
 |
 |
 |
 |
 |
Educação
| Hybrid atomic orbitals in organic chemistry. Part 2: critique of practical aspects |
|
Guy Lamoureux*,I; and John F. OgilvieI,II
I. Universidad de Costa Rica, Escuela de Química, San Pedro, San José, Costa Rica Recebido em 13/04/2019 *e-mail: guy.lamoureux@ucr.ac.cr The importance of hybrid atomic orbitals, in both general and organic chemistry, is reviewed. Every contemporary textbook of organic chemistry introduces the directed-valence (e.g. sp3, sp2, sp) model, but the suitability of these hybrid orbitals for use in the teaching of molecular structure has been increasingly questioned. Based on a critical survey of the literature, we submit seven practical criteria that deprecate the use of hybrid orbitals in a pedagogical context. We suggest how the teaching of organic chemistry without hybrid orbitals will provide students with an enhanced education. INTRODUCTION In Part 1, we present a critique of hybrid atomic orbitals (which we abbreviate HAO) from a logical perspective (Lamoureux & Ogilvie, 2019). In Part 2 here, we examine the practical problems with this concept as a pedagogical model, focusing on the HAO in only organic chemistry with their limitations as delineated in Part 1, and on how to overcome these challenges. We list seven practical criteria that deprecate their use in a pedagogical context. On the basis of these two articles, we maintain that the HAO model is seriously flawed from a mathematical, logical, experimental, practical and, thus necessarily, pedagogical point of view.
USE OF HYBRIDIZATION IN GENERAL CHEMISTRY As most instructors and undergraduate students of introductory general and organic chemistry recognize, trying to understand a unified picture of molecular bonding and structure has reached a critical point. Most students are instructed to follow the following flowchart1 to unveil the plausible bonding as they learn about molecular structure:
Astute students rapidly learn the limitations of this scheme. There is no 'correct method' to describe, to predict or to explain molecular structures. Lewis structures are models suitable for a prediction of molecular properties such as connectivity and number of lone pairs, but they bear little relation to the electronic structure;4 the emphasis can vary from elimination of the formal charge to a preservation of the octet rule. VSEPR has simple rules and predictive capability, but there are many exceptions to the rules5 as the procedure lacks theoretical justification and, hence, explanatory power.6 The LCP scheme (vide supra) takes into consideration the non-bonded interactions among atoms but not lone-pair interactions. Hybridization schemes require a prior knowledge of the general molecular geometry before assigning the type of orbitals. Moreover, each scheme uses a disparate nomenclature to describe the geometry. "Note also that the molecular geometry is not the same as the hybrid orbital type. The oxygen atom in the H2O molecule utilizes sp3 hybrid orbitals, but the molecular geometry is certainly not tetrahedral!".7 Using both systems of nomenclature - 'a bent molecule with approximately tetrahedral H-O-H bond angle' and 'using tetrahedral sp3 HAO forming the two O-H bonds and the two lone pairs' - clarifies the description of the structure of H2O but with a loss of simplicity. In general chemistry, there is a modern tendency, if not to remove hybridization completely, to de-emphasize its use. A recent textbook states "For some molecules, describing bond formation in terms of hybrid orbitals is not appropriate."8 Galbraith indicated that "hybridization is shown not to be an all-inclusive phenomenon" and suggested "eliminating the need for spd hybridization in any form."9 In research articles, the use of hybrids for any elements except those in the second period has been questioned. "The model of hybridization already becomes less straightforward for heavier atoms than for the first-row elements C-Ne.".10 Even among the first-row elements, one might set aside the elements fluorine and neon, for which hybridization is never necessary in introductory chemistry. Oxygen and nitrogen have also been considered without hybridization: "The limiting angle in this case [H2O and NH3] would be 90° which is considerably smaller than the observed angles. Here, repulsion between the hydrogens would in part lead to the observed angles."11 This periodic-table ablation of hybridization leaves only carbon. Carbon hybridization is so entrenched in organic chemistry as to have become almost a dogma; organic chemists have universally adopted the use of HAO, typically in a simplified form. "Hybridization is particularly important in carbon, which tends to form four bonds in its compounds and therefore always hybridizes".1 We restrict the rest of this article to the teaching of undergraduate organic chemistry, instead of a more general approach that would encompass all chemistry courses. As general chemistry and organic chemistry are generally taught in parallel or tandem, the de-emphasis of the use of HAO should start in general chemistry and continue throughout the undergraduate education. We first show the reasoning behind the use of HAO before seeking to reform the chemical curriculum. What is the ultimate purpose of using HAO in organic chemistry? Has it altered over the years?
THE THREE SYSTEMS OF HYBRID ATOMIC ORBITALS (HAO) There are at least three models for the hybridization, or directedvalence, scheme. The original presentation of hybridization in organic chemistry used only sp3 hybridized atomic orbitals for all carbon single, double and triple bonds. The calculated relative bond energies for sp3 (2.000), sp2 (1.991) and sp (1.932) hybrids indicate that the 'best' hybrid is sp3 for maximum bond strength in any organic compound.12 This selection required the use of banana or tau (τ) bonds (Figure 1) for multiple bonds. The τ model was fiercely defended by Pauling;13 its utility has been suggested by some authors,14,15 but has never gained ground in textbooks of organic chemistry.
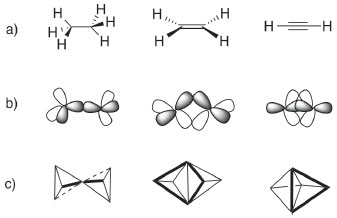
Much more common is the use of sp3, sp2 and sp hybrids (Figure 2) collectively - the special atomic hybrids.16 In this system, a student learns to search the Lewis structure for four single (sp3), one double and two single (sp2), and one triple and one single (sp) or two double bonds around carbon and then to correlate the structure to the orbital hybridization, which then serves as further evidence of the geometry (tetrahedral, trigonal planar and linear, respectively) around the central atom.
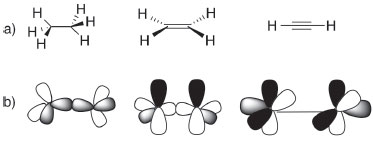
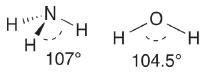
In these images of 'orbitals', the electron density appears to be concentrated along the axis containing adjacent nuclei; this simplification is gross because accurate plots of calculated hybrids show a more nearly spherical distribution (as shown in Part 1). In a third system used, hybrids are constructed in an infinite series.17 According to this system, the bond angle (θ) is directly correlated with the hybridization index i, represented by spi and calculated with the formula 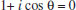 This extension of orbital hybridization has been termed non-integral, isovalent or second-order hybridization. For example (Figure 3), in ammonia (NH3) the H-N-H (bond angle ~107°) hybrids have been calculated as sp3.4 and for water H-O-H (bond angle ~104.5°) sp4 hybrids are typically presented. In this system the original meaning of the hybridization index, viz. the integer number of p orbitals mixing in the linear combination, is obviously lost. We have encountered no introductory organic textbook that presents this non-integral system.
Let us investigate the use of the three systems of hybridization - sp3 τ hybrids, special hybrids and non-integral hybrids - and compare their utility. Much ink has been spilt over the mathematical equivalency of τ and σ-π bonds18 and their use in various contexts.19 We remind the reader, however, that these models are modern constructs, using modern computational software; there is no conflict with the use of τ or σ-π bonds in any advanced research project. We continue in this section to examine how each system is limited in its own way. If one applies the special hybrid model, the bond angles are limited to 109.5°, 120° and 180°. To accommodate these results for all organic molecules within the hybridization model, further 'explanations' must be appended to the model for any deviation away from these bond angles. We must thus suppose that hybridization (as measured from a fixed bond angle) depends on, inter alia, the electronegativity of directly bonded atoms (Bent's Rule, vide infra), bond length,20 the curvature of the bond21 and non-bonded interactions.22 This layering of explanations is not only inelegant but also confusing. "It is necessary to graft on extra hypotheses without experimental support to explain each effect as it arises. The existence of the phenomenon then becomes the proof of the explanation".23 If one uses the non-integral hybrid scheme, the relation between bond angle and hybrid number cannot be independently tested. We are left with the inescapable conclusion that this scheme can serve as neither explanandum nor explanans (i.e. one can state neither that bond angle is determined by hybridization nor that hybridization determines bond angle). There is no advantage in creating an infinitely flexible hybridization scheme. "It is found to be about five-fold less costly in loss of overlap to fix the hybridization of the carbon orbitals at an optimum value and 'bend' the C-H bonds than it is to vary the carbon hybridization to follow the angular displacements".22 Moreover, the direct relation between the two values is doubtful. "The derivation of simple bond angle/sp ratio formulae is shown by calculation to be incorrect".23
HYBRID ATOMIC ORBITALS (HAO) IN ORGANIC CHEMISTRY In the twenty-first century, every textbook of organic chemistry that we have examined introduces at least one hybridization model, with neither critical assessment nor explanation, to provide information about the orientation of chemical bonds in molecules of organic compounds. Another common lore in chemistry is that HAO must be taught, generally at the beginning, in every introductory course of organic chemistry. In 2013, the American Chemical Society (ACS) provided a list of anchoring concepts to outline a map of the content for organic chemistry in an undergraduate curriculum.24 The main points of the map related to HAO follow.
A cursory examination of these three main points invokes further queries. What does "linear combinations of atomic orbitals' hybridized orbitals" mean? How are they better representations and for what reasons are they necessary? What is the relation of HAO to the molecular-orbital or valence-bond theories of bonding? With the related term hybridization, HAO have been used traditionally to describe, to explain and to predict concepts in organic chemistry. Since its inception, the original concept of hybridization has been used for explanation. "Slater and Pauling both argued for the necessity of using an s orbital as well as three p orbitals in the carbon atom in order to explain [emphasis added] carbon's four valences and the tetrahedral structure of methane".25 The continued use of HAO to explain various aspects of bonding, especially organic chemistry, is evident from a perusal of contemporary publications of chemical research. A recent review states "The usefulness of the concept of hybridization as a simple way of explaining experimental observations is apparent to anyone who has taught undergraduate organic chemistry".26 These authors state also that "The concept of hybridization explains many chemical and physical observables".26 We summarize three explanations, taken from textbooks, that involve hybridization.
We have demonstrated in Part 1 that an attempt to use hybrids as pseudo-explanation is never warranted. Furthermore, hybrid orbitals have been invoked for their predictive power -- a tool so basic to predict trends that it requires no further description or justification. Some examples follow.
SPECIFIC CRITIQUE OF HYBRID ATOMIC ORBITALS IN ORGANIC CHEMISTRY We contend that the use of HAO should be debated not on the prospective utility of the model, but rather on the mathematical constructs used to create the model. "...a model which is based on assumptions that are falsified by accurate calculations becomes questionable even if it offers the advantage of simplicity".10 Recalling that most chemists use HAO to describe, to explain and to predict concepts without examining the paradigm itself, one must obviously address several refinements. These objections apply equally to dsp2 or d2sp3 or any other hybrid function that involves more than one p or d component. First, there is no need for hybridization as a consequence of quantum mechanics. As Lewars clearly showed, the formation of HAO is never necessary, and, whether or not one invokes sp3 hybridization for calculations on methane, one obtains the same tetrahedral structure.40 Paradoxically, one of the most common supporting arguments of HAO is that they show the student the mathematical nature of chemistry and lend quantum credence to the Lewis structure. "The chemical bond is not a causal agent for hybridization as textbooks often labor to explain. Nor is hybridization a consequence of quantum laws."41 "On the other hand, 'hybridization effects' as presented to students masquerade as if they are based on firm quantum-mechanical principles. Beginning students have no way of perceiving that, as generally taught, hybridization implies little more than the already known structure of the molecule under consideration".42 Second, the hybrid concept implies only the wave function of the (atomic) electrons. Notwithstanding the confusion between geometry and hybridization, describing a) an atomic center in a molecule or b) a lone pair or c) a bond as a hybrid is completely unacceptable. Many organic molecules contain unequal bond angles around the central atom (e.g. propane); it is thus impossible to assign only one hybridization to any atom. A search of Internet (scholar.google. com) in 2019 January revealed 2947 instances of "(sp3 or sp2 or sp) hybridized carbon atom" in published scientific books, articles and patents, indicating that the confusion is rampant. Hybrids are atomic orbitals and, by definition, are perturbed during the formation of a molecule; one cannot thus apply a hybridization label for lone pairs and bonds in molecules.43 Furthermore, discussing hybridization in a diatomic molecule, or for a terminal atom in a polyatomic molecule, is never justifiable. Third, one cannot claim hybridization as the cause of an observation.44 Regarding experimental support of hybridization, correlation is not causation.45 In one book the authors claimed that "non-integral hybridization is more than just an after-the-fact rationalization, and has experimental support".17 In another book the authors contended that "This [overlap of hybrid orbitals] is substantiated by experimental results".46 These statements are in direct conflict with another critical viewpoint: "The objective of a hybridization scheme is an after-the-fact rationalization of the experimentally observed shape of a molecule. Hybridization is not an actual physical phenomenon."8 Fourth, no direct experimental evidence for HAO exists. HAO, as indicated in an article celebrating 50 years of hybridization, are not experimentally observable phenomena but are rather "a 'Linnean classification' of molecular observables."47 Even though the concept of hybridization of CH4, NH3 and H2O shows "pedagogical significance in chemistry and molecular physics",48 the authors of the most recently published articles48,49 demonstrated that contemporary experimental data are inconsistent with the HAO portrayed in organic chemistry: "[the] hybrid orbital model descriptions fail to reproduce the observed physical behavior of the [purported] valence electrons in CH4, NH3 and H2O."48 Fifth, HAO constitute a flawed model, limited by simple trigonometry, that nevertheless continues to influence the contemporary teaching of structural organic chemistry. A simple model of sp3, sp2, sp hybrids describes no real molecule. One might compare this model to wooden or plastic models of molecules, which are also simple models but which are not designed to reproduce the complicated reality of molecular structure, even though they illustrate some salient features. Whereas we have found no claimant that insists that wooden models are mathematically and experimentally correct, the same cannot be said of the HAO model. Why should one continue with this, or other, flawed model? Mislow succinctly stated, "Scientific concepts or categories are therefore fuzzy unless they are expressed in the language of mathematics."50 Please continue to use plastic models -- they can be useful. As HAO have, however, a mathematical basis, their precision or imprecision is based on mathematics. A flawed model based on flawed mathematics must be unacceptable. Sixth, the method of applying sp3, sp2 and sp HAO is not generally applicable to all organic molecules. The exceptions are highly symmetric (Td , D3h and D∞h) systems, which correspond to organic molecules in a small proportion. Simple examples of hybridization are not extensible convincingly to more complicated organic structures, even with small alterations of structure. For example, the C-C-H angles of ethane (a small molecular deviation from methane) determined by electron diffraction are 111.0±0.2° (not 109.5°)51 and the H-C-H bond angle 117.2±1.2° of ethene (the simplest alkene) differs significantly from the sp2 angle, 120°.52 These results led an eminent structural chemist, who reported those angles, to proclaim "Hybridization is a fraud!".42 Seventh, how can one easily understand systems in which the bond angle is less than the minimum hybrid bond angle, 109.5°? For example, for cyclobutane and cubane, the bond angles approximately 90° seem to imply unhybridized carbons. "Cubane's structure demonstrates the limitations of Pauling's hybrid-orbital theory, a potent reminder that molecules don't know about Pauling's hybrid orbitals, so are in no way constrained by them".53 The case of cyclopropane (and other three-membered rings with bond angles ~60°) is certainly even more confusing, even if one includes the use of bent bonds to describe the inter-orbital angle instead of the bond angle.54 Cyclopropane is notably conspicuous by its absence from Pauling's quintessential presentation of his advocacy of the doctrine of hybridization.55 These seven arguments should effectively eliminate the concept of HAO in organic chemistry. It is counterproductive to describe, to explain or to predict structure or properties based on a flawed concept such as 'hybridization' or '% s character'.56
PREVIOUS CRITIQUE OF HYBRID ORBITALS VERSUS OUR PERSPECTIVE In the Journal of Chemical Education, authors of papers in a series debated the role of hybrid atomic orbitals in a discussion of the structures and shapes of molecules and their reactions. The following arguments are completely separate from our critique (vide supra). Grushow argued to retire the hybrid atomic orbital as a useful model,57 whereas other authors defended the practice of invoking hybrid atomic orbitals on the grounds that valence-bond theory is still useful,58 that localized bond orbitals can be used to interpret photoelectron spectra of molecules,59 that localized bonds formed on covalent linking of hybrid orbitals are a viable representation of molecules,60 and that the hybridization model is useful in conjunction with both valence-bond and molecular-orbital theories.61 Landis and Weinhold summarized62 four premises about 'hybrid orbitals' discussed by Grushow: i) hybrid orbitals do not exist [they have no tangible existence and are merely arbitrary mathematical constructs], ii) hybrid atomic orbitals are inappropriate models, iii) electron density is best described using delocalized rather than localized approaches, iv) bonding cannot be characterized properly using a localized electron model; Landis and Weinhold considered those premises to be unsupported in Grushow's article. In previous discussions, both proponents and opponents of this approach seem to have been unaware of the fundamental deficiencies in the original formulation of these HAO, which we have outlined above. Tro claimed "because valence-bond theory has limited value without hybridization, it seems that a call to eliminate one implies the elimination of both".58 We refrain from calling for an elimination of 'hybridization' from valence-bond calculations; the qualitative application of hybrid orbitals in teaching organic chemistry is completely separate from explicit valence-bond calculations. Whether HAO (as they are used in organic chemistry) are a replacement for 'localized' orbitals in valence-bond calculations is irrelevant, thus avoiding the critiques of DeKock59 and Truhlar.60 The premise that "significant experimental evidence and theoretical advances ... indicate that hybrid orbitals do not exist and do not appropriately describe molecular bonding" that Landis and Weinhold tried to undermine62 becomes self-evident when viewed from a perspective of modern experimental data (vide supra). Moreover, despite the contention of Hiberty et al. that the "hybrid atomic orbital model is correct, useful, free of conflicts with experiment",61 these characteristics are inaccurate and simply inapplicable to HAO. With regard to the teaching of HAO, the many misconceptions63 and the frustration64-67 faced by students show that hybridization is among the most difficult concepts to understand. The fact that one can view hybrid orbitals on a computer68 or print hybrids on a 3D printer69 fails to relieve this frustration. It is important to expand on the reply of Grushow to specific comments in the Journal of Chemical Education:70 "The reality is that hybrids are not properly understood and [are] incorrectly used by students, because of the mathematical difficulties"; we add but also that the textbooks and the instructors are basing their arguments on a false pretense. Based on this perspective, we strongly advocate the retirement of HAO from the teaching of chemistry.
TEACHING WITHOUT HYBRID ATOMIC ORBITALS Is it possible to teach organic chemistry without hybridization? Absolutely, and in some regards this change provides the students with a superior grasp of organic chemistry, according to our experience in the classroom. A modified flowchart is shown below to teach molecular structure for students of the future.
For a description of a geometrical system, once the sp3, sp2, sp model is eliminated, one might continue to use VSEPR or LCP, with the caveat that these schemes fail to explain the molecular structure and are also prone to error and exception. To label a system or to compare it with others, it would be preferable to use simple point groups according to group theory (without the underlying mathematics) to assign the symmetry of the molecular system. CH4, NH3 and H2O can hence be described as Td, C3v and C2v instead of (the sometimes confusing) sp3, bent or 'approximately tetrahedral' labels. Is it necessary to describe or to assign individual atoms in a complicated molecule with a hybridization designation? Innovative chemists of the future should be able to identify a specific atom X using known labels (e.g. aliphatic X vs. aromatic X, cyclic X vs. acyclic X) or a combination of atom symbol and number (e.g. X2). A complicated explanation of reactivity is best left to contemporary calculations based on molecular-orbital or valencebond or density-functional theory (DFT), which are all inevitably approximate. After a student has the geometric and electronic structure, a prediction of properties (e.g. acid-base) becomes the main objective of his or her education, instead of memorization and rote application of obsolete concepts. To show the possibility of teaching without hybridization, in a recent undergraduate organic chemistry course we used the freely available molview.org to bypass the VSEPR and hybridization steps. In a molview structure, the bond angle around a central atom can be directly measured as a numerical value, instead of being estimated as tetrahedral or trigonal or linear. We indicated at the beginning of the semester that any use of hybrids or hybridization can be replaced with geometric arguments (i.e. after comparing the bond angles in two or more molecules, how does the geometry affect the reactivity?). The geometry of an organic molecule is observable (if obtained from experiment) and generally free of arbitrary interpretation. We thus replaced every description, explanation and prediction that were once ascribed to hybridization in organic chemistry to focus on the structure and how the structure affects the reactivity.
CONCLUSIONS Regarding the second principal question that we posed, we have demonstrated that HAO clearly incur serious problems in a pedagogical setting; based on the fallacy of HAO demonstrated above, we contend that HAO should be phased out from the teaching of organic chemistry. As many authors have declared over the years, hybridization is essential for no description, no explanation nor prediction.
CODA Even with this overwhelming evidence, instructors have generally refused to judge what is best for modern students in learning the complicated subject of organic chemistry, preferring to use what was taught in the past. Textbook authors should take into consideration the caducity of HAO and reform the organic program. All these arguments presented in this article are not merely our opinion but have strong support in the literature, in more eloquent words than we can compose, a few of which we cite here. "This reviewer recommends, to the extent that it is still used in freshman chemistry courses, that the directed valence analysis be abandoned completely in the teaching of chemistry. It is inelegant, wrong in some respects and leads to unnecessary confusions...".23 "...the directed-valence theory often gives an incorrect impression of the actual forces at work in a molecule. Future generations of chemical educators may wish to reconsider the appropriateness of teaching this model of chemical bonding to introductory chemistry students."6 "We think that 'atomic orbitals' are more a problem than a help in teaching chemistry. Perhaps getting rid of all these lexical false friends is a good starting point for designing an integrated chemistry curriculum."72 "Attempts to simplify the presentation of this material [hybridization] result in the presentation in elementary text-books of specious arguments, which are at best misleading and at worst incorrect. In these the author attempts to persuade, or perhaps a better word would be to hoodwink, students into thinking that they understand a number of difficult and subtle ideas."73
ACKNOWLEDGMENTS El Centro de Investigaciones en Productos Naturales (CIPRONA) and la Escuela de Química, Universidad de Costa Rica provided support. We thank many professors and students at UCR for helpful discussion.
REFERENCES 1. Tro, N. J.; Principles of Chemistry, Pearson: Boston, 2017, p. 388. 2. Gillespie, R.; Chem. Soc. Rev. 1992, 21, 59. 3. Bartell, L. S.; J. Chem. Phys. 1960, 32, 827. 4. Purser, G. H.; J. Chem. Educ. 1999, 76, 1013. 5. Myers, R. T.; Monatsh. Chem. 1992, 123, 363. 6. See, R. F.; Dutoi, A. D.; McConnell, K. W.; Naylor, R. M.; J. Am. Chem. Soc.2001, 123, 2839. 7. House, J. E.; House, K. A. L.; Descriptive Inorganic Chemistry, Academic Press: London, 2016, p. 31. 8. Petrucci, R. H.; Herring, F. G.; Madura, J. D.; Bissonnette, C.; General Chemistry: Principles and Modern Applications, Pearson: Toronto, 2017; p. 474. 9. Galbraith, J. M.; J. Chem. Educ. 2007, 84, 783. 10. Frenking, G.; Fröhlich, N.; Chem. Rev. 2000, 100, 717. 11. Wiberg, K. B.; Acc. Chem. Res. 1996, 29, 229. 12. Maccoll, A.; Trans. Faraday Soc. 1950, 46, 369. 13. Bursten, J. R.; Ann. Sci. 2012, 69, 69. 14. Wintner, C. E.; J. Chem. Educ. 1987, 64, 587. 15. Deslongchamps, G.; Deslongchamps, P.; Org. Biomol. Chem. 2011, 9, 5321. 16. Bingel, W. A.; Lüttke, W.; Angew. Chem. Int. Ed. Engl. 1981, 20, 899. 17. Anslyn, E. V.; Dougherty, D. A.; Modern Physical Organic Chemistry, University Science Books: Sausalito, CA, 2006, p. 10. 18. Palke, W. E.; J. Am. Chem. Soc. 1986, 108, 6543. 19. Dunning, T. H.; Xu, L. T.; Takeshita, T. Y.; Lindquist, B. A.; J. Phys. Chem. A 2016, 120, 1763. 20. Coulson, C. A.; Proc. R. Soc. Lond. A 1951, 207, 63. 21. Wiberg, K. B.; Murcko, M. A.; J. Mol. Struct. 1988, 169, 355. 22. Bartell, L. S.; Tetrahedron 1962, 17, 177. 23. Gilheany, D. G.; Chem. Rev. 1994, 94, 1339. 24. Raker, J.; Holme, T.; Murphy, K.; J. Chem. Educ. 2013, 90, 1443. 25. Park, B. S.; Studies in History and Philosophy of Science Part B: Studies in History and Philosophy of Modern Physics 2000, 31, 451. 26. Alabugin, I. V.; Bresch, S.; J. Phys. Org. Chem. 2015, 28, 147. 27. Bent, H. A.; Chem. Rev. 1961, 61, 275. 28. Bent, H. A.; J. Chem. Educ. 1984, 61, 421. 29. Laing, M.; J. Chem. Educ. 1987, 64, 124. 30. Clauss, A. D.; Nelsen, S. F.; Ayoub, M.; Moore, J. W.; Landis, C. R.; Weinhold, F.; Chem. Educ. Res. Pract. 2014, 15, 417. 31. Hiberty, P. C.; Danovich, D.; Shaik, S.; Chem. Educ. Res. Pract. 2015, 16, 689. 32. Clauss, A. D.; Ayoub, M.; Moore, J. W.; Landis, C. R.; Weinhold, F.; Chem. Educ. Res. Pract. 2015, 16, 694. 33. Bent, H. A.; Can. J. Chem. 1960, 38, 1235. 34. Politzer, P.; Murray, J. S.; J. Mol. Model. 2018, 24, 214. 35. Brown, M. G.; J. Chem. Phys. 1960, 33, 1881. 36. Maksic, Z. B.; Randic, M.; J. Am. Chem. Soc. 1970, 92, 424. 37. Dicks, A. P.; J. Chem. Educ. 2003, 80, 1322. 38. Bauer, L.; Anderson, H. J.; J. Chem. Educ. 1999, 76, 1151. 39. Baldwin, J. E.; Chem. Commun. 1976, 734. 40. Lewars, E. G.; Computational Chemistry: Introduction to the Theory and Applications of Molecular and Quantum Mechanics, 3rd ed., Springer International Publishing: Switzerland, 2016, pp 121-123. 41. Vemulapalli, G. K. In Philosophy of Chemistry; Baird, D., Scerri, E., McIntyre, L., eds.; Springer: Dordrecht, 2006; pp. 191-204. 42. Bartell, L. S.; Coord. Chem. Rev. 2000, 197, 37. 43. Taber, K. S.; Sci. Educ. 2005, 89, 94. 44. Mosher, M. D.; Ojha, S.; J. Chem. Educ. 1998, 75, 888. 45. Gil, V. M. S.; J. Chem. Educ. 2001, 78, 31. 46. March, N. H.; Mucci, J. F.; Chemical Physics of Free Molecules, Springer: New York, 1993, pp 161-217. 47. Barbier, C.; Berthier, G.; Adv. Quantum Chem. 2000, 36, 1. 48. Brion, C. E.; Wolfe, S.; Shi, Z.; Cooper, G.; Zheng, Y. J.; Can. J. Chem. 2017, 95, 1314. 49. Yuan, L.; Ling-Fung, C.; Chuan-Gang, N.; Chin. Phys. B 2014, 23, 063403. 50. Mislow, K.; Chirality 2002, 14, 126. 51. Bartell, L. S.; Higginbotham, H. K.; J. Chem. Phys. 1965, 42, 851. 52. Bartell, L. S.; Roth, E. A.; Hollowell, C. D.; Kuchitsu, K.; Young Jr, J. E.; J. Chem. Phys. 1965, 42, 2683. 53. Francl, M.; Nat. Chem. 2018, 10, 1. 54. Bernett, W. A.; J. Chem. Educ. 1967, 44, 17. 55. Pauling, L.; The Nature of the Chemical Bond and the Structure of Molecules and Crystals: an Introduction to Modern Structural Chemistry; Cornell University Press: Ithaca, NY, USA, 1960. 56. Pomerantz, M.; Liebman, J. F.; Tetrahedron Lett. 1975, 16, 2385. 57. Grushow, A.; J. Chem. Educ. 2011, 88, 860. 58. Tro, N. J.; J. Chem. Educ. 2012, 89, 567. 59. DeKock, R. L.; Strikwerda, J. R.; J. Chem. Educ. 2012, 89, 569. 60. Truhlar, D. G.; J. Chem. Educ. 2012, 89, 573. 61. Hiberty, P. C.; Volatron, F.; Shaik, S.; J. Chem. Educ. 2012, 89, 575. 62. Landis, C. R.; Weinhold, F.; J. Chem. Educ. 2012, 89, 570. 63. Gillespie, R. J.; J. Chem. Educ. 2004, 298. 64. Nakiboglu, C.; Chem. Educ. Res. Pract. 2003, 443. 65. Salah, H.; Dumon, A.; Chem. Educ. Res. Pract. 2011, 12, 443. 66. Stefani, C.; Tsaparlis, G.; J. Res. Sci. Teach. 2009, 46, 520. 67. Bouayad, A.; Kaddari, F.; Lachkar, M.; Elachqar, A.; Procedia - Social and Behavioral Sciences 2014, 116, 4612. 68. Allendoerfer, R. D.; J. Chem. Educ. 1990, 67, 37. 69. de Cataldo, R.; Griffith, K. M.; Fogarty, K. H.; J. Chem. Educ. 2018, 1601. 70. Grushow, A.; J. Chem. Educ. 2012, 89, 578. 71. Shusterman, A. J.; Shusterman, G. P.; J. Chem. Educ. 1997, 74, 771. 72. Sánchez-Gómez, P. J.; Martín, F.; Chem. Educ. Res. Pract. 2003, 4, 131. 73. Gillespie, R.; Chem. Can. 1976, 23. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access