
 |
 |
 |
 |
 |
Revisão
| Design of immobilized enzyme biocatalysts: drawbacks and opportunities |
|
Carla Luzia Borges ReisI; Emerson Yvay Almeida de SousaII; Juliana de França SerpaI; Ravena Casemiro OliveiraI; José Cleiton Sousa dos Santos*,III
I. Departamento de Engenharia Química, Universidade Federal do Ceará, Campus do Pici, 60455-760 Fortaleza – CE, Brasil Recebido em 19/01/2019 *e-mail: jcs@unilab.edu.br The enzymatic processes are increasingly highlights, especially in the synthesis of chemical products with high added value. The enzyme immobilization can improve industrial biocatalytic processes. The immobilization of enzymes provides the production of efficient, stable biocatalysts, possibility of reuse and easy purification of the products, when compared to the free enzymes. There is a growing research for more efficient methods of enzyme immobilization. In this context, the choice of support and immobilization strategy can significantly improve the final enzymatic properties. In this review paper, we aimed to discuss the versatility of biocatalysts immobilized enzymes design, focusing on the opportunities and disadvantages for each method presented. They discussed the recent development of enzyme immobilization methods and applications relating the final properties of the produced biocatalysts with the desired goals. ENZYME IMMOBILIZATION Enzymes have been remarkably accepted as biocatalysts in diverse sectors owing to green chemistry and their substrate specificity.1-8 The enzymes are used industrially, among others, in food products, cellulose and paper, animal feed, textile cosmetics, detergents and the pharmaceutical industry.9-16 To increase the use of enzymes as industrial biocatalysts, it is necessary to obtain stable preparations of the enzymes with an improved operational stability,17 their application renders into lower demand for raw materials, chemicals and energy, heir specificity and high selectivity, and they drive into cleaner industrial processes.18 However, immobilization is one of the important ways for enzymes to become stable.19-23 Many methods of immobilization are described and used in the literature to circumvent the possible instability problems of the enzymes as well as to optimize the various applications. In recent years, the empirical use of these immobilization techniques (for example, covalent bonding,24 physical adsorption,25 ionic adsorption,26 crosslinking,27 encapsulation,28 etc) and their influences on the specificity, activity and stability of the enzymatic molecules, as well as the usability of the biocatalysts for application-related29-31 reactions.29,32-35 These immobilized enzymes, bound to the solid supports, can simply be removed from the reaction mixture. Thus, by minimizing the contamination of the final product containing the enzyme, it will be possible to reuse the recovered enzyme.36 This reuse of the immobilized enzymes will help reduce the cost of the industrial process, and the immobilization can also improve the enzyme’s stability against extreme temperatures and pH values.37-39 However, there is a persistent need to synthesize new enzyme vehicles that are effective due to meaning of the immobilization process, readily available at a reasonable and non-toxic cost.40 Enzymes can be immobilized by different methods, such as those shown in Figure 1: (A) covalent attachment; (B) multipoint covalent attachment; (C) multipoint covalent attachment of an enzyme to a functionalized support; (D) immobilization of enzymes by cross-linking; (E) matrix entrapment; (F) encapsulation; (G) immobilization by adsorption; (H) magnetic nanoparticles hybrids formation; (I) enzyme immobilization via formation mechanism of hybrid nanoflowers. These methods were chosen for the development of this review work. It can be said that there is not only single immobilization method or support applicable to all enzymes and their various applications due to the different properties of the substrates, the diverse applications of the products obtained and the different physicochemical characteristics of each enzyme.41,32
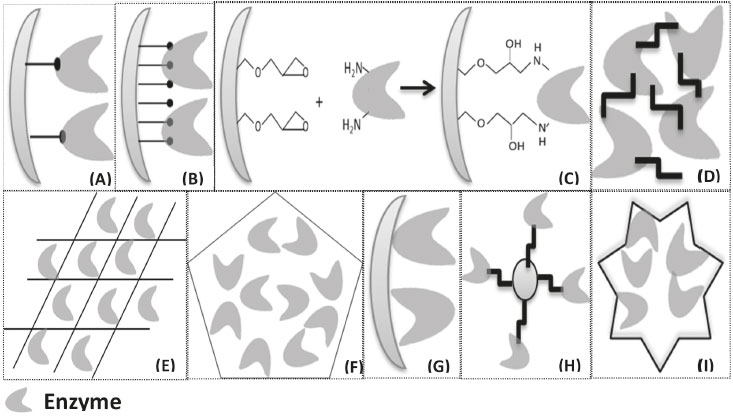 Figure 1. (A) Covalent Attachment; (B) Multipoint Covalent Attachment; (C) Multipoint covalent attachment of an enzyme to a functionalized support; (D) Immobilization of enzymes by cross-linking; (E) Matrix entrapment; (F) Encapsulation; (G) Immobilization by adsorption; (H) Magnetic nanoparticles hybrids; (I) Proposed formation mechanism of hybrid nanoflowers
IMMOBILIZATION OF ENZYMES VIA COVALENT ATTACHMENT METHOD The immobilization of enzymes via covalent attachment provides a strong chemical interaction between the carrier material and the enzyme.42-49 The most commonly used covalent immobilization techniques are based on the Schiff or carbodiimide chemists which require initial support regularization with aldehyde and carboxyl or amine groups, respectively.50 In this point, the Table 1 shows the data on the immobilization of enzymes by the covalent method drawn from the literature are presented as a summary. Generally, glutaraldehyde and glyoxyl are two of the reagents most used in the activation of supports and/or as a spacer arm, due to the simplicity of the methods of activation and obtaining active and stable enzymatic preparations. Thus, examples are shown in the examples of Table 1, where some types of enzyme (β-galactosidase widely used in the literature) (such as glutaraldehyde and glyoxyl) which facilitates the covalent attachment of the enzyme to the support, thereby obtaining a favorable immobilization result, as shown in the above Table 1.
In this context, studies with calcium pectinate (CP) gel beads, for example, they have been treated with glutaraldehyde (GA) and polyethyleneimine (PEI), greatly benefiting the mechanical strength of the material with this treatment.51 Furthermore, it enabled the CP beads to covalently immobilize enzymes, such as β-d-galactosidase (β-gal).51 Was applied the central composite design (CCD) in order to improve the treatment of PEI / GA, while applying the observed activity of immobilized β-gal in response. For the treatment of the CP beads with a 3.49 % PEI solution of pH 10.55 followed by a 5.66% GA solution in 0.1 mol L-1 phosphate buffer pH 7.02 the CCD predicted that would allow for the immobilization of 6.25 U g-1 gel of the β-gal. This experiment was performed under optimum conditions obtaining a result of 6.285 ± 0.22 U g-1 of immobilized gel.51 The reusability study revealed that 79.34% of initial activity the β-gal immobilized onto the treated CP beads was retained after being used for fourteen times.51 According to Sulaiman et al.,52 a great potential to be used in enzymatic immobilization is the cellulose nanofiber (CNF) of the Kenena fiber. The preparation of CNF, cyclodextrin glucanotranferase (CGTase) immobilization in CNF is described by the chemical coupling and its application through the ultrafiltration membrane. More than 62% of the binding yield and more than 45% of the residual activity were obtained through the efficiency of the immobilized CGTase.52 This study shows the great capacity of CGTase immobilization in CNF via covalent bonding, with the reuse profile of the immobilized CGTase that contaminated the membrane surface and was able to retain up to 50-60% of CGTase activity in the 10º cycle. Several factors contributed to the covalent immobilization of the enzyme in the CNF support, such as enzymatic production, recycling cycles, productivity, thermal stability and the significant increase in enzyme loading and its residual activity.52 Cieh et al.,42 evaluated the immobilized CGTase properties, such as thermal stability, storage stability, and reuse. The effect of different coupling agents (spacer arms and ligands) on immobilization of cyclodextrin glucanotransferase (CGTase) on bleached kenaf microfiber as support matrix was investigated. According to the results, CGTases immobilized on microfibers resulted in 0.162-0.24 U mg-1 fiber when during immobilization 55.6 U mL-1 CGTase activity was initially added.42 The highest storage stability (60 °C) was shown by CGTase which was immobilized with ethylenediamine and ophthalaldehyde, where after 15 days its activity remained at 60%. CGTase immobilized using glutaraldehyde and ethylenediamine showed the best retention of the enzymatic activity after 12 batch reaction cycles up to 72.72%.42 These results show that the kenaf microfiber has the potential to be applied as support to the enzymatic immobilization and its enzymatic properties were affected by the coupling agents.42 Multipoint covalent attachment The multivalent covalent attachment of enzymes to activated substrates is considered a very important tool for stabilizing proteins.53-60 The number of chemical bonds between support and enzyme is what defines the stabilization factor of the immobilized enzyme.53,54 The formation of multiple chemical covalent bonds is one that maintains the relative positions of all chemical groups involved in the immobilization unaltered during the conformational change induced by any distorted agent (organic solvents, heat, and extreme pH values).53,54 It is generally used for immobilization of enzymes in aldehyde functionalized supports.53,54 An example is shown in Figure 1 (C) of a multi-point covalent bond, where the enzyme is attached to a functionalized support, and the Table 2 presents data from the literature on enzymatic immobilization using the covalent multipoint method.53,54
Table 2 indicates favorable results in the use of some types of lipase, as well as in the use of agarose as support that were submitted to multipoint covalent immobilization. In general, the presented results do not show distortions in relation to the optimal conditions of temperature and pH, presenting excellent properties for future industrial application. It is reported, for example, a work in which the multivalent covalent attachment of Rhizopus oryzae lipase (ROL) on silica and silica nanoparticles is functionalized with chemical epoxy groups (MCM-41 and SBA-15). Immobilization in these multi-point carriers of enzymes is generally accomplished by the reaction between several epoxy groups of the support and various Lys residues on the outer surface of the enzymatic molecules at pH 10.54 According to results, there was a higher thermal stability and co-solvent for immobilized derivatives of amino ROL compared to the results obtained for the non-amino ROL derivatives and free ROL. Thus, the influence of the immobilization procedure on the selectivity of the immobilized preparations in selective hydrolysis of fish oil under three different conditions was studied. The selectivity and reuse of ROL were greatly improved after immobilization.54 Another example illustrating immobilization via multipoint covalent attachment is the work by Rivero et al.,61 where Escherichia coli ATP 4157 PPM phosphopentomutase was overexpressed, purified, stabilized at alkaline pH and immobilized on through various supports. Reactions catalysed by this enzyme are useful for the production of nucleoside analogs. However, PPM is unstable when it is outside its natural environment and its stability is affected by parameters such as temperature and pH.61 Therefore, to irreversibly immobilize this enzyme, it needs to be stabilized. The PPM maintained 86% of its initial activity at pH 10 after 18 h of incubation, which allowed the additional covalent immobilization of this enzyme in high yield glyoxyl agarose. This is the first time that through multivalent covalent binding PPM has been immobilized on the glyoxyl carrier, a derivative capable of biosynthesizing ribavirin from a-d-ribose-5-phosphate.61
ENZYMATIC IMMOBILIZATION BY THE AFFINITY METHOD The affinity immobilization aims to explore the specificity of the enzyme in its support through different physiological conditions.62 This occurs in two ways: either the enzyme is conjugated to an entity which develops affinity towards the matrix or the matrix is pre-coupled to an affinity ligand for the target enzyme.62 The affinity adsorbents were also used for simultaneous purification of enzymes.62 Supporters of complex affinities, such as agarose bound multilayer concanavalin A and porous silica granules coated with chitosan and alcohols, have been shown to have higher amounts of enzymes that lead to greater stability and efficiency.63,64 An improvisation of this technique is the bioaffinity layer that exponentially increases the ability to bind to the enzyme and reuse due to the presence of non-covalent forces such as coulombi hydrogen bonding, Van der Waals forces, etc.64-66 An example of immobilization by the affinity method is the engineering platform prepared to produce low weight heparin controllable molecule (LMWH) using the chitin affinity interaction between heparinase I three-phase (Hep I) and chitin. Such affinity absorption is achieved by genetically engineered the protein to carry a binding domain that binds specifically to cognitive affinity carriers. The improved soluble protein called ChBD-SUMO-Hep I (CSH-I) was fermented in a 3 L batch with high bioactivity of 2.5 × 103 IU L-1.67 The chitin binding domain (ChBD) can be specifically bind to it in a non-covalent manner, which leads to a single step immobilization and purification of the enzyme.67 However, obtaining protease molecules from natural sources requires simple purification protocols, economically and efficiently. The main focus is to acquire affinity matrices through the covalent immobilization of dipeptidyl peptidase IV (DPP-IV) and papain on cellulose membranes, previously activated with formalin (FCM) or glyoxyl (GCM) groups.68 The highest activation rate of GCM was shown with 10.2 μmol aldehyde cm-2. According to data analyzed at the optimum pH of 9.0, protein immobilization can occur through agglomerates of few reactive groups. The highest immobilized DPP-IV CGM protein load was 2.1 μg cm-2), 91% immobilization percentage and possible covalent multipoint binding probability.68 The results showed the ability of the matrices synthesized for affinity purification of protease inhibitors.68
CROSS-LINKED ENZYME AGGREGATES (CLEA) Cross-linked enzyme aggregates (CLEA) is a method of free standing immobilization, which allows recyclable and stable biocatalysts with high retention of activity, it can be applied to the immobilization of almost any enzyme and have many economic and environmental benefits in the context of industrial biocatalysis.69-73 They are produced by cross-linking enzymatic aggregates resulting from mixing an aqueous solution of proteins with organic solvents, polymers or anionic salts, followed by cross-linking with a chemical bifunctional reagent, as shown in Figure 1 (D).74-76 The crosslinking agent is a chemical molecule having at least two reactive ends, which bind to specific groups of amino acids on the surface of the enzyme, providing the physical aggregation of enzyme molecules into supermolecular structures, in some cases without disturbance of the original three-dimensional structure of the protein.74,77 The crosslinking reagents can be classified in relation to the reactive groups present at their ends (homobifunctional and heterobifunctional), specificity of their reactive groups, the length of the spacer arm, solubility and also the reactivity.78 Glutaraldehyde has been used for decades as a protein crosslinking chemical agent, and also a bifunctional agent commonly used for cross-link enzyme aggregates, due to its low value and available commercially. However, there are several others crosslinking agent, such as (3-aminopropyl) triethoxysilane, (3-aminopropyl) trimethoxysilane, (3-Chloropropyl) trimethoxysilane, 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide methiodide, epichlorohydrin, glyoxal, formaldehyde, ethylenediamine, glycidol e carbonyldiimidazole.79,80 In some cases glutaraldehyde is able to modify some amine groups, resulting in a significant loss of biological activity, leading to the need of a key variable, the crosslinking agent, making it necessary to optimize the crosslinking concentration, crosslinking time, mass ratio (crosslinking agent: enzymatic protein).71,79 The CLEA technique is attractive due to its simplicity and robustness, is based essentially on the purification and immobilization in a single step.17,72,76 They present nuances that characterize them as a superior immobilization method, presenting highly concentrated enzyme activity in the catalyst, considerable space time yield, high operational stability, storage and are easily prepared from raw enzyme extracts and there is the possibility of co-immobilize different enzymes, besides, they present decrease of the costs of production for not needing a solid support.17,71,75,81,82 The development of CLEA emerged from the inherent disadvantage of commercialization into industrial biocatalysts of cross-linked enzyme crystals CLECs, which is often a laborious procedure requiring enzyme of high purity.83 Actually, there are any numerous companies that market CLEAs. Bifunctional agents provide strong bonding between the carrier and the clusters present in the enzyme (hydroxyl, mercapto or amine), leading to greater conformational flexibility, where the amine content of the enzyme is relatively low, crosslinking may be inefficient, leading to need to use additives to implement the lack of amine groups.80,84 Alternatively, there is the use of bovine serum albumin (BSA) which has a large number of amine groupings, its use may increase the activity and stability of the CLEA.84-86 When studying the CLEA-protease obtained from catfish viscera Mahmod et al.,87 analyzed its activity, stability and recyclability, in which it obtained a specific activity of 4.512 U mg-1 protein, the highest activity was reached at pH 6.8 and temperature of 45 °C. The CLEA-protease retained 28% of its original activity after six cycles. In the study by Rehman et al.,75 a Pencillium Notatum Lipase (PNL) was immobilized by cross-linked using glutaraldehyde (GLA) and ethylene glycol-bis [succinic acid N-hydroxysuccinimide (EG-NHS) as crosslinking agents. The aggregates of EG-NHS presented higher hydrolytic activity (52.08 ± 2.52%) and esterification (64.42%) in comparison with GLA aggregates that obtained only 23.8 ± 1.86% and 34.54% of hydrolytic activity and esterification, respectively. The GLA and EG-NHS CLEAs reticulated lipase retained, respectively, 63.62% and 70.9% of their original activities after 10 cycles of reuse in aqueous medium.75 Studies of Zhou et al.,81 used for the first time CLEAs of nitrile hydratase (NHase) ES-NHT-118 of Escherichia coli prepared with ammonium sulfate as the precipitating agent, with later cross-linking with dextran polyaldehyde. Where got around 50% of activity recovery with dextran polyaldehyde as cross-linker, while with glutaraldehyde was obtained only 13.79% of recovered activity.81 Hybrid magnetic CLEAs differ from traditional magnetic CLEAs by the fact that in the preparation of the latter, the enzymes are generally immobilized on nano particles through covalent bonds, already hybrid magnetic CLEAs the enzymatic aggregates formed cross-links between each other and were introduced into nanoparticle aggregates of no covalent bonds.87,88 Applying the magnetic CLEAs technique, Peirce et al.,89 developed biocatalysts based on carbonic anhydrase (CA), bovine carbonic anhydrase (bCA) e magnetic nanoparticles (NPs). The maximum immobilization yield obtained by Peirce et al., was 84% and the maximum activity (1268 WAU mg-1 CLEA) was measured for CLEAs prepared with 100 mmol L-1 glutaraldehyde in 4 °C, after 16 h of cross-linking and 0.5 gNPs gbCA-1.89 By the addition of nanoparticles of Fe3O4 and subsequent crosslinking with glutaraldehyde, Cui et al.,88 obtained positive results in the activity of CLEAs hybrids of phenylalanine ammonia lyase (HM-PAL-CLEAs), presenting Vmax of HM-PAL-CLEAs 1.75 higher than in CLEAs, however, there was no significant variation of the Km of the enzymes in CLEAs and HM-PAL-CLEAs. When comparing CLEAs without NPs (nano particles of nano-TiO2, nano-MgO, nano-Ni, nano-Cu e nano-Fe3O4) and with NPs, Wang et al.,90 observed that the value of km of NTiO2-CLEAs decreased by 24.9% and the value of Vmax increased by 57.6%, indicating that the affinity and activity towards the substrate of CLEAs were increased by the addition of nano-TiO2.90 In contrast, the other four NPs decreased the activity of CLEAs due to less amorphous cavities and larger or smaller particle sizes when compared to CLEAs without NP.90 Briefly, some papers in the literature that use CLEAs as a method of immobilization are shown in Table 3. Where the utilized enzymes, the crosslinking agent, the optimum pH and temperature are analyzed, and the results of the yield/activity recovery/relative activity. Thus, CLEAs technique is a carrier-free immobilization method and can be applicable to the immobilization of many enzymes.
Combi-CLEAs CLEAs combined are a new perspective for the immobilization of a mixture of enzymes, elucidating cases of cascade reactions or reactions in which the catalyst must attack several substrates by different enzymes.74 Immobilization on pre-existing substrates may be a problem when using a mixture of enzymes because the protein with the largest size will determine the pore diameter of the support, thereby controlling the final specific area.74,92 As can be observed in the study by Chen et al.,93 in which neutrase (EC 3.4.24.4) and papain (EC 3.4.22.2) were immobilized together by cross-linked enzyme aggregates (N-P-CLEAs) and their properties characterized. The best ratio crosslinking enzyme:glutaraldehyde was 1:5 (v v-1) and the optimized crosslinking time was 4 h.93 The relative activity of papain and free neutrase was 94.58% and 33.42%, respectively, while the activity with ammonium sulphate was obtained 83.81% and 20.39%, respectively, for papain and neutrase.93 Neutrase and papain immobilized by cross-linking showed better thermal stability (with cross-linking presenting 70.55% relative activity, while with free enzyme only 3.78% relative activity), pH stability (when compared with free enzymes NP-CLEAs showed higher relative activity over a wide range of pH, except for pH of 2 and 4), and showed high retention of activity in apolar and hydrophilic solvents without loss of activity for more than six months (4 ºC ).93 Assuming that cascade reactions involving unstable intermediates are often found in biological systems, Nguyen and Yang94 have developed combi-CLEAs to catalyze a cascade reaction involving hydrogen peroxide as an unstable intermediate. A co-axial flow reactor for the production of combi-CLEA with two enzymes, glucose oxidase (GOx) and horseradish peroxidase (HRP).94 Based on the fact that the first GOx enzyme catalyzes the oxidation of glucose and produces hydrogen peroxide, which is used by the second HRP enzyme to oxidize 2-2-azino-bis (3-ethylbenzothiazoline 6-sulphonicacid) (ABTS).94 The apparent reaction rate of the cascade is 10.5 ± 0.5 μmol L-1 min-1, at the enzyme ratio of 150:1 (GOx: HRP), it being verified that even in the presence of catalase, an enzyme that rapidly decomposes hydrogen peroxide only decreases by 18.7% to 8.3 ± 0.3 μmol L-1 min-1 the reaction rate.94 Nguyen and Yang, showed that intermediate hydrogen peroxide is not decomposed by catalase due to a short diffusion distance between GOx and HRP in the combi-CLEA. 94
ENTRAPMENT OR ENCAPSULATION OF ENZYMES Enzyme entrapment or encapsulation of enzymes is based on the entrapment of the enzyme in a polymer network (gel network), which may be organic polymers, silica sol-gel (the sol-gel method is given by the formation of an inorganic polymer network by gelling reactions at low temperatures) or a membrane device (a hollow fiber or a microcapsule).95-98As shown in the Figures 1 (E) and Figure 1 (F). The entrapment disadvantages such as leakage and high resistance to mass transfer to the substrate, on this, Adhikari et al.,99 proposed the entrapment using a special cationic polymer, the poly (2-(dimethylamino) ethyl methacrylate) (MADQUAT) on a single-wall carbon nanotube and reduced graphene oxide (SWCNT-r GO) nanohybrid thin film.99 The nanocomposites/hybrids can present improvements in properties by combining the advantages of their constituent components.99-101 Adhikari et al.,99 used alcoholdehydrogenase (ADH) in the entraptment for the design of an electrochemical biosensor for the detection of ethanol, were tested in real samples of wine, beer and alcohol in the blood, presenting as promising in the analytical and biomedical applications. In the literature, there are numerous applications of enzyme immobilization by encapsulation/entrapment. The Table 4 brings a summarizes of the encapsulation/entrapment enzymes and this industrial applications, for example, promising biocatalyst for industries, Biodiesel production, among others. More detailed information can be found in the following table.

Hydrogels and enzymatic applications Hydrogels are based on cross-linked hydrophilic polymers, among their main characteristics is the increase of volume when coming in contact with water or biological fluids.109-111 The microenvironment of free enzyme catalysis can be imitated by the hydrated hydrogel matrix, benefiting the mobility and flexibility of the immobilized enzymes, causing a high catalytic enzymatic activity.111 Hydrogels gain space in biologically important areas such as drug delivery and release,112-114 release of DNA,115,116 entrapment and release of enzymes117,118 and biosensor.119 However, enzymes immobilized in hydrogels tend to pour out of the gel, because the enzyme is basically encapsulated in the small pores of the gel, against this, functional hydrogels utilize functional building blocks, such as proteins.96,102,120 The encapsulation efficiency and controlled release of the enzymes from the gel matrix can be optimized by the addition of chitosan, modified polymers or proteins.104,105 Alginate is one of the most compatible polymers for immobilization and microencapsulation due to advantages such as: hydrophilic nature, presence of carboxylic acid groups, natural, mechanical stability and stability under extreme operating conditions.103,105 Avnir et al.,106 used sol-gel matrices for enzymatic immobilization through the encapsulation formed by hydrolytic polymerization of metal alkoxides or tetraethoxysilane. The wide use of this type of matrix is due to the fact that it is a procedure performed at room temperature (biomolecules can withstand) and that the operating conditions are soft so that there is no denaturation of the encapsulated biomolecules.95,106 According to Zhang et al.,107 the use of encapsulated enzymes increased juice clarification activity and removal of polyphenol compounds. Zhang et al., studied the encapsulation of enzymes (β-galactosidase) in hydrogel beads of carrageenan as a way to optimize its use and activity in food. The activity of the free enzyme was only 63 μmol min-1, while for the encapsulated enzyme it was 266 μmol min-1 at pH 7.121 Encapsulation of enzymes with chitosan Chitosan appears as an applicable support for the encapsulation of enzymes due to its unique characteristics, such as: non-toxic, biocompatible, physiological inertia, cheap and biodegradable.32,122-127 In view thereof, Bilal et al.,128 studied the immobilization by encapsulation of horseradish peroxidase (HRP) in chitosan pearls aiming at the degradation of textile dyes, the granules of chitosan (2.5% of chitosan) presented maximum immobilization yield of approximately 92.54%, the relative free and encapsulated HRP activities were decreased after preincubation above 30 °C and 50 °C, respectively, after 120 min at 70 °C, encapsulated HRP retained 48.3% of activity, while free HRP retained 19.35% of activity. Long et al.,129 also used chitosan, in which the sol-gel encapsulation of pullulanase was carried out in the presence of chitosan/Fe3O4 magnetic nanoparticles, the enzyme immobilized by sol-gel encapsulation retained 52% of its initial activity (after 5 h at 62 °C), while the free enzyme retained only 11% activity. They chose this type of process for the immobilization of pullulanase because the mechanical entrapment of the enzymes using sol-gel materials allows the stabilization of the tertiary structure of the protein and the high retention of activity.129-132 Due to this, in recent years the interest in using bioencapsulation, in which the possibility of immobilizing bioactive molecules within silica gels appears as an alternative route for the development of biosensors and/or bioreactors.129 Smart support Special attention can be given to intelligent polymers in which special resources are added to the colloidal compounds, making them sensitive to the stimuli employed, changing their properties, such as temperature, ionic strength, solvent polarity, electric/magnetic field, light or biomolecules. 133,134 Using smart polymers responding to stimuli are delivery vehicles that are being considered as promising for controlled encapsulation and release of drugs.135,136 In the study, gadolinium oxide and europium-encapsulated temperature/pH-responsive polymeric particles (PLTPPs) were synthesized by emulsifier-free emulsion polymerization presented excellent biocompatibility with C6 cellulases and anticancer drug loading capacity (doxorubicin, DOX).137 Encapsulation by alginate The most applied and studied polymer for cell encapsulation is alginate, which is an unbranched heteropolysaccharide of 1-4 glycosidically linked β-d-mannuronic (M) and α-l-guluronic (G) acids in different sequences and compositions.138 In the work of Takenaka et al.,139 the Pyruvate Ferredoxin Oxidoreductase was purified from Citrobacter sp. S-77 (PFORS77) with the aim of developing a method for producing acetyl-CoA. Takenaka et al., were able to immobilize PFORS77 in ceramic hydroxyapatite (PFORS77-HA) obtaining an immobilization efficiency superior to 96%. After the encapsulation of PFORS77-HA in the alginate, the catalytic production rate of acetyl-CoA was highly reduced to 36% when compared to the free enzyme, however, it maintained more than 68% of the initial activity (even after 10 cycles), thus exhibiting a high operational stability of PFORS77-HA in alginate hydrogels.139 In order to optimize the reaction of biodiesel production, Ferreira et al.,140 immobilized the lipase from Pseudomonas fluorescens in mixtures of silk fibroin with calcium alginate beads (FA-LPf) which was applied to the transesterification of soybean oil with ethanol to obtain ethyl biodiesel (fatty acid ethyl esters, FAEE), where the maximum yield of FAEE (63%) was achieved after 96 h at 32 °C and 400 rpm in 30% (v v-1) n-hexane, which utilized 20% (w w-1) immobilized lipase, thereby showing the efficacy of an enzyme immobilized on a carrier biodegradable LPFf.
ADSORPTION Enzymatic adsorption is the simplest method of immobilization. This technique refers to the interaction between the support and the enzyme from ionic interactions or hydrophobic interactions. 93 The type of interaction that will be formed will depend on the surface chemistry of the carrier and the type of amino acid present on the surface of the enzyme. The disadvantage of this technique is the weak interactions (ionic and hydrophobic interactions, hydrogen bonds and van der Waals forces) between the enzyme and the carrier, making the compound produced unstable compared to the compounds produced by other techniques.141 Figure 1 (G) shows the scheme of the general adsorption and desorption process. Many studies have used adsorption as a method of enzyme immobilization and some studies are reported in Table 5. It can be noticed that the adsorption is very used for the enzyme stabilization achieving quite significant results. This result can be seen through the high number of recycles that were performed without having a loss of catalytic activity of the enzyme. In addition, the Table 5 shows that the reactions mentioned occur at neutral pH or close to neutral.
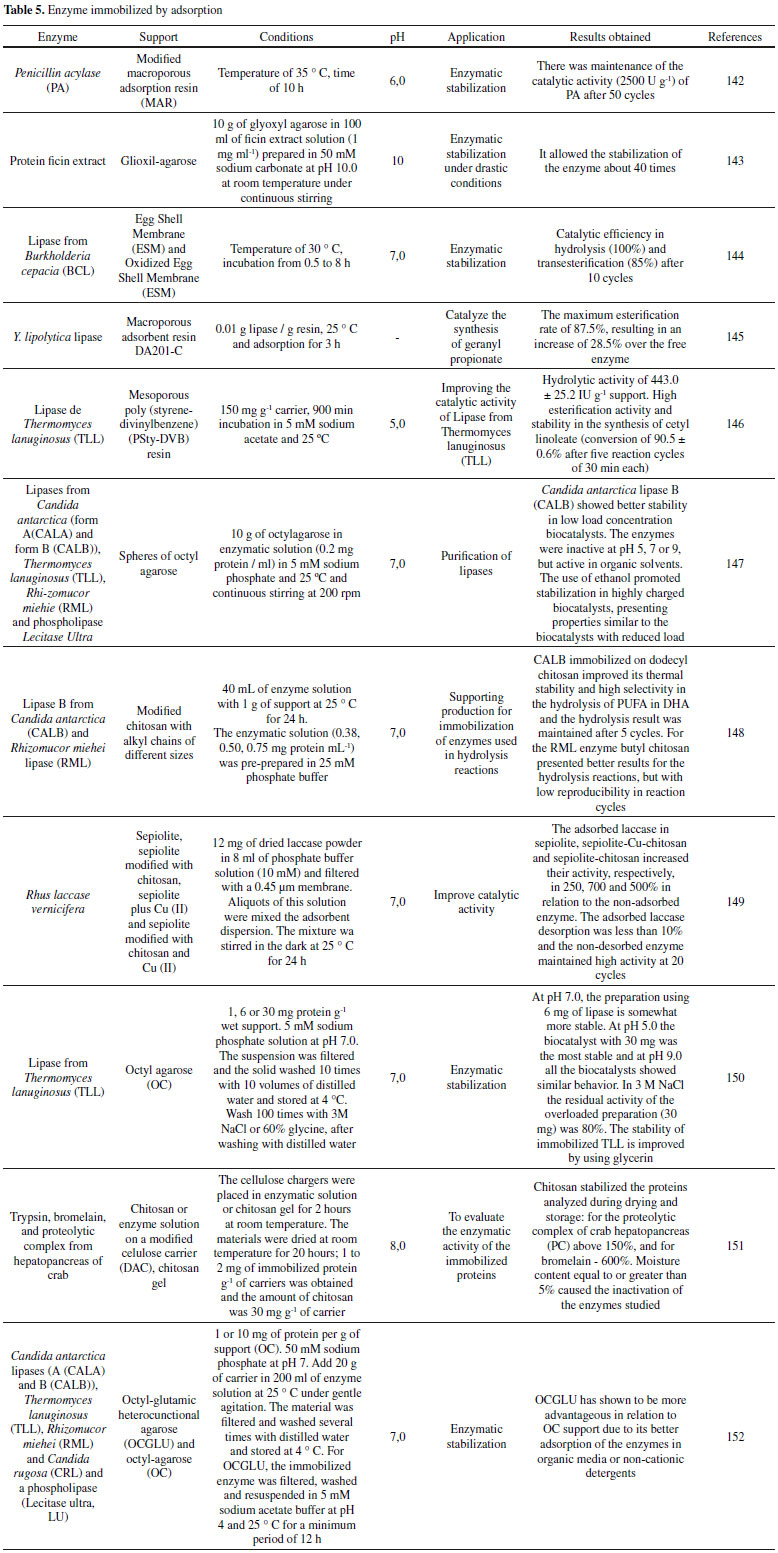
For López-Gallego et al.,153 the supports are very diverse and help to improve the reactional yields of the immobilized enzyme. A carrier must possess some essential properties for the immobilization of enzymes, such as physical resistance to compression, hydrophilicity, biocompatibility, resistance to microbial attacks and low cost. Some papers are presented using some support, inorganic and organic.32,154,155 Silica Zhou et al.,156 performed immobilization studies of CALB lipase on silica by adsorption in order to improve the soluble expression of CALB in E. coli. The optimal condition was achieved with N-terminal 6-histidine-labeled CALB lipase and C-terminal 10-lysine (6 His-CALB-10Lys), which showed high solubility (0.1 mg mL-1) and specific activity (10.1 U mg-1). This system, CALB and silica, has high affinity in immobilization processes, resulting in high values of immobilized material.157 The kinetics of Michaelis-Menten indicate that the affinity, with respect to the type of conformation of the enzyme-substrate complex,158,159 enabling better enantioselectivity of immobilized 6 His-CALB-10Lys enzymes in relation to other enzymes commercial in the resolution of (S)-N-(2-ethyl-6-methylphenyl) alanine ((S)-NEMPA). 156 In the work of Soldatkina et al.,160 a new amperometric biosensor was developed for the detection of glutamate by the immobilization of glutamate oxidase (GlOx) on silicalite particles by adsorption. Biosensors of glutamate oxidase or decarboxylase adsorbed on silica showed high sensitivity to glutamate.142,160 These biosensors exhibit excellent specificity, being able to detect glutamate in small amounts, reproducibility of results for several hours and stability to operate for several days.143 Silica nanoparticles can be used as support for photochromic derivatives of 1-vinylidene-naphtho furan by direct adsorption and covalent interaction. The immobilization technique directly influenced the activity results, the adsorbents being immobilized by the ones that showed photochromic activity, revealing intense staining at pH 6.0. This activity was maintained for up to 8 test cycles of exposure to sunlight or radiation.144 Different materials have adsorption characteristics, but to remove nitrobenzene using this technique the activated carbon is very used because it has a porous structure that allows a larger surface for interaction.145,146 In an attempt to obtain a compound with better adsorption properties, a new formed material was obtained using silica doped with activated charcoal, this material showed stability at the absorption values around 5 minutes.147 Nanomaterials Borlido et al.,148 studied the incorporation of monoclonal antibodies into magnetic nanoparticles sensitive to magnetic stimuli used to increase the adsorption/desorption efficiency of these antibodies and to be easily separated and purified from the culture medium. These particles showed superparamagnetic behavior, exhibiting saturation magnetization of 12.6 emu g-1. 148 Jiang et al.,149 functionalized Fe3O4 nanoparticles with polyethyleneimine to remove lead (Pb+ 2) from aqueous medium, being reused for several cycles. Some parameters were modified to optimize the process, obtaining high absorption (143 mg g-1) when compared to other similar works.150,151 Many studies are designed to minimize the release of silver into the environment, since this metal is widely used in several products because it has antimicrobial potential.152,161-163 A study was carried out to investigate the controlled release of immobilized silver nanoparticles in thin layer of aluminum oxide. A 15 nm layer of this oxide is capable of inhibiting the release of silver for up to 48 h, since a 2 nm layer delayed the release for 4 h.164 Another release study using nanoparticles was the work done by Mohammady et al.,165 obtaining nanoparticles with a mean size of 120 to 300 nm from the poly-lactide-co-glycolide polymer (PLGA). The transport of the drug irinotecan, an anticancer that inhibits topoisomerase I, has been investigated and has shown cytotoxic activity in vitro.165 Biopolymers Chitosan is a very abundant natural polymer, composed of β-1,4-linked glucosamine and N-acetylglucosamine, and can be obtained by N-deacetylation of chitin. This biopolymer has several amine groups which gives it many chemical and biological applications in various areas.166-170 Chitosan can be used as a support matrix in enzyme immobilization processes, such as chitosan beads produced with 1-butyl-3-methylimidazolium acetate (A) and 1-butyl-3-methylimidazolium chloride (B) for removal of the Malachite green dye in aqueous solutions. The chitosan beads were used in the work because they present a larger contact surface compared to pure chitosan. The optimum reaction conditions at pH 4.0 revealed absorption of 8.07 mg g-1 of A and 0.24 mg g-1 of B.171 Other works have reported the use of chitosan for application of removal of substances from aqueous solutions. Zhu et al. developed a composite of chitosan and cerium to remove fluoride from the water. Removal analysis of this composite showed absorption of 153 mg g-1 at 20 °C and pH 3.0, a much higher value when compared to crude chitosan (13.2 mg g-1). Fluoride is removed from the medium by adsorbing fluoride on its surface by electrostatic attraction and obtained good reproducibility after 3 cycles.172 Bacillus subtilis bacteria were immobilized on chitosan beads to remove copper (II) from aqueous solutions. The study revealed that the optimum pH in the process for copper absorption is 6.0. The chitosan beads with copper ions were removed with 0.1 mol L-1 NaOH solution and washed with water to pH 7.0 for reuse, maintaining 76% yield after 5 cycles.173 Many diseases are administered by drugs174 and chitosan can be applied for this purpose as a carrier for drug loading. One study studied the effect of methotrexate used for treatment of intraocular lymphoma using a chitosan-mediated poly (lactic-co-glycolic acid) support, obtaining support with drug release for 3 to 5 months.175
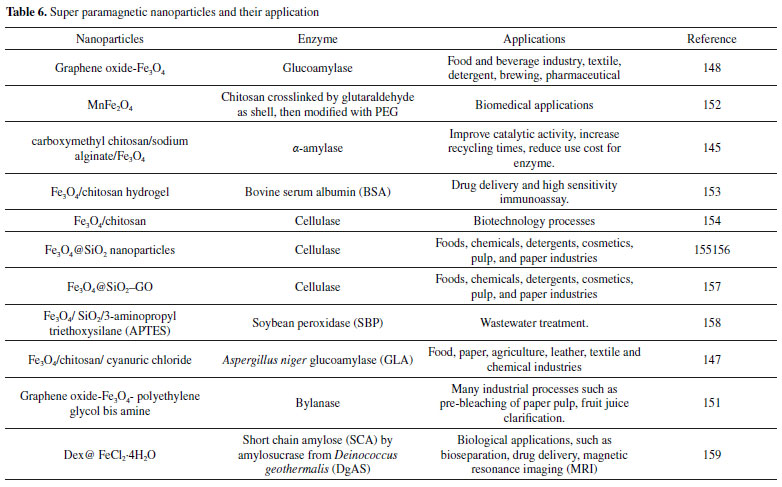
HYBRIDS The industrial application of the soluble enzyme as a biocatalyst is economically unattractive because of its high cost and inconvenience in separation, recycling, and reusing.176,177 For these reasons, enzyme immobilization on hybrids materials shows interesting advantages over these problems, including high stability and reusability and highly concentrated enzymatic activity.126,178-181 Magnetic nanoparticles hybrid The use of nanotechnology in biology and medicine is expected to produce main advances in molecular diagnostics, therapeutics and bioengineering.182 Magnetite (Fe3O4) is currently the most commonly used among the various nano-oxides exhibiting magnetic properties. This high level of concern about this substance is caused by its high magnetic saturation, low cytotoxicity, good biocompatibility and stability in a variety of physiological conditions.183-185 Magnetite has recently been commonly combined with natural polymers or with other materials such as graphene, graphene oxide or carbon nanotubes as well as poly (3-thiophene acetic acid), L-carnosine and alginic acid to produce functional hybrid nanomaterials or nanocomposites183,186-190 as shown in Figure 1 (H). Partially phosphonated polyethylenimine (PEIP) has been studied as a functionalisable coating agent for iron oxide nanoparticles. For example, Monteil et al.,191 synthetized a magnetic nanoparticles (NP) suitable for biofunctionalization. The PEIP was used for coating nanoparticles (NP-PEIP) for trypsin immobilization. The NP-PEIP was conserved at room temperature at neutral pH during few months without important loss of activity confirming the fact that coating is important for immobilized enzyme performance providing higher stability.191 You et al.,192 also used polyethylenimine in their studies. Magnetic chitosan-polyethylenimine (Fe3O4/CS-PEI) polymer composite was synthesized. The porous magnetic Fe3O4/CS-PEI composite demonstrated ultrahigh capacity (1876 m g-1) for one of the watersoluble anionic dyes, Congo Red (CoR) removal. It removed over 99.3% of CoR (100 mg L-1) when the dosage was over 1.4 g L-1. They proved that a higher temperature was benefit to CoR removal. The Fe3O4/CS-PEI composite was effective for CoR removal in a wide pH range (3-13).192 Super paramagnetic nanoparticles hybrid In the earlier years, magnetic nanoparticles have been observed as a potential carrier materials for the preparation of heterogeneous catalysts.193-195 The properties of magnetic nanostructuredmaterials such as superparamagnetic behavior, low cost, high chemical and physical stability in a large range of operational conditions, biocompatibility, low toxicity and eco-friendly characteristic, ease of separation and high capacity for loading biomacromolecules make them useful as supports for the immobilization of protein.193-195 Nano Fe3O4 possesses superparamagnetism and it can be effectively concentrated, separated and reused in a magnetic field.196,197 Amkirbandeh et al.,198 studied the use of magnetic nanoparticles for a simple covalent immobilization procedure. It showed a high loading capacity, and high catalytic activity, thermal stability and easily reusability of the immobilized glucoamylases (GLA), which can be used to the immobilization of other industrial enzymes. In the work, aspergillus niger glucoamylase (GLA) was covalently immobilized on 1-3-5-triazine-functionalized chitosan coated superparamagnetic nanoparticles (MNPCh-CC). The GLA immobilized on nanocarrier showed great catalytic activity at pH 4.5 and 60 °C.198 It could be noticed that the immobilized GLA showed quite impressive stability, even after 10 reaction cycles, it could still retain about 70% of the initial activity.198 The results of this work showed that immobilization process could not significantly inhibit enzyme-substrate interaction and subsequently retained its effective catalytic activity.198 For the authors, the substantial improvement of reactivity, reusability, and stability of this biocatalyst system may confer it a wider range of applications in industrial processes.198 In another study Inagaki et al.,199 reported a simple way to synthesize grapheme oxide nanosheets decorated with functionalized Fe3O4 magnetic nanoparticles as unique and convenient nanoplatforms for immobilization of glucoamylase. The immobilizated glucoamylase on triazine-functionalized Fe3O4/graphene oxide nanocomposite (GO/MNP-CC/GLA) showed great catalytic activity at pH 6.5 and 60 °C and keep more than 96% of the activity of free glucoamylase.199 Notably, GO/MNP-CC/GLA exhibited quite impressive stability, even after 20 reaction cycles and it could retain more than 56% of the initial activity.199 Supperparamagnetic graphene oxide (GO/MNP) has been received major attention especially in enzyme engineering researches due to its unique magnetic properties, two-dimensional structure, easy fabrication, low toxicity, great reusability, large surface area, simple manipulation of surface modification, and large enzyme loading capacity.199-201 The Table 6 shows some examples of superparamagnetic nanoparticles hybrid and their application. It can be seen in the Table 6 that this type of configuration has a wide industrial application, and can be used, for instance, from the food industry to the production of drugs and paper. It may also be noted that Fe3O4 is most commonly used to give the magnetic property to the carrier.
Organic – Inorganic hybrid nanoflower In recent years, organic-inorganic hybrid nanoflowers technology has been considered as an effective immobilization method.202,203 This method has motivated a major interest in exploiting them as a potential matrix for biomolecule immobilization because of their simple synthesis, high efficiency, great promise of enhancing biomolecule stability, activity and even selectivity.204-206 The formation of the organic-inorganic hybrid nanoflowers comprehend the following steps: nucleation, growth and completion.207,208 The formation of hybrid nanoflowers is shown in Figure 1 (I). The hybrid organic-inorganic nanoflowers were first developed by He et al.,209 using copper (II) ions as the inorganic component and enzyme as the organic component. Since then, the attention in synthesizing protein molecule-metal phosphate hybrid materials has been majorly increased.210,211 As demonstrated by Nadar et al.,212 the copper phosphate was used to prepare an organic-inorganic hybrid glucoamylase nanoflower. The aqueous CuSO4 solution was added to phosphate buffered saline (PBS) containing enzyme.212 The hybrid nanoflowers of this work showed 204% enhanced activity recovery and two folds improvement in thermal stability in terms of half-life (in the range of 50-70 °C) with respect to the free form. Besides that, it showed higher storage stability with retention of 91% activity after 25 days of incubation.212 Copper phosphate also was used to synthesize lactoperoxidase (LPO).213 Altinkaynak and coworkers developed a hybrid nanoflowers (HNF) formed of LPO enzyme purified from bovine milk and copper ions (Cu2+) were synthesized at two different temperatures (+4 °C and 20 °C) in PBS (pH 7.4). LPO-copper phosphate HNF has upper activity than free LPO. LPO-copper phosphate HNFs exhibited ~160% and ~360% increase in activities at pH 6 and pH 8, respectively, when compared with free LPO. 213 On the other hand Chung et al.,214 studied the immobilization of α-acetolactate decarboxylase (ALDC) using enzymeinorganic hybrid nanoflowers (Ca3(PO4)2-ALDC) and the results obtained show only a small increase.214 The activity of Ca3(PO4)2-ALDC nanoflowers increased by 10% compared with that of free ALDC.214 He et al.,209 demonstrated that the activity of laccase-incorporated copper phosphate nanoflowers was valued to exhibit 4.5-6.5 times more active at oxidizing catecholamine and syringaldazine than free laccase. In the meantime, the laccase-incorporated Cu3(PO)4 nanoflowers showed exceptional storage stability and reusability. In addition, Chung and collaborators used copper phosphate to develop a mediatorless glucose biofuel cell based on hybrid nanoflowers incorporating enzymes including glucose oxidase (GOx), laccase, or catalase with copper phosphate.215 A higher power density up to 200 W cm-2 were obtained using the enzyme nanoflowers-based biofuel cell system without mediator, which was almost 80% to that from the biofuel cell system prepared with the corresponding free enzymes.215 They affirm that by applying enzyme nanoflowers to the biofuel cell, they achieved good improved performance stability. Based on the application results to biofuel cell, it is notable that enzyme nanoflowers can be utilized for various enzyme catalysis-based applications such as biosensors and biocatalysis.215 Different metal ions can form hybrid nanoflowers.202 The preparation of different hybrid nanoflowers based on the type of metal ions and biomolecules used is shown in the Table 7.
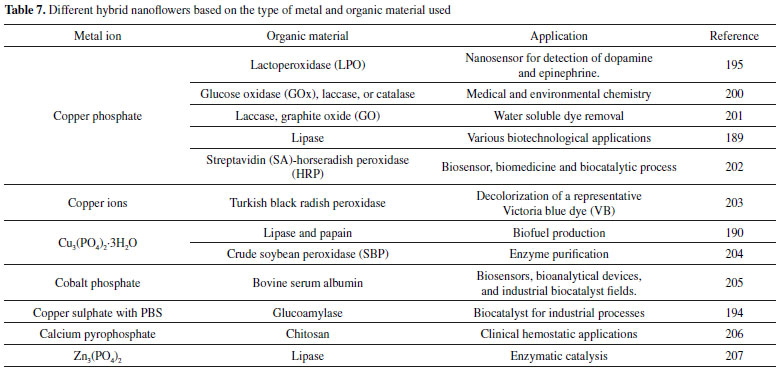
Table 7 shows different combinations for the formation of hybrid nanoflowers, as well as their possible applications. It is noticed that there is a wide industrial application being an advantage when compared to other types of immobilization since it has no restrictions. It can also be seen that various materials are capable of forming hybrids, thus making a good method of immobilization. Another metal ion is the zinc ion that has no destructive action on proteins.108,216 Zhang et al.,108 developed a papain/Zn3(PO4)2 hybrid nanoflower by the precipitation method. They proved that the catalytic properties of papain immobilized on hybrid nanoflowers are enhanced compared with that of free papain. Zhang also studied a lipase/Zn3(PO4)2 hybrid nanoflower.217 The catalytic performance of lipase/Zn3(PO4)2 hybrid nanoflower was measured and the optimal catalytic conditions have been found. The maximum enzyme activity was 855 ± 13 U g-1. In comparison with the free lipase, the enzyme activity increment of hybrid nanoflower is 147%. For them, the results indicate that the well-designed materials should be useful in industrial enzyme catalysis.108 Smart polymer hybrid Conjugated polymers (CPs) are vastly used in the field of biosensors due to their electric, electronic, magnetic and optical properties.218,219 Immobilization of enzymes onto organic or inorganic polymer matrices has been developed to overcome some drawbacks associated to their routine use, such as the lack of longterm stability and the difficulty in their recovery and reuse.220,221 The most extensively studied conducting polymers are polyacetylene (PA), polyaniline (PANI), polypyrrole (PPy), polythiophenes (PTh), polyparaphenylene (PPPh), polyparaphenylene vinylene (PPV) and polyorthotoluidine (POT) and their derivatives.222 Liu et al.,223 demonstrated the new concept of enzyme-hybrid poly (3-4-ethylenedioxythiophene) (PEDOT) microspheres (PEDOT-MSs) as an advanced processable bio-conducting interface material for the facile fabrication of electrochemical biosensors.223 The microstructure of PEDOT-MSs supply a larger active conducting surface for intimate immobilization of enzyme molecules (i.e. glucose oxidase-GOx).223 Their results showed that the GOx-PEDOT-MS showed a good sensitivity of 116.25 μAm (mol L-1 cm-2) -1, a limit of detection of 1.55 μmol L-1, and retained 97% of the sensitivity after 12 days storage at room temperature.223
CONCLUSIONS In the present study, we demonstrated the versatility and main strategy used for the preparation of immobilized biocatalysts. In this context, recent years were dedicated to the development of new immobilized enzymes for industrial application. The strategies presented are an efficient way of the possibility of immobilization and enzymatic purification. This revision includes the relevant questions that will help the researcher to make a decisive decision in choosing the best enzyme immobilization strategy. In this way, new ideas, different support, chemical modification of proteins, and others solutions have been proposed in recent years for the immobilization of enzymes and it can be expected that this trend increase. As a result, there is a significant progress in chemical and biotechnological processes aiming to expand the performance of industrial enzymes.
ACKNOWLEDGMENTS We gratefully acknowledge the financial support of Brazilian Agencies for Scientific and Technological Development, Fundação Cearense de Apoio ao Desenvolvimento Científico e Tecnológico - FUNCAP (project number BP3-0139-00005.01.00/18), Conselho Nacional de Desenvolvimento Científico e Tecnológico – CNPq (project number 422942/2016-2) and Coordenação de Aperfeiçoamento de Ensino Superior (CAPES).
REFERENCES 1. Pinho, G. P.; Matoso, J. R. M.; Silvério, F. O.; Mota, W. C.; Lopes, P. S. N.; Ribeiro, L. M.; J. Braz. Chem. Soc. 2014, 7, 1246. 2. Luca, G. C.; Reis, B. F.; Quim. Nova 2001, 24, 191. 3. Madhu, A.; Chakraborty, J. N.; J. Clean. Prod. 2017, 145, 114. 4. Fatibello-Filho, O.; Vieira, I. da C.; Quim. Nova 2002, 25, 455. 5. Rebelo, L. P.; Netto, C. G. C. M.; Toma, H. E.; Andrade, L. H.; J. Braz. Chem. Soc. 2010, 21, 1537. 6. Freitas, L.; Perez, V. H.; Santos, J. C.; De Castro, H. F.; J. Braz. Chem. Soc. 2007, 18, 1360. 7. Ruiz, M. I.; Sanchez, C. I.; Torrresa, R. G.; Molina, D. R.; J. Braz. Chem. Soc. 2011, 12, 2337. 8. Melo, W. C.; Santos, A. S. dos; Santa Anna, L. M. M.; Pereira Jr., N.; J. Braz. Chem. Soc. 2008, 19, 418. 9. Wells, A.; Meyer, H.; ChemCatChem. 2014, 6, 918. 10. Machado, A.; Liria, C. W.; Proti, P. B.; Remuzgo, C.; Miranda, M. T. M.; Quim. Nova 2004, 27, 781. 11. Marques, P. R. B. D. O.; Yamanaka, H.; Quim. Nova 2008, 31, 1791. 12. Schuchardt, U.; Sercheli, R.; Vargas, R. M.; J. Braz. Chem. Soc. 1998, 9, 199. 13. Alvim, J.; Dias, R. L. A.; Castilho, M. S.; Oliva, G.; Corrêa, A. G.; J. Braz. Chem. Soc. 2005, 16, 763. 14. Neto, S. A.; De Andrade, A. R.; J. Braz. Chem. Soc. 2013, 24, 1891. 15. Mantovani, S. M.; De Oliveira, L. G.; Marsaioli, A. J.; J. Braz. Chem. Soc. 2010, 21, 1484. 16. Lima, M. L. S. O.; Gonçalves, C. C. S.; Barreiro, J. C.; Cass, Q. B.; Marsaioli, A. J.; J. Braz. Chem. Soc. 2015, 26, 319. 17. Sheldon, R. A.; Van Pelt, S.; Chem. Soc. Rev. 2013, 42, 6223. 18. Raman, K.; Henning, P.; J. Clean Prod. 2013, 42, 228. 19. Hanefeld, U.; Gardossi, L.; Magner, E.; Chem. Soc. Rev. 2009, 38, 453. 20. Liese, A.; Hilterhaus, L.; Chem. Soc. Rev. 2013, 42, 6236. 21. Pinheiro, B. B.; Rios, N. S.; Rodríguez Aguado, E.; Fernandez-Lafuente, R.; Freire, T. M.; Fechine, P. B. A.; dos Santos, J. C. S.; Gonçalves, L. R. B.; Int. J. Biol. Macromol. 2019, 130, 798. 22. Rios, N. S.; Pinheiro, B. B.; Pinheiro, M. P.; Bezerra, R. M.; dos Santos, J. C. S.; Barros Gonçalves, L. R.; Process Biochem. 2018, 75, 99. 23. Yan, Y.; Zhang, X.; Chen, D.; Bioresour. Technol. 2013, 131, 179. 24. Rios, N. S.; Pinheiro, M. P.; dos Santos, J. C. S.; de S. Fonseca, T.; Lima, L. D.; de Mattos, M. C.; Freire, D. M. G.; da Silva, I. J.; Rodríguez-Aguado, E.; Gonçalves, L. R. B.; J. Mol. Catal. B: Enzym. 2016, 133, 246. 25. Polyák, P.; Urbán, E.; Nagy, G. N.; Vértessy, B. G.; Pukánszky, B.; Enzyme Microb. Technol. 2019, 120, 110. 26. Adamczyk, Z.; Curr. Opin. Colloid Interface Sci. 2019, 41, 50. 27. Hu, W.; Liu, M.; Yang, X.; Zhang, C.; Zhou, H.; Xie, W.; Fan, L.; Nie, M.; Carbohydr. Polym. 2019, 206, 468. 28. Drout, R. J.; Robison, L.; Farha, O. K.; Coord. Chem. Rev. 2019, 381, 151. 29. Snoch, W.; Tataruch, M.; Zastawny, O.; Cichoń, E.; Gosselin, M.; Cabana, H.; Guzik, M.; Bioorg. Chem. 2019, 280, 1. 30. Liu, D.; Chen, Z.; Long, J.; Zhao, Y.; Du, X.; Adv. Polym. Technol. 2016, 0, 1. 31. Zhang, Z.; Li, D.; Zhang, X.; Bioresour. Technol. 2019, 280, 165. 32. Melo, A.; Silva, F.; dos Santos, J.; Fernández-Lafuente, R.; Lemos, T.; Dias Filho, F.; Molecules. 2017, 22, 2165. 33. Kumar, A.; Park, G. D.; Patel, S. K. S.; Kondaveeti, S.; Otari, S.; Anwar, M. Z.; Kalia, V. C.; Singh, Y.; Kim, S. C.; Cho, B. K.; Chem. Eng. J. 2019, 359, 1252. 34. Jun, L. Y.; Yon, L. S.; Mubarak, N. M.; Bing, C. H.; Pan, S.; Danquah, M. K.; Abdullah, E. C.; Khalid, M.; J. Environ. Chem. Eng. 2019, 7, 102961. 35. Darwesh, O. M.; Matter, I. A.; Eida, M. F.; J. Environ. Chem. Eng. 2019, 7, 102805. 36. Elnashar, M. M. M.; Wahba, M. I.; Amin, M. A.; Eldiwany, A. I.; J. Appl. Polym. Sci. 2014, 131, 40295. 37. Lima, G. V.; da Silva, M. R.; de Sousa Fonseca, T.; de Lima, L. B.; de Oliveira, M. da C. F.; de Lemos, T. L. G.; Zampieri, D.; dos Santos, J. C. S.; Rios, N. S.; Gonçalves, L. R. B.; Appl. Catal., A 2017, 546, 7. 38. Verdasco-Martín, C. M.; Villalba, M.; dos Santos, J. C. S.; Tobajas, M.; Fernandez-Lafuente, R.; Otero, C.; Biochem. Eng. J. 2016, 111, 75. 39. Villalba, M.; Verdasco-Martín, C. M.; dos Santos, J. C. S.; Fernandez-Lafuente, R.; Otero, C.; Enzyme Microb. Technol. 2016, 90, 35. 40. Wahba, M. I.; Int. J. Biol. Macromol. 2017, 105, 894. 41. Bommarius, A. S.; Chem. Soc. Rev. 2013, 42, 6534. 42. Cieh, N. L.; Sulaiman, S.; Mokhtar, M. N.; Naim, M. N.; Process Biochem. 2017, 56, 81. 43. Albuquerque, T. L. D.; Rueda, N.; Dos Santos, J. C. S.; Barbosa, O.; Ortiz, C.; Binay, B.; Özdemir, E.; Gonçalves, L. R. B.; Fernandez-Lafuente, R.; Process Biochem. 2016, 51, 865. 44. Dos Santos, J. C. S.; Rueda, N.; Gonçalves, L. R. B.; Fernandez-Lafuente, R.; Enzyme Microb. Technol. 2015, 77, 1. 45. Dos Santos, J. C. S.; Rueda, N.; Sanchez, A.; Villalonga, R.; Gonçalves, L. R. B.; Fernandez-Lafuente, R.; RSC Adv. 2015, 5, 35801. 46. Rueda, N.; Dos Santos, J. C. S.; Torres, R.; Barbosa, O.; Ortiz, C.; Fernandez-Lafuente, R.; RSC Adv. 2015, 5, 55588. 47. Lyu, J.; Li, Z.; Men, J.; Jiang, R.; Tang, G.; Zhou, Y.; Gao, R.; Process Biochem. 2019, 81, 63. 48. Kashefi, S.; Borghei, S. M.; Mahmoodi, N. M.; J. Mol. Liq. 2019, 276, 153. 49. Cheng, G.; Xing, J.; Pi, Z.; Liu, S.; Liu, Z.; Song, F.; Chin. Chem. Lett. 2018, 30, 656. 50. Meller, K.; Szumski, M.; Buszewski, B.; Sens. Actuators, B 2017, 244, 84. 51. Wahba, M. I.; Int. J. Biol. Macromol. 2016, 91, 877. 52. Sulaiman, S.; Cieh, N. L.; Mokhtar, M. N.; Naim, M. N.; Kamal, S. M. M.; Process Biochem. 2017, 55, 85. 53. Rodrigues, R. C.; Godoy, C. A.; Volpato, G.; Ayub, M. A. Z.; Fernandez-Lafuente, R.; Guisan, J. M.; Process Biochem. 2009, 44, 963. 54. Ashjari, M.; Mohammadi, M.; Badri, R.; J. Mol. Catal. B: Enzym. 2015, 115, 128. 55. Dos Santos, J. C. S.; Rueda, N.; Barbosa, O.; Fernández-Sánchez, J. F.; Medina-Castillo, A. L.; Ramón-Márquez, T.; Arias-Martos, M. C.; Millán-Linares, M. C.; Pedroche, J.; Yust, M. D. M..; RSC Adv. 2015, 5, 20639. 56. Dos Santos, J. C. S.; Rueda, N.; Barbosa, O.; Millán-Linares, M. D. C.; Pedroche, J.; Del Mar Yuste, M.; Gonçalves, L. R. B.; Fernandez-Lafuente, R.; J. Mol. Catal. B: Enzym. 2015, 117, 38. 57. Fernandez-Lopez, L.; Rueda, N.; Bartolome-Cabrero, R.; Rodriguez, M. D.; Albuquerque, T. L.; Dos Santos, J. C. S.; Barbosa, O.; Fernandez-Lafuente, R.; Process Biochem. 2016, 51, 48. 58. Pinheiro, M. P.; Rios, N. S.; Fonseca, T. de S.; Bezerra, F. de A.; Rodríguez-Castellón, E.; Fernandez-Lafuente, R.; Carlos de Mattos, M.; dos Santos, J. C. S.; Gonçalves, L. R. B.; Biotechnol. Prog. 2018, 34, 878. 59. Sharifi, M.; Robatjazi, S. M.; Sadri, M.; Mosaabadi, J. M.; Chin. J. Chem. Eng. 2019, 27, 191. 60. De Souza, T. C.; De Fonseca, T. S.; Da Costa, J. A.; Rocha, M. V. P.; De Mattos, M. C.; Fernandez-Lafuente, R.; Gonçalves, L. R. B.; Dos Santos, J. C. S.; J. Mol. Catal. B: Enzym. 2016, 130, 58. 61. Rivero, C. W.; De Benedetti, E. C.; Gallego, F. L.; Pess, ela, B. C.; Guisán, J. M.; Trelles, J. A.; J. Biotechnol. 2017, 249, 34. 62. Ho, L. F.; Li, S. Y.; Lin, S. C.; Hsu, W. H.; Process Biochem. 2004, 39, 1573. 63. Shi, Q. H.; Tian, Y.; Dong, X. Y.; Bai, S.; Sun, Y.; Biochem. Eng. J. 2003, 16, 317. 64. Sardar, M.; Gupta, M. N.; Enzyme Microb. Technol. 2005, 37, 355. 65. Haider, T.; Husain, Q.; Int. J. Pharm. 2008, 359, 1. 66. Datta, S.; Christena, L. R.; Rajaram, Y. R. S.; 3 Biotech 2013, 3, 1. 67. Xu, S.; Zhang, X.; Duan, B.; Chen, J.; Carbohydr. Polym. 2017, 177, 297. 68. Del Monte-Martínez, A.; González-Bacerio, J.; Cutiño-Avila, B.; Rojas, J.; Chappé, M.; Salas-Sarduy, E.; Pascual, I.; Guisán, J. M.; Prep. Biochem. Biotechnol. 2017, 47, 745. 69. Nadar, S. S.; Muley, A. B.; Ladole, M. R.; Joshi, P. U.; Int. J. Biol. Macromol. 2016, 84, 69. 70. Nadar, S. S.; Rathod, V. K.; Enzyme Microb. Technol. 2016, 83, 78. 71. Sheldon, R. A.; Appl. Microbiol. Biotechnol. 2011, 92, 467. 72. Sheldon, R. A.; Biochem. Soc. Trans. 2007, 35, 1583. 73. Piligaev, A. V.; Sorokina, K. N.; Samoylova, Y. V.; Parmon, V. N.; Energy Convers. Manage. 2018, 156, 1. 74. Dal Magro, L.; Hertz, P. F.; Fernandez-Lafuente, R.; Klein, M. P.; Rodrigues, R. C.; RSC Adv. 2016, 6, 27242. 75. Rehman, S.; Bhatti, H. N.; Bilal, M.; Asgher, M.; Int. J. Biol. Macromol. 2016, 91, 1161. 76. Prabhavathi Devi, B. L. A.; Guo, Z.; Xu, X.; JAOCS, J. Am. Oil Chem. Soc. 2009, 86, 637. 77. Barbosa, O.; Ortiz, C.; Berenguer-Murcia, Á.; Torres, R.; Rodrigues, R. C.; Fernandez-Lafuente, R.; RSC Adv. 2014, 4, 1583. 78. La Rotta Hernandez, C. E.; Lütz, S.; Liese, A.; Bon, E. P. S.; Enzyme Microb. Technol. 2005, 37, 582. 79. Cui, J. D.; Jia, S. R.; Crit. Rev. Biotechnol. 2015, 35, 15. 80. Carvalho, N. B.; Lima, Á. S.; Soares, C. M. F.; Quim. Nova 2015, 38, 399. 81. Zhou, L.; Mou, H.; Gao, J.; Ma, L.; He, Y.; Jiang, Y.; Chin. J. Chem. Eng. 2017, 25, 487. 82. Cui, J.; Cui, L.; Jia, S.; Su, Z.; Zhang, S.; J. Agric. Food Chem. 2016, 64, 7179. 83. Schoevaart, R.; Wolbers, M. W.; Golubovic, M.; Ottens, M.; Kieboom, A. P. G.; Rantwijk, F. Van; Wielen, L. A. M. Van Der; Sheldon, R. A.; Biotechnol. Bioeng. 2004, 87, 754. 84. Khanahmadi, S.; Yusof, F.; Amid, A.; Mahmod, S. S.; Mahat, M. K.; J. Biotechnol. 2015, 202, 153. 85. Dong, T.; Zhao, L.; Huang, Y.; Tan, X.; Bioresour. Technol. 2010, 101, 6569. 86. Shah, S.; Sharma, A.; Gupta, M. N.; Anal. Biochem. 2006, 351, 207. 87. Mahmod, S. S.; Yusof, F.; Jami, M. S.; Khanahmadi, S.; Bioresour. Bioprocess. 2016, 3, 3. 88. Cui, J. D.; Cui, L. L.; Zhang, S. P.; Zhang, Y. F.; Su, Z. G.; Ma, G. H.; PLoS One 2014, 9. 89. Peirce, S.; Russo, M. E.; Isticato, R.; Lafuente, R. F.; Salatino, P.; Marzocchella, A.; Biochem. Eng. J. 2017, 127, 188. 90. Wang, S.; Zheng, D.; Yin, L.; Wang, F.; Enzyme Microb. Technol. 2017, 107, 22. 91. Zhou, Z.; Piepenbreier, F.; Marthala, V. R. R.; Karbacher, K.; Hartmann, M.; Catal. Today 2015, 243, 173. 92. Santos, J. C. S. D.; Barbosa, O.; Ortiz, C.; Berenguer-Murcia, A.; Rodrigues, R. C.; Fernandez-Lafuente, R.; ChemCatChem. 2015, 7, 2413. 93. Chen, Z.; Wang, Y.; Liu, W.; Wang, J.; Chen, H.; Int. J. Biol. Macromol. 2017, 95, 650. 94. Nguyen, L. T.; Yang, K. L.; Enzyme Microb. Technol. 2017, 100, 52. 95. Hwang, E. T.; Gu, M. B.; Eng. Life Sci. 2013, 13, 49. 96. Matsumoto, T.; Isogawa, Y.; Tanaka, T.; Kondo, A.; Biosens. Bioelectron. 2018, 99, 56. 97. Jochems, P.; Satyawali, Y.; Diels, L.; Dejonghe, W.; Green Chem. 2011, 13, 1609. 98. Beniwal, A.; Saini, P.; Kokkiligadda, A.; Vij, S.; LW. Food Sci. Technol. 2018, 87, 553. 99. Adhikari, B.-R.; Schraft, H.; Chen, A.; Analyst 2017, 142, 2595. 100. Zhang, R.; Wang, X.; Chem. Mater. 2007, 19, 976. 101. Adhikari, B. R.; Govindhan, M.; Chen, A.; Sensors (Switzerland). 2015, 15, 22490. 102. Guan, D.; Ramirez, M.; Shao, L.; Jacobsen, D.; Barrera, I.; Lutkenhaus, J.; Chen, Z.; Biomacromolecules. 2013, 14, 2909. 103. Arica, M. Y.; Kaçar, Y.; Genç, Ö.; Bioresour. Technol. 2001, 80, 121. 104. Han, J.; Guenier, A.; S, S.; Lacroix, M.; J. Agric. Food Chem. 2008, 56, 2528. 105. Matto, M.; Husain, Q.; J. Mol. Catal. B: Enzym. 2009, 57, 164. 106. Avnir, D.; Braun, S.; Lev, O.; Ottolenghi, M.; Chem. Mater. 1994, 6, 1605. 107. Gassara-Chatti, F.; Brar, S. K.; Ajila, C. M.; Verma, M.; Tyagi, R. D.; Valero, J. R.; Food Chem. 2013, 137, 18. 108. Zhang, B.; Li, P.; Zhang, H.; Fan, L.; Wang, H.; Li, X.; Tian, L.; Ali, N.; Ali, Z.; Zhang, Q.; RSC Adv. 2016, 6, 46702. 109. Kloxin, A. M.; Kasko, A. M.; Salinas, C. N.; Anseth, K. S.; Science 2009, 324, 59. 110. Appel, E. A.; Loh, X. J.; Jones, S. T.; Biedermann, F.; Dreiss, C. A.; Scherman, O. A.; J. Am. Chem. Soc. 2012, 134, 11767. 111. Ying, H.; Zhang, L. F; Wu, D.; Lei, Q. F; Guo, Y. S; Fang, W. J.; Energy Convers. Manage. 2017, 144, 303. 112. Dadsetan, M.; Liu, Z.; Pumberger, M.; Giraldo, C. V.; Ruesink, T.; Lu, L.; Yaszemski, M. J.; Biomaterials 2010, 31, 8051. 116. Wu, T.; Huang, J.; Jiang, Y.; Hu, Y.; Ye, X.; Liu, D.; Chen, J.; Food Chem. 2018, 240, 361. 114. Ma, G.; Lin, W.; Yuan, Z.; Wu, J.; Qian, H.; Xu, L.; Chen, S.; J. Mater. Chem. B 2017, 5, 935. 115. Agarwal, A.; Unfer, R. C.; Mallapragada, S. K.; Biomaterials. 2008, 29, 607. 116. Gačanin, J.; Kovtun, A.; Fischer, S.; Schwager, V.; Quambusch, J.; Kuan, S. L.; Liu, W.; Boldt, F.; Li, C.; Yang, Z.; Adv. Health. Mater. 2017, 6, 1700392. 117. Qian, Y.-C.; Chen, P.-C.; Zhu, X.-Y.; Huang, X.-J.; RSC Adv. 2015, 5, 44031. 118. Yuan, D.; Jacquier, J. C.; O’Riordan, E. D.; Food Chem. 2018, 239, 1200. 119. Fusco, G.; Chronopoulou, L.; Galantini, L.; Zerillo, A.; Rasik, Z. M.; Antiochia, R.; Favero, G.; D’Annibale, A.; Palocci, C.; Mazzei, F.; Microchem. J. 2018, 137, 105. 120. Ramirez, M.; Guan, D.; Ugaz, V.; Chen, Z.; J. Am. Chem. Soc. 2013, 135, 5290. 121. Zhang, Z.; Zhang, R.; Chen, L.; McClements, D. J.; Food Chem. 2016, 200, 69. 122. Monier, M.; Ayad, D. M.; Wei, Y.; Sarhan, A. A.; Int. J. Biol. Macromol. 2010, 46, 324. 123. Asgher, M.; Ramzan, M.; Bilal, M.; Chin. J. Catal. 2016, 37, 561. 124. De Oliveira, U. M. F.; Lima de Matos, L. J. B.; de Souza, M. C. M.; Pinheiro, B. B.; dos Santos, J. C. S.; Gonçalves, L. R. B.; Appl. Biochem. Biotechnol. 2018, 184, 1263. 125. Dos Santos, J. C. S.; Bonazza, H. L.; de Matos, L. J. B. L.; Carneiro, E. A.; Barbosa, O.; Fernandez-Lafuente, R.; Gonçalves, L. R. B.; de Sant’ Ana, H. B.; Santiago-Aguiar, R. S.; Biotechnol. Rep. 2017, 14, 16. 126. Gonçalves, L. R. B.; Lima de Matos, L. J. B.; dos Santos, J. C. S.; Pinheiro, B. B.; de Oliveira, U. M. F.; de Souza, M. C. M.; Mol. Biol. Rep. 2018, 46, 597. 127. Wang, D.; Jiang, W.; Int. J. Biol. Macromol. 2019, 126, 1125. 128. Bilal, M.; Iqbal, H. M. N.; Hu, H.; Wang, W.; Zhang, X.; Sci. Total Environ. 2017, 575, 1352. 129. Long, J.; Li, X.; Zhan, X.; Xu, X.; Tian, Y.; Xie, Z.; Jin, Z.; Bioprocess Biosyst. Eng. 2017, 40, 821. 130. Avnir, D.; Coradin, T.; Lev, O.; Livage, J.; J. Mater. Chem. 2006, 16, 1013. 131. Sahin, O.; Erdemir, S.; Uyanik, A.; Yilmaz, M.; Appl. Catal., A 2009, 369, 36. 132. Kauffmann, C.; Mandelbaum, R. T.; J. Biotechnol. 1998, 62, 169. 133. Ulijn, R. V.; J. Mater. Chem. 2006, 16, 2217. 134. Strozyk, M. S.; Jimenez de Aberasturi, D.; Liz-Marzán, L. M.; Chem. Rec. 2017, doi: 10.1002/adfm.201701626. 135. Yu, J.; Chu, X.; Hou, Y.; Chem. Commun. 2014, 50, 11614. 136. Wu, X.; Wang, Z. Y.; Zhu, D.; Zong, S. F.; Yang, L. P.; Zhong, Y.; Cui, Y. P.; ACS Appl. Mater. Interfaces. 2013, 5, 10895. 137. Liu, R.; Liang, S.; Jiang, C.; Zhang, L.; Yuan, T.; Li, P.; Xu, Z.; Xu, H.; Chu, P. K.; J. Mater. Chem. B 2016, 4, 1100. 138. Simó, G.; Fernández-Fernández, E.; Vila-Crespo, J.; Ruipérez, V.; Rodríguez-Nogales, J. M.; Carbohydr. Polym. 2017, 170, 1. 139. Takenaka, M.; Yoon, K. S.; Matsumoto, T.; Ogo, S.; Bioresour. Technol. 2017, 227, 279. 140. Ferreira, I. M.; de S. Ganzeli, L.; Rosset, I. G.; Yoshioka, S. A.; Porto, A. L. M.; Catal. Lett. 2017, 147, 269. 141. Costa, S. A.; Azevedo, H. S.; Reis, R. L.; Biodegrad. Syst. Tissue Eng. Regener. Med. 2004, 17, 301. 142. Akladious, A.; Azzam, S.; Hu, Y.; Feng, P.; CNS Neurosci. Ther. 2018, 6, 549. 143. Ma, X.; Fang, C.; Yan, J.; Zhao, Q.; Tu, Y.; Talanta 2018, 186, 206. 144. Pinto, T. V.; Sousa, C. M.; Sousa, C. A. D.; Aboelhassan, M. M.; Peixoto, A. F.; Pereira, C.; Coelho, P. J.; Freire, C.; Dalton Trans. 2017, 46, 9076. 145. Piazzoli, A.; Antonelli, M.; Water Air Soil Pollut. 2018, 229, 193. 146. Li, C.; Kumar, S.; Biomass Convers. Biorefinery 2016, 6, 407. 147. Nezampour, F.; Ghiaci, M.; Masoomi, K.; J. Chem. Eng. 2018, 63, 1977. 148. Borlido, L.; Moura, L.; Azevedo, A. M.; Roque, A. C. A.; Aires-Barros, M. R.; Farinha, J. P.; S. Biotechnol. J. 2013, 8, 709. 149. Jiang, H.; Sun, M.; Xu, J.; Lu, A.; Shi, Y.; CLEAN -- Soil, Air, Water 2016, 44, 1146. 150. Khoobi, M.; Motevalizadeh, S. F.; Asadgol, Z.; Forootanfar, H.; Shafiee, A.; Faramarzi, M. A.; Mater. Chem. Phys. 2015, 149, 77. 151. Chen, B.; Zhao, X.; Liu, Y.; Xu, B.; Pan, X.; RSC Adv. 2015, 5, 1398. 152. Muhammad, G.; Hussain, M. A.; Amin, M.; Hussain, S. Z.; Hussain, I.; Abbas Bukhari, S. N.; Naeem-ul-Hassan, M.; RSC Adv. 2017, 7, 42900. 153. López-Gallego, F.; Guisán, J. M.; Betancor, L.; Methods Mol. Biol. 2013, 1051, 33. 154. Zdarta, J.; Jędrzak, A.; Klapiszewski, Ł.; Jesionowski, T.; Catalysts 2017, 7, 374. 155. Ema, T.; Miyazaki, Y.; Kozuki, I.; Sakai, T.; Hashimoto, H.; Takada, J.; Green Chem. 2011, 13, 3187. 156. Zhou, X.; Han, Y.; Lv, Z.; Tian, X.; Li, H.; Xie, P.; Zheng, L.; J. Biotechnol. 2017, 249, 1. 157. Matuella, A.; Ficanha, M.; Nyari, N. L. D.; Levandoski, K.; Luis, M.; Quim. Nova 2015, 38, 364. 158. Piephoff, D. E.; Wu, J.; Cao, J.; J. Phys. Chem. Lett. 2017, 8, 3619. 159. Kari, J.; Andersen, M.; Borch, K.; Westh, P.; ACS Catal. 2017, 7, 4904. 160. Soldatkina, O. V.; Soldatkin, O. O.; Kasap, B. O.; Kucherenko, D. Y.; Kucherenko, I. S.; Kurc, B. A.; Dzyadevych, S. V.; Nanoscale Res. Lett. 2017, 12, 260. 161. Hamed, S.; Emara, M.; Shawky, R. M.; El-domany, R. A.; Youssef, T.; J. Basic Microbiol. 2017, 57, 659. 162. Tabassum, N.; Vidyasagar, G. M.; Int. J. Chemtech. Res. 2016, 9, 352. 163. Perni, S.; Hakala, V.; Prokopovich, P.; Colloids Surf., A 2013, 460, 219. 164. Brobbey, K. J.; Haapanen, J.; Gunell, M.; Toivakka, M.; Mäkelä, J. M.; Eerola, E.; Ali, R.; Saleem, M. R.; Honkanen, S.; Bobacka, J.; Thin Solid Films 2018, 645, 166. 165. Mohammady, H.; Dinarvand, R.; Manesh, M. E.; Ebrahimnejad, P.; Nanomed. J. 2016, 3, 159. 166. Lee, M. H.; Kim, S. Y.; Park, H. J.; Food Hydrocolloids 2018, 84, 58. 167. Wang, H.; Qian, J.; Ding, F.; J. Agric. Food Chem. 2018, 66, 395. 168. Yang, J.; Xiong, L.; Li, M.; Sun, Q.; J. Agric. Food Chem. 2018, 66, 6104. 169. Campos, E. V. R.; Oliveira, J. L.; Fraceto, L. F.; Front. Chem. 2017, 5, 1. 170. Kashyap, P. L.; Xiang, X.; Heiden, P.; Int. J. Biol. Macromol. 2015, 77, 36. 171. Naseeruteen, F.; Hamid, N. S. A.; Suah, F. B. M.; Ngah, W. S. W.; Mehamod, F. S.; Int. J. Biol. Macromol. 2018, 107, 1270. 172. Zhu, T.; Zhu, T.; Gao, J.; Zhang, L.; Zhang, W.; J. Fluor Chem. 2017, 194, 80. 173. Liu, Y. G.; Liao, T.; He, Z. B.; Li, T. T.; Wang, H.; Hu, X. J.; Guo, Y. M.; He, Y.; Trans. Nonferrous Met. Soc. 2013, 23, 1804. 174. Sadaka, A.; Sisk, R. A.; Osher, J. M.; Toygar, O.; Duncan, M. K.; Riemann, C. D.; Clin. Ophthalmol. 2016, 10, 1811. 175. Manna, S.; Donnell, A. M.; Kaval, N.; Al-rjoub, M. F.; Augsburger, J. J.; Banerjee, R. K.; Int. J. Pharm. 2018, 547, 122. 176. Xiao, A.; Xu, C.; Lin, Y.; Ni, H.; Zhu, Y.; Cai, H.; Electron. J. Biotechnol. 2016, 19, 1. 177. Kanimozhi, S.; Perinbam, K.; Mater. Res. Bull. 2013, 48, 1830. 178. Klapiszewski, Ł.; Zdarta, J.; Jesionowski, T.; Colloids Surf., B 2018, 162, 90. 179. Bezerra, R. M.; Neto, D. M. A.; Galvão, W. S.; Rios, N. S.; Carvalho, A. C. L. d. M.; Correa, M. A.; Bohn, F.; Fernandez-Lafuente, R.; Fechine, P. B. A.; de Mattos, M. C.; dos Santos, J.C.S.; Gonçalves, L. R. B.; Biochem. Eng. J. 2017, 125, 104. 180. Adeel, M.; Bilal, M.; Rasheed, T.; Sharma, A.; Iqbal, H. M. N.; Int. J. Biol. Macromol. 2018, 120, 1430. 181. Bilal, M.; Zhao, Y.; Rasheed, T.; Iqbal, H. M. N.; Int. J. Biol. Macromol. 2018, 120, 2530. 182. Majouga, A.; Sokolsky-Papkov, M.; Kuznetsov, A.; Lebedev, D.; Efremova, M.; Beloglazkina, E.; Rudakovskaya, P.; Veselov, M.; Zyk, N.; Golovin, Y.; Colloids Surf., B 2015, 125, 104. 183. Klapiszewski, Ł.; Zdarta, J.; Antecka, K.; Synoradzki, K.; SiwińskaStefańska, K.; Moszyński, D.; Jesionowski, T.; Appl. Surf. Sci. 2017, 422, 94. 184. Wang, F.; Zhang, X.; Liu, Y.; Lin, Z. Y. W.; Liu, B.; Liu, J.; Angew. Chemie, Int. Ed. 2016, 55, 12063. 185. Zhang, W.; Shen, F.; Hong, R.; Particuology 2011, 9, 179. 186. Lasheen, M. R.; El-Sherif, I. Y.; Tawfik, M. E.; El-Wakeel, S. T.; El-Shahat, M. F.; Mater. Res. Bull. 2016, 80, 344. 187. Honarmand, D.; Ghoreishi, S. M.; Habibi, N.; Nicknejad, E. T.; J. Appl. Polym. Sci. 2016, 133, 43556. 188. Shen, J.; Li, Y.; Zhu, Y.; Hu, Y.; Li, C.; J. Environ. Chem. Eng. 2016, 4, 2469. 189. Tayyebi, A.; Outokesh, M.; Moradi, S.; Doram, A.; Appl. Surf. Sci. 2015, 353, 350. 190. Tang, Y.; Guo, H.; Xiao, L.; Yu, S.; Gao, N.; Wang, Y.; Colloids Surf., A 2013, 424, 74. 191. Monteil, C.; Bar, N.; Retoux, R.; Henry, J.; Bernay, B.; Villemin, D.; Sens. Actuators. Chem. 2014, 192, 269. 192. You, L.; Huang, C.; Lu, F.; Wang, A.; Liu, X.; Zhang, Q.; Int. J. Biol. Macromol. 2018, 107, 1620. 193. Abdollahi, K.; Yazdani, F.; Panahi, R.; Int. J. Biol. Macromol. 2017, 94, 396. 194. Yazdani, F.; Fattahi, B.; Azizi, N.; J. Magn. Magn. Mater. 2016, 406, 207. 195. Ni, H.; Sun, X.; Li, Y.; Li, C.; J. Mater. Sci. 2015, 50, 4270. 196. Jiang, J.; Chen, Y.; Wang, W.; Cui, B.; Wan, N.; Carbohydr. Polym. 2016, 151, 600. 197. Wei, M. H.; Li, B.; David, R. L. A.; Jones, S. C.; Sarohia, V.; Schmitigal, J. A.; Kornfield, J. A.; Science 2015, 350, 72. 198. Amirbandeh, M.; Taheri-Kafrani, A.; Soozanipour, A.; Gaillard, C.; Biochem. Eng. J. 2017, 127, 119. 199. Amirbandeh, M.; Taheri-Kafrani, A.; Int. J. Biol. Macromol. 2016, 93, 1183. 200. Inagaki, M.; Kang, F.; J. Mater. Chem. A 2014, 2, 13193. 201. Soozanipour, A.; Taheri-Kafrani, A.; Landarani Isfahani, A.; Chem. Eng. J. 2015, 270, 235. 202. Cui, J.; Jia, S.; Coord. Chem. Rev. 2017, 352, 249. 203. Zhang, H.; Guo, L. Y.; Jiao, J.; Xin, X.; Sun, D.; Yuan, S.; ACS Sustain. Chem. Eng. 2017, 5, 1358. 204. He, X.; Chen, L.; He, Q.; Xiao, H.; Zhou, X.; Ji, H.; Chinese J. Chem. 2017, 35, 693. 205. Lee, H. R.; Chung, M.; Kim, M. Il; Ha, S. H.; Enzyme Microb. Technol. 2017, 105, 24. 206. Li, Y.; Fei, X.; Liang, L.; Tian, J.; Xu, L.; Wang, X.; Wang, Y.; J. Mol. Catal. B: Enzym. 2016, 133, 92. 207. Somturk, B.; Yilmaz, I.; Altinkaynak, C.; Karatepe, A.; Özdemir, N.; Ocsoy, I.; Enzyme Microb. Technol. 2016, 86, 134. 208. Altinkaynak, C.; Tavlasoglu, S.; Özdemir, N.; Ocsoy, I.; Enzym. Microb. Technol. 2016, 93-94, 105. 209. Ge, J.; Lei, J.; Zare, R. N.; Nat. Nanotechnol. 2012, 7, 428. 210. He, G.; Hu, W.; Li, C. M.; Colloids Surf., B 2015, 135, 613. 211. Rong, J.; Zhang, T.; Qiu, F.; Zhu, Y.; ACS Sustain. Chem. Eng. 2017, 5, 4468. 212. Nadar, S. S.; Gawas, S. D.; Rathod, V. K.; Int. J. Biol. Macromol. 2016, 92, 660. 213. Altinkaynak, C.; Yilmaz, I.; Koksal, Z.; Özdemir, H.; Ocsoy, I.; Özdemir, N.; Int. J. Biol. Macromol. 2016, 84, 402. 214. Zhao, F.; Wang, Q.; Dong, J.; Xian, M.; Yu, J.; Yin, H.; Chang, Z.; Mu, X.; Hou, T.; Wang, J.; Process Biochem. 2017, 57, 87. 215. Chung, M.; Nguyen, T. L.; Tran, T. Q. N.; Yoon, H. H.; Kim, I. T.; Kim, M. Il.; Appl. Surf. Sci. 2018, 429, 203. 216. Kochańczyk, T.; Drozd, A.; Krężel, A.; Metallomics 2015, 7, 244. 217. Zhang, B.; Li, P.; Zhang, H.; Wang, H.; Li, X.; Tian, L.; Ali, N.; Ali, Z.; Zhang, Q.; Chem. Eng. J. 2016, 291, 287. 218. Barbosa, C. G.; Caseli, L.; Péres, L. O.; J. Colloid Interface Sci. 2016, 476, 206. 219. Simon, D. T.; Gabrielsson, E. O.; Tybrandt, K.; Berggren, M.; Chem. Rev. 2016, 116, 13009. 220. Cirillo, G.; Nicoletta, F. P.; Curcio, M.; Spizzirri, U. G.; Picci, N.; Iemma, F.; React. Funct. Polym. 2014, 83, 62. 221. Singh, R. K.; Tiwari, M. K.; Singh, R.; Lee, J. K.; Int. J. Mol. Sci. 2013, 14, 1232. 222. Iqbal, S.; Ahmad, S.; J. Ind. Eng. Chem. 2017, 53. 223. Liu, Y.; Turner, A. P. F.; Zhao, M.; Mak, W. C.; Biosens. Bioelectron. 2018, 100, 374. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access