation of aluminum in the materials after the purification, probably due to the pH of the reaction medium, which was not suitable for the full hydrolysis of the aluminum precursor (aluminum-tri-sec-butoxide),36 resulting in a higher zirconium content in both materials. It must be emphasized that it is extremely difficult to obtain ternary oxides in the expected weight proportions due to the differences between the hydrolysis kinetics of the precursors in the same reaction medium. Nevertheless, the sol-gel process has been recognized as an extremely useful and reproducible method for the synthesis of this material.37 Only samples calcined at 1273 and 1073 K showed crystalline phases sensitive to XRD. Samples calcined to 873 K and the samples without heat treatment exhibited only a halo-amorphous peak of silica, evidencing the high thermal stability of these materials. No crystalline phase containing aluminum was observed. The crystallographic data of these samples were evaluated using values of unit cell parameters, crystallite size and merit figures of Rietveld refinements, which are summarized in Table 1.
The data shown in Table 1 and Figure 1 are consistent with the ZrO2 (zirconia - HT) tetragonal crystal system of the space group 137 (P42/nmcZ), inorganic crystal structure database ICSD#66781.38 The ZrO2 polymorph indexed as monoclinal phase of baddeleyite phase (9.9%) was observed in SiAlZr-A samples treated at 1073 K. When this sample was treated at 1273 K, the content of the baddeleyite phase decreased to 8.7%. The badeleyite content in SiAlZr-B samples treated at 1073 K was 7.1% and only 3.0% for those treated at 1273 K. The baddeleyite phase is a monoclinic crystal system of space group 14 (P121/c1), as described in the inorganic crystal structure database ICSD #18190.39
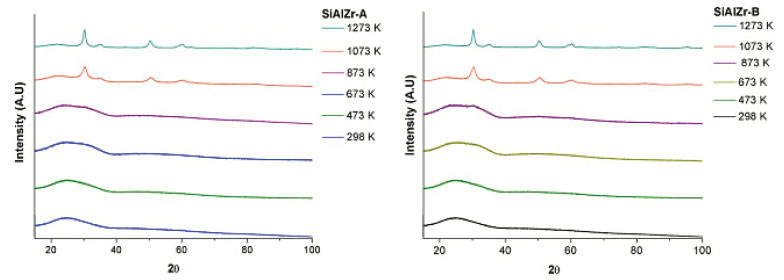 Figure 1. XRD for non-calcined and heated (473-1273 K) SiAlZr-A and SiAlZr-B samples
Textural properties BET specific surface area (SBET) was 349 m2 g-1 for SiAlZr-A and 376 m2 g-1 for SiAlZr-B. As both materials present a high specific surface area, they are eligible as adsorbents of metal ions, electrochemical sensors or heterogeneous catalysts.7 The mean average pore size is 33.4 Å for both materials, according to the Brunauer-Emmett-Teller (BJH) method, indicating that these ternary oxides are predominantly mesoporous;40 the mean pore volume was 0.021 cm-3 g-1 for SiAlZr-A and 0.018 cm-3 g-1 for SiAlZr-B, thus correlating with their surface area. These data suggest a good accessibility to the active adsorption sites and are comparable to those reported in the literature for other mixed oxides prepared by the sol-gel process.1,41-43 Only one type of adsorption isotherm was found. Both oxides presented type IV(b) isotherms. This is typical of mesoporous materials, according to the 2015 IUPAC classification.44,45 Pore volume distribution in the mesopore zone determined by BJH method34 shows the existence of a large population of mesopores centered around 20 and 40 nm for both samples. Thermal stability Thermogravimetric analysis (TGA) data of the ternary oxides showed a somewhat distinct behavior. The SiAlZr-A oxide exhibits a total weight loss of approximately 44%, while the SiAlZr-B sample lost 35%. In relation to the SiAlZr-A material, a first weight loss zone (approximately 15%) up to 438 K corresponds to loss of water from hydrated oxides.46,47 It is followed by an endothermic peak at 348 K. The second weight loss zone (about 25%) occurred at 438-738 K and was accompanied by an endothermic peak at 473 K. This region corresponds to the structural water of the silanol groups (Si-OH) or similar ones (-Al-OH, -Zr-OH),47 -Si-O-Si- (siloxane groups)47 and/or their corresponding analogues (-Si-O-Al-, -Si-O-Zr- and -Al-O-Zr-). The third weight loss was approximately 4% and occurred between 738 and 1173 K, corroborating with the high thermal stability observed by XRD. It was followed by an exothermic peak at 783 K, which may be ascribed to organic matter aggregated or encapsulated in the pores of the sample.48 For the SiAlZr-B material, a weight loss up to 438 K (weight of mass of 17%) is due to loss of water from hydrated oxides.46,47 It is accompanied by an endothermic peak at 338 K. The second weight loss (about 11%) occurred at 438-738 K and was followed by an endothermic peak at 473 K, being similar to the SiAlZr-A sample. The third weight loss was approximately 7% and occurred between 738 and 1173 K, again comparable to SiAlZr-A sample. Surface properties Figures 2(a) and 2(b) shows the scanning electron micrographs (SEM) and their respective element mapping images (Si, Al, Zr, Cl and O) made by EDS. The particles do not present uniform size and regular spherical shapes, being characterized by a flat surface with rough structure. This result points to a very porous material, as expected from the textural data presented earlier. Elemental mapping by EDS shows that the metal oxide particles are dispersed without phase segregation in the silica matrix. Moreover, at the magnification levels used (550 x), the ternary oxides are very homogeneous, thus suggesting a high uniform distribution of the metal oxides on their surfaces. This dispersion is very important because it tends to increase the number of acidic sites on the surface or pores of the SiAlZr ternary oxides. It can be attributed to strong interactions of other metals with the siloxane groups of the silica matrix, forming strong covalent bonds (for example -Si-OH, -Al-OH, Si-O-Zr-OH).7 Table 2 shows the weight ratio on the surface of the samples, calculated using the EDS point spectra. The SiAlZr-B sample has a slightly higher amount of zirconium than the SiAlZr-A, according to XPS data, however, in disagreement with EDXRF results. Probably, the different amounts of elements on the surface of materials by the EDS e XPS techniques (surface analysis), regarding of the EDXRF technique (bulk analysis), can be associated with the washing process, leading to removal of species that were not covalently bound to the silica matrix.
 Figure 2. Scanning electron micrograph of SiAlZr-A (a) and SiAlZr-B (b) ternary oxides. Magnification of 550x
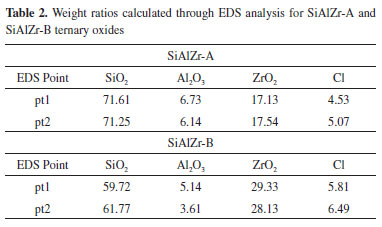
From XPS curves, all the expected elements were found in the samples, including chlorine. Table 3 reports the atomic percentage of each element in the ternary oxides. It should be noted that the washing method in two steps (Procedure B) is removed more chlorine because its amount in sample B was lower. Moreover, its aluminum content is about three times lower than in the sample A. This difference can be attributed to the removal of some aluminum during the washing process. As the EDXRF (bulk analysis) data show that both samples present similar Al2O3 contents, the different amounts of this element on the surface can be associated with the washing process as observed by the XPS (surface analysis).
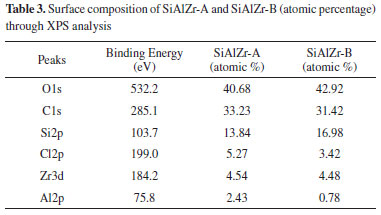
High resolution spectra of Al2p, Si2p and Zr3d for SiAlZr-A and SiAlZr-B are presented on Figure 3. As already observed by EDS, there is a mixture of Al2O3, SiO2 and ZrO2 oxides. The presence of chlorine in both samples is associated with the crystalline array of all oxides. After washing with water (Procedure B), this array did not change for silicon and aluminum oxides, however, a significant decrease in the Zr3d3/2 peak at 184.8 eV, attributable to a chlorinated zirconium oxide suggests the removed chlorine was only associated with zirconium oxide. High resolution Si2p spectrum has a profile attributed to the Si-C groups, thus suggesting the presence of a tetraethylorthosilicate residue in both ternary oxides. Therefore, the synthesis process was not fully effective in removing the organic part of the silicon precursor. Table 4 shows the relative proportion of each element on the surface.
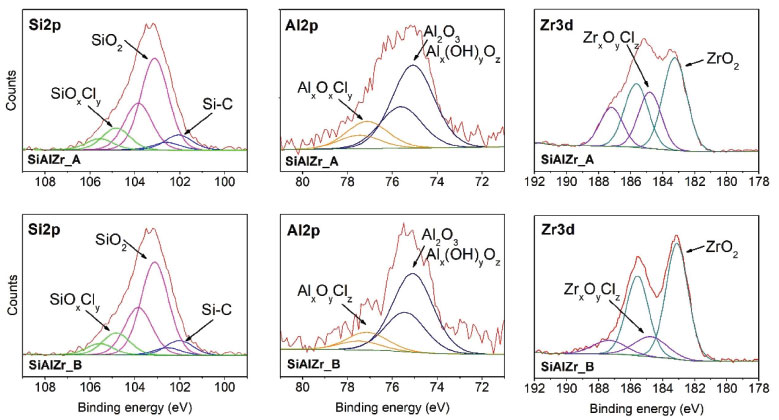 Figure 3. High resolution spectra of Si2p, Al2p and Zr3d core levels for both ternary oxides
Diffuse reflectance spectroscopy (DRS) is used to describe the absorption property for the obtained oxides. Since only the samples treated from the 1073 K temperature showed crystalline phases observable in the powder X ray diffractogram, only the diffuse reflectance UV VIS spectroscopy data for SiAlZr-A and SiAlZr-B samples treated at 873, 1073 and 1273 K will be discussed. The optical band gap was obtained using the Kubelka-Munk function [F(R)] from the diffuse reflectance data of the heat treated SiAlZr-A and SiAlZr-B (Figure 4). The optical band gap values for SiAlZr-A and SiAlZr-B are shown in Table 5.
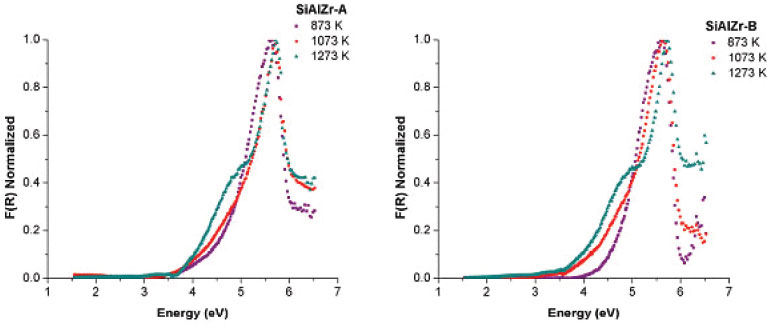 Figure 4. DRS [Kubelka-Munk function, [F(R)] Normalized] of the SiAlZr-A and SiAlZr-B samples treated at 873, 1073 and 1273 K
The values of the band gap (Eg) were obtained by extrapolating the linear section of the graphics to the abscissa axis.7 The crystalline structure plays an important role in the electronic structure of ZrO2. It was also been reported that the pure monoclinic ZrO2 particle showed energy band gap of 3.5 eV.49 Navio et al.50 have reported band gaps of 3.7 eV for monoclinic ZrO2 powders prepared by the sol-gel method. The differences among the band gap values shown in Table 5 can be attributed to a structural disorder resulting from the experimental conditions used in preparation and the heating of the samples.50 Absorptions in the range of 5 to 3.5 eV were attributed to O2- → Zr4+ in addition to the charge transfer in SiO2 and Al2O3 matrices.51 The crystal structure of ZrO2 markedly influences the d-electron-derived conduction band (CBs). The monoclinic ZrO2 displayed hybridization of Zr 4d CBs in a new single Zr 4d CB.51 Regarding the materials spectra at room temperature, the vibrational frequencies relative to pure silica (SiO2) are easily identifiable from the FTIR spectra (figure not shown). A broadband (3700-3200 cm-1) present in all ternary oxides is due to OH axial deformation in Si-OH groups (νO-H) involving hydrogen bonding. The band at 1630-1640 cm-1 corresponds to the vibrational mode deformation of O-H bonds and the band at 1680 cm-1 is due to adsorbed water molecules (δO-H). The siloxane groups (Si-O-Si) are characterized by a band in the 1130-1000 cm-1 region due to axial deformation of this group (νSi-O-Si). The band in the 950-830 cm-1 region is attributed to the stretching of the Si-OH bond (νSi-OH). The band around 799 cm-1 represents the stretching mode of Si-O-Si linkage (νSi-O-Si). Another characteristic silica region is found between 1193-1071 cm-1, corresponding to the axial vibration of the Si-O group (νSi-O). The angular deformation Si-O-Si bond (δSi-O-Si) appears at around 460 cm-1.52-55 Acid-base properties TPD desorption profiles of the CO2-TPD curves on the surface of the ternary oxides examined are shown in Figure 5(a). In the SiAlZr-A sample the CO2 desorption started at approximately 323 K, with three peaks centered at 402.5, 553.0 and 627.6 K. They are relative to weak basic sites. Another peak centered at 1085.9 K is relative to stronger basic sites. The SiAlZr-B sample also showed three peaks centered at 377.4, 410.1 and 539.6 K, but the peaks related to strong basic sites are absent. Table 6 describes the total number of basic sites (weak and strong) and the corresponding maximum temperatures. Washing with ultra-pure water reduced the overall basicity of the ternary oxides, as shown by the reduction of the total CO2 uptake by approximately 50% and the removal of strong basic sites.
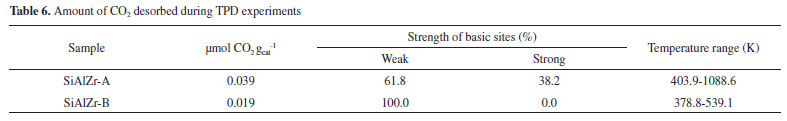
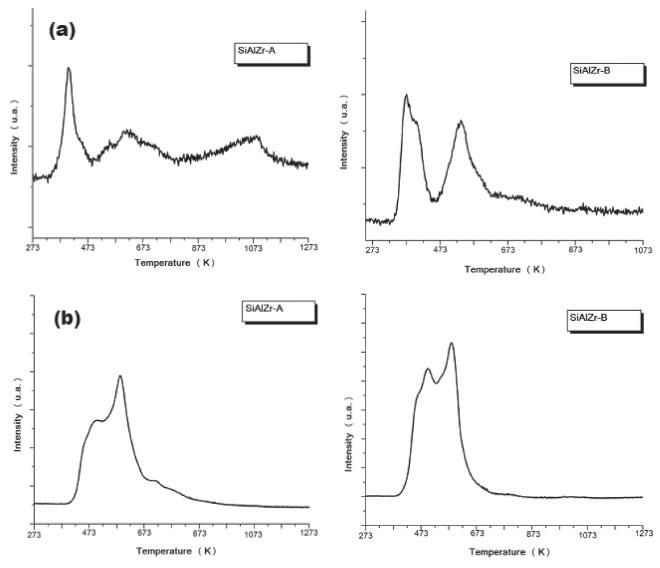 Figure 5. Profile of temperature-programmed desorption of CO2 (above) and NH3 (below) adsorbed of SiAlZr-A (left) and SiAlZr-B (right) ternary oxides
Figure 5(b) shows the TPD desorption profiles of the NH3-TPD curves of the ternary oxides. In the SiAlZr-A sample, the ammonia desorption started at approximately 423 K, with two peaks centered at 495.2 and 585.8 K, both related to weak acidic sites. The SiAlZr-B sample also showed two peaks centered at 497.4 and 583.6 K. The ternary oxides prepared in this work present only weak acidic sites. The acidic strength distribution appears to be quite similar, although the amount of acidic sites increased considerably after washing with water. The amount of ammonia desorbed during TPD experiments was 0.039 µmols NH3 gcat-1 for SiAlZr-A and 2.131 µmols NH3 gcat-1 for SiAlZr-B, both in the temperature range 495.2-585.8 K. The NH3-TPD profile (Figure 5(b)) of the ternary oxides shows that washing significantly increased by about 25% the amount of weak acidic sites on the surface of SiAlZr-B compared to SiAlZr-A probably through the formation of Al-OH and Si-OH species. Ammonia is a smaller molecule than carbon dioxide, which favors access to the micropores.56-59 Moreover, ammonia basicity is stronger than acidity of carbon dioxide.56-59 Adsorption may via both NH3 and NH4+ forms, but it is not possible to distinguish Lewis (LAS) and Brønsted (BAS) acidic sites in a NH3-TPD profile.59 FTIR after pyridine adsorption (Figure 6) is compared to the spectra of pyridine-free adsorbed materials on the surface of each material. Spectra are limited in the wavelength range of 1700-1400 cm-1, where vibrational modes of pyridine are normally observed.7,60
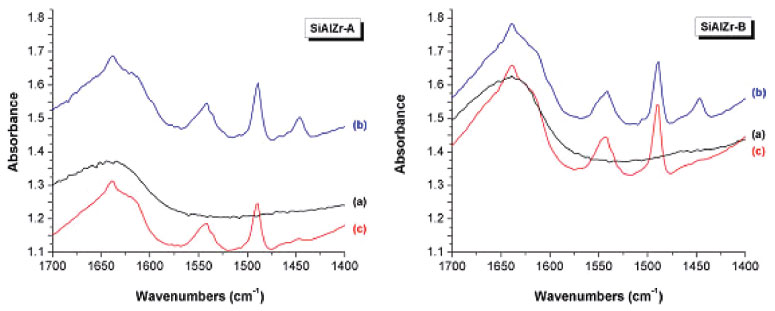 Figure 6. Infrared spectra of pyridine adsorbed on SiAlZr-A and SiAlZr-B ternary oxides at different temperatures under vacuum: (a) pure SiAlZr, (b) 298.15 K, (c) 423 K
Brønsted acidic sites are due to the -ZrOH or -AlOH groups present on the surface of the silica matrix.61-63 In contrast, coordinately unsaturated zirconium or aluminum ions are responsible for the presence of Lewis acidic sites.7,64,65 All spectra exhibited typical bands of Brønsted and Lewis type surface acidic sites, but with different intensities, as observed by the EDXRF and EDS data. SiAlZr-A has a higher percentage of ZrO2 and almost the same amount of Al2O3, thus explaining its FTIR spectrum (more intense bands in the presence of immobilized pyridine, corroborating with EDXRF and EDS results). The SiAlZr-A sample presents a higher number of acidic sites on the surface than the SiAlZr-B one (Table 6), leading to a greater pyridine adsorption. The band at about 1450 cm-1 indicates the presence of Lewis acidic sites (LAS). It refers to the 19b vibration mode of pyridine, indicating the onset of the molecularly coordinated C-C stretch of pyridine.64 This band is more intense for the SiAlZr-A sample, but it shows low interaction with the ternary oxide in both samples. It disappeared with increasing temperature. The band around 1541 cm-1 indicates the presence of Brønsted acidic sites (BAS) due to the 8a vibration mode of liquid pyridine adsorbed by van der Waals forces.66-68 A band around 1639 cm-1 was found in all samples, which is generally attributed to the physically adsorbed pyridine by hydrogen bonding to the hydroxyl groups on the oxide surface.69,70 The 1490 cm-1 band describes a mixture of the two types of acidic sites.35,56,69,71-73 After heating, the samples shows mainly Brønsted acidic sites. There is a progressive reduction of the bands corresponding to Lewis and Brønsted sites after release of pyridine at 423 K. Concentration of BAS and LAS expressed in milligrams of pyridine (Py) adsorbed per gram of material (mg Py g-1), were determined by the integration of the peaks at 1541 and 1447 cm-1 related to the same vibrational mode 19b of pyridine adsorbed at the Brønsted and Lewis sites, respectively, of the spectra collected after the release of pyridine at 423 K68 according to the procedure described by Emeis.60,74 The results are summarized in Table 7. The SiAlZr-A material presents about 1.38 times more LAS than SiAlZr-B. On the other hand, SiAlZr-B has about 1.37 times more BAS than SiAlZr-A.
It can be argued that the nature of the weak sites predicted by NH3-TPD is a mixture of Lewis and Brønsted acidic sites and physisorption for hydrogen bonding. The presence of the Lewis and Brønsted acidic sites on the surface of the ternary oxide allows the absorption of electroactive species, for example. Too, this feature is very interesting in the area of catalysis.75
CONCLUSIONS Synthesis of SiAlZr materials by the sol-gel method proved efficient. Although the composition of these ternary oxides did not exactly match the expected one, the zirconium and aluminum contents are still relatively high. In addition, they displayed high specific surface areas (SBET). According to XPS and SEM-EDS data, the synthesized materials are homogeneous, show no phase segregation and suggest insertion of aluminum and zirconium in the silica matrix. These materials present good thermal stability according to TGA-DTA data. Crystalline phases in the ternary oxides were found only after heating at 1073 K, through XRD. CO2-TPD showed the presence of weak and strong basic sites for the SiAlZr-A material and only weak basic ones for the SiAlZr-B sample. NH3-TPD pointed to the presence of weak acid sites only, but pyridine adsorption detected both Lewis (LAS) and Brønsted (BAS) acidic sites. The detailed characterization of SiAlZr ternary oxides showed that these samples are promising for testing as electrochemical sensors, adsorbent for metal ions in effluents, dye removal, heterogeneous catalysis, electrocatalysis, photocatalysis, among others.
ACKNOWLEDGMENTS The authors thank to Fundaçao de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ), Coordenaçao de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and Agência Nacional do Petróleo, Gás Natural e Biocombustíveis (ANP) for financial support. The authors thank to NUCAT ((Nucleus of Catalysis of the Chemical Engineering Program of PEQ-COPPE-UFRJ) for MEV-EDS analysis and Institute of Nuclear Engineering (IEN) for EDXRF analysis.
SUPPLEMENTARY MATERIAL Figure 1S presents the nitrogen adsorption-desorption isotherm (above) and BJH pore volume distribution plot (below) of SiAlZr-A (a and c) and SiAlZr-B (b and d) ternary oxides. Figure 2S presents thermogravimetric analysis of SiAlZr-A and SiAlZr-B ternary oxides: DTA data (dashed lines) and relative weight loss (full lines). Figure 3S presents typical XPS survey spectra for SiAlZr ternary oxides. All figures are available for download at http://quimicanova.sbq.org.br in pdf format with free access.
REFERENCES 1. Graham, T.; J. Chem. Soc. 1864, 17, 318. 2. Ebelmen, M.; Ann. Chim. Phys. 1846, 16, 129. 3. Brinker, C. J.; Scherer, G. W.; Sol-Gel Science, Academic Press: San Diego, 1990. 4. Walcarius, A.; Electroanalysis 1999, 10, 1217. 5. Bouchmella, K.; Mutin, P. H.; Stoyanova, P. M.; Eloy, C.; P.; Rodemerck, U.; Gaigneaux, E. M.; Debecker, D. P.; J. Catal. 2013, 301, 233. 6. Sava, B. A.; Diaconu, A.; Elisa, M.; Grigorescu, C. E. A.; Vasiliu, I. C.; Manea, A.; Superlattices Microstruct. 2007 42, 314. 7. da Fonseca, B. T.; D’Elia, E.; Junior, J. M. S.; de Oliveira, S. M.; dos Santos Castro K. L.; Ribeiro, E. S.; J. Mater. Sci.: Mater. Electron. 2018, 29, 2159. 8. Ribeiro, M. S.; de Vasconcellos Jr., F. J.; da Fonseca, B. T.; de Souza, F. C.; Soares, F. D. R.; Lima, E. C.; Cabral, M. F.; Ribeiro, E. S.; D’Elia, E.; Anal. Methods 2014, 6, 521. 9. Diniz, K. M.; Gorla, F. A.; Ribeiro, E. S.; do Nascimento, M. B. O.; Correa, R. J.; Cesar Tarley R. T.; Segatellia, M. G.; Chem. Eng. J. 2014, 239, 233. 10. Jung, K. Y.; Park, S. B.; and Ihm, S.-K.; Appl. Catal., B 2004, 51, 239. 11. Ardizzone, S.; Bianchi, C. L.; Cappelletti, G.; Ionita, M.; Minguzzi, A.; Rondinini, S.; Vertova, A.; J. Electroanal. Chem. 2006, 589, 160. 12. Pal, U.; Sandoval, A.; Madrid, S. I. U.; Corro, G.; Sharma, V.; Mohanty, P.; Chemosphere 2016, 163, 142. 13. Pauly, N.; and Tougaard, S.; Surf. Sci. 2008, 602, 1974. 14. Bansal, N. P.; Doremus, R. H.; Handbook of Glass Properties, Academic Press: San Diego, 1986. 15. Kawachi, M.; Opt. Quant. Electron. 1990, 22, 391. 16. Feldman, R. F.; Cheng-Yi, H.; Cem. Concr. Res. 1985, 15, 765. 17. Wiberg, E.; Wiberg, N.; Holleman, A. F.; Inorganic Chemistry, Academic Press: San Diego, 2001. 18. Greenwood, N. N.; Earnshaw, A.; Chemistry of the elements, 2nd ed., Oxford: Boston, 1997. 19. Karatzas, X.; Jansson, K.; Gonzalez, A.; Dawody, J.; Pettersson, L. J.; Appl. Catal., B 2011, 106, 476. 20. Hudson, L. K.; Misra, C.; Perrotta, A. J.; Wefers, K.; Williams, F. S. In Ullmann’s Encyclopedia of Industrial Chemistry, John Wiley & Sons, Inc.: Hoboken, 2000, vol. 2, 607. 21. Vissers, J. P. R.; Scheffer, B.; de Beer, V. H. J.; Moulijn, J. A.; Prins, R.; J. Catal. 1987, 105, 277. 22. Topsoe, N.-Y.; Topsoe, H.; J. Catal. 1982, 75, 354. 23. Hind, A. R.; Bhargava, S. K.; and Grocott, S. C.; Colloids Surf., A 1999, 146, 1, 359. 24. U. S. G. Survey, Mineral commodity summaries 2018: https://minerals.usgs.gov/minerals/pubs/mcs/2018/mcs2018.pdf, accessed May 2019. 25. Nogami, M.; J. Non-Cryst. Solids 1994, 178, 320. 26. Nielsen, R. H.; Wilfing, G. In Ullmann’s Encyclopedia of Industrial Chemistry, John Wiley & Sons, Inc.: Hoboken, 2010, 39, 753. 27. Nielsen, R. H.; Schlewitz, J. H.; Nielsen, H. In Kirk - Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, Inc.: Hoboken, 2007, 26, 621. 28. Drese, R. J.; Wuttig, M.; J. Appl. Phys. 2006, 99, 123517. 29. Nagarajan, V. S.; Rao, K. J.; J. Solid State Chem. 1990, 88, 419. 30. Damodaran, K. V.; Nagarajan, V. S.; Rao, K. J.; J. Non-Cryst. Solids 1990, 124, 233. 31. Milliken, T. H.; Mills, G. A.; Oblad, A. G.; Discuss. Faraday Soc. 1950, 8, 279. 32. da Silva, E. L.; Budziak, D.; and Carasek, E.; Anal. Lett. 2004, 37, 1909. 33. de Farias, R. F.; Arnold, U.; Martı́nez, L.; Schuchardt, U.; Jannini, M. J. D. M.; Airoldi, C.; J. Phys. Chem. Solids 2003, 64, 2385. 34. Barrett, E. P.; Joyner, L. G.; Halenda, P. P.; J. Am. Chem. Soc. 1951, 73, 373. 35. Topaloǧlu Yazıcı D.; Bilgic, C.; Surf. Interface Anal. 2010, 42, 959. 36. Moran-Pineda, M.; Castillo, S.; Lopez, T.; Gomez, R.; Cordero, B.; Novaro, O.; Appl. Catal., B 1999, 21, 79. 37. Brinker, C. J.; Scherer, G. W.; Sol-gel science: the physics and chemistry of sol-gel processing, Academic Press: Boston, 1990. 38. Bondars, B.; Heidemane, G.; Grabis, J.; Laschke, K.; Boysen, H.; Schneider, J.; Frey, F.; J. Mater. Sci. 1995, 30, 1621. 39. Smith, D. K.; Newkirk, W.; Acta Crystallogr. 1965, 18, 983. 40. Teixeira, V. G.; Coutinho, F. M. B.; Gomes, A. S.; Quim. Nova 2001, 24, 808. 42. Arguello, J.; Magosso, H. A.; Ramos, R. R.; Canevari, T. C.; Landers, R.; Pimentel, V. L.; Gushikem, Y.; Electrochim. Acta 2009, 54, 1948. 42. Umpierres, C. S.; Prola, L. D. T.; Adebayo, M. A.; Lima, E. C.; dos Reis, G. S.; Kunzler, D. D. F.; Dotto, G. L.; L. Arenas T.; Benvenutti, E. V.; Environ. Technol. 2017, 38, 566. 43. Hench, L. L.; West, J. K.; Chem. Rev. 1990, 90, 33. 44. Sing, K. S. W.; IUPAC Technical Report 1985, 57, 603. 45. Thommes, M.; Kaneko, K.; Neimark, A. V.; Olivier, J. P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K. S. W.; IUPAC Technical Report 2015, 87, 1051. 46. Brinker, C. J.; Keefer, K. D.; Schaefer, D. W.; Ashley, C. S.; J. Non-Cryst. Solids 1982, 48, 47. 47. Montoya, I. A.; Viveros, T.; Dominguez, J. M.; Canales, L. A.; Schifter, I.; Catal. Lett. 1992, 15, 207. 48. Pabon, E.; Retuert, J.; Quijada, R.; Zarate, A.; Microporous Mesoporous Mater. 2004, 67, 195. 49. Rashad, M. M.; Baioumy, H. M.; J. Mater. Process. Technol. 2008, 195, 178. 50. Navio, J. A.; Hidalgo, M. C.; Colon, G.; Botta, S. G.; Litter, M. I.; Langmuir 2001, 17, 202. 51. French, R. H.; Glass, S. J.; Ohuchi, F. S.; Xu, Y. N.; Ching, W. Y.; Phys. Rev. B 1994, 49, 5133. 52. Nakamoto, K.; Infrared and Raman Spectra of Inorganic and 54. Silverstein R.; Webster, F.; Spectrometric Identification of Organic Compounds, 7th ed., John Wiley and Sons Inc, New York, 2005. 55. Lin, M. H.; Wang, M. C.; J. Mater. Sci. 1995, 30, 2716. 56. Al-Dughaither, A. S.; de Lasa, H.; Ind. Eng. Chem. Res. 2014, 53, 15303. 57. Al-Ghamdi, S.; Volpe, M.; Hossain, M. M.; de Lasa, H.; Appl. Catal., A 2013, 450, 120. 58. Forni, L.; Vatti, F. P.; Ortoleva, E.; Microporous Mater. 1995, 3, 367. 59. Moreno, E. L.; Rajagopal, K.; Quim. Nova 2009, 32, 538. 60. Emeis, C. A.; J. Catal. 1993, 141, 347. 61. Damyanova, S.; Grange, P.; Delmon, B.; J. Catal. 1997, 168, 421. 62. Damyanova, S.; Petrov, L.; Centeno, M. A.; Grange, P.; Appl. Catal., A 2002, 224, 271. 63. Zaki, M. I.; Hasan, M. A.; Al-Sagheer, F. A.; Pasupulety, L.; Colloids Surf., A 2001, 190, 261. 64. Denofre, S.; Gushikem, Y.; de Castro, S. C.; Kawano, Y.; J. Chem. Soc. Faraday Trans. 1993, 89, 1057. 65. Campos, E. A.; Gushikem, Y.; do Carmo Goncalves, M.; de Castro, S. C.; J. Colloid Interf. Sci. 1996, 180, 453. 66. Gallo, J. M. R.; Alonso, D. M.; Mellmer, M. A.; Dumesic, J. A.; Green Chem. 2013, 15, 85. 67. Lefrancois M.; Malbois, G.; J. Catal. 1971, 20, 350. 68. Morterra C.; Cerrato, G. Langmuir 1990, 6, 1810. 69. Tonetto, G.; Atias, J.; de Lasa, H.; Appl. Catal., A 2004, 270, 9. 70. Al-Bogami, S. A.; de Lasa, H. I.; Fuel 2013, 108, 490. 71. Jin, F.; Li, Y.; Catal. Today 2009, 145, 101. 72. Topsoe, N.-Y.; Pedersen, K.; Derouane, E. G.; J. Catal. 1981, 70, 41. 73. Matsuura, H.; Katada, N.; Niwa, M.; Microporous Mesoporous Mater. 2003, 66, 283. 74. Catrinck, M. N.; Ribeiro, E. S.; Monteiro, R. S.; Ribas, R. M.; Barbosa, M. H. P.; Teofilo, R. F.; Fuel 2017, 210, 67. 75. Corma A.; Garcia, H.; Chem. Rev. 2002, 102, 3837. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access