
 |
 |
 |
 |
 |
Artigo
| Raman studies of bis(phenylterpyrazine)iron(II) and supramolecular species with pentacyanidoferrate(II) ions |
|
Ana P. Mangoni; Bruno B. N. S. Brandão; Jorge S. Shinohara; Alceu T. Silveira; Marcelo Nakamura; Henrique E. Toma* Instituto de Química, Universidade de São Paulo, 05508-000 São Paulo – SP, Brasil Recebido em 02/04/2019 *e-mail: henetoma@iq.usp.br The chemistry and spectroscopy of the iron(II) phenylterpyrazine complex, [Fe(phtpz)2]2+, were investigated in comparison with the analogous phenylterpyridine species, Fe(phtpy)2]2+, and the results indicated a strong electronic delocalization on the Fe(II) terimine chromophore encompassing the lateral pyrazyl groups. The delocalized nature of the molecular orbitals involved in the electronic excitation precluded a direct assignment of the pyridine and pyrazine vibrations in the complex. However, the capability of the terpyrazine complex to bind transition metal ions, such as the [Fe(CN)5]3- species, allowed to generate pentanuclear complexes displaying characteristic charge-transfer and resonance Raman spectra, revealing the pyrazine vibrations in the complex. The [Fe(CN)5]3- containing species also yielded polymeric compounds in presence of transition metal ions, exhibiting intervalence transfer bands and chemical similarities with the Prussian Blue complex. INTRODUCTION Iron(II) terpyridine complexes are classical examples of coordination compounds exhibiting a typical (terimine)iron(II) chromophore in a low spin configuration.1-3 The complexes have been extensively employed in analytical chemistry, coordination polymers4 and devices5 because of their strong red-violet colors associated with metal-to-ligand charge-transfer transitions in the visible region. However, the analogous iron(II) terpyrazine complexes remain poorly understood, and according to the best of our knowledge, they have never been investigated before from the point of view of their electronic, Raman and supramolecular properties in aqueous solution. As a matter of fact, the terpyrazine ligands exhibit a central pyridine moiety attached to two pyrazine groups, which possess strong π-acceptor characteristics and are also able to bind metal ions by acting as bridging ligands. These aspects depart from the typical behavior of the iron(II) terpyridine complexes. Since their electronic and vibrational spectra are expected to be strongly influenced by the π-acceptor properties of the pyrazine group, a detailed investigation is required for improving the understanding of the chemistry of the iron(II) terimine complexes, and their possible application in photo-induced molecular pathways.6 In this paper, the chemistry and spectroscopy of the bis(phenylterpyrazine)iron(II) complex, [Fe(phtpz)2]2+, have been investigated in comparison with the related bis(phenylterpyridine)iron(II), [Fe(phtpy)2]2+, analogue7 (Figure 1).
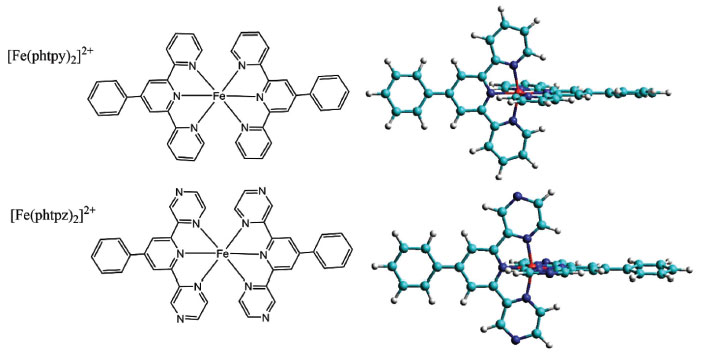 Figure 1. Structural representation of the [Fe(phtpy)2]2+ and [Fe(phtpz)2]2+ complexes
The four pyrazine moieties are very accessible to the interaction with π-donor complexes such as the pentacyanidoferrate(II) ions, and can generate pentanuclear complexes displaying a symmetric supramolecular structure. Formation of such species perturbs the pyrazine moieties and this aspect allows to discriminate the aromatic pyrazine vibrations from the pyridine ones, located in the same spectral region of the Raman spectra. This is actually a critical point in the analysis of the vibrational spectra of the metal-terimine complexes from the experimental point of view. On the other hand, the pentanuclear species can react with transition metal ions (M), generating novel supramolecular structures containing iron(II)-cyanide-M bonds, analogous to the Prussian Blue complex. Such species are quite novel, and their chemistry is reported in this paper.
EXPERIMENTAL SECTION Materials The (NH4)2Fe(SO4)2.6H2O (Mohr's salt) and the organic reagents benzaldehyde, 2-acetylpyridine and 2-acetylpyrazine were obtained from Sigma/Aldrich. All other reagents were of analytical grade and used as supplied. Synthesis of the phenylterpyridine and phenylterpyrazine ligands The 4'-phenyl-2,2':6',2"terpyridine (phtpy) and 4'-phenyl-2,2':6:2'terpyrazine (phtpz) ligands were synthesized according to the procedure reported by Wang and Hanan:8 1.0 mL of benzaldehyde (10 mmol) was added to 30 mL of ethanol solution containing 1.5 g KOH. Then 2.3 mL of acetylpyridine (20 mmol) or acetylpyrazine (20 mmol) was added, followed by 24 mL of NH4OH (18 mol L-1). The mixture was kept under stirring for 6 h at room temperature. The white precipitate was collected on a filter, and washed with water until obtaining a neutral pH. The phtpy ligand was recrystallized from an ethanol/water mixture (1:1) and the phtpz species recrystallized from chloroform/ethanol (1:1). They were kept dried under vacuum in a desiccator. Anal: C21H15N3 (MM 309.36 g mol-1) CHN exp (calc)%: 80.27 (81.53); 4.72 (4.89); 13.21(13.58). ESI-MS (m/z) exp(calc) [phtpy-H]+ : 310.1 (310.3); and C19H13N5 (MM = 311.17 g mol-1), CHN exp(cal)% 74.47(73:30); 4.22(4.21); 22.30(22.49). ESI-MS (m/z) exp(calc) [phtpz-H]+: 312.12(312.17). Synthesis of the [Fe(phtpy)2]2+ and Fe(phtpz)2]2+ complexes The [Fe(phtpy)2](PF6)2 and [Fe(phtpz)2](PF6)2 complexes were prepared by dissolving 80 mg of (NH4)2Fe(SO4)2.6H2O (0.2 mmol) in 30 mL of water, followed by the addition of 130 mg of the phtpy or phtpz ligands (0.4 mmol) dissolved in 4 mL trifluoroethanol. The mixture was stirred for 3 h under a nitrogen atmosphere. Then, 1 mL of a saturated NH4PF6 solution was added to precipitate the complexes, which were collected on a filter, washed with water and kept under vacuum, in a desiccator. C42H30N6P2F12Fe (MM 964.12 g mol-1), CHN exp(cal)% 51.47(52.30); 3.22(3.14); 8.56(8.71), C38H26N10P2F12Fe (MM 968.1 g mol-1), CHN exp(cal)%: 46.49(47.13); 2.96 (2.71); 14.49(14.46). For the experiments performed in aqueous solution, the [Fe(phtpy)2]Cl2 and [Fe(phtpz)2]Cl2 complexes were employed because of the poor solubility of the hexafluorophosphate analogues. They can be obtained from the corresponding PF6- compounds by dissolving into a minimum amount of acetonitrile and adding LiCl in excess. Under this condition, the chloride salts which are poorly soluble in the organic medium, precipitate as a fine powder material. The solids were isolated by centrifugation at 5000 rpm, washed with small amounts of acetone and kept in the desiccator. Generation of polynuclear pentacyanidoferrate(II) complexes with the [Fe(phtpz)2]2+ core In general, the substituted pentacyanidoferrate(II) complexes which are extremely soluble in water, generate untreatable oils when processed with organic solvents such as ethanol, acetone, or acetonitrile. However their chemistry in solution is quite well behaved and the complexes have been extensively investigated in this way.9-11 An important aspect to mention is the monodentate binding characteristics of the [Fe(CN)5]3- moiety, which leads to complexes of general formula [Fe(CN)5L]3- with aromatic N-heterocyclic ligands. For this reason, in this work the pentacyanidoferrate(II) complexes have been generated and studied in aqueous solution. Isolation of the solid forms, e. g. by drastic evaporation, leads to impure products, since of the precursor oily material can carry many impurities and are susceptible to contamination with coordination polymers formed in the drying process. Generation of Prussian Blue derivatives of the metallated polynuclear pentacyanidoferrate(II) complexes containing the [Fe(phtpz)2]2+ core The pentanuclear pentacyanidoferrate(II) complexes precipitate in the presence of transition metals ions, such as Mn2+, Co2+, Ni2+, Cu2+, Zn2+ and Fe3+, generating colloidal solutions as observed for the analogous Prussian Blue complexes.12 The colloidal suspensions can be broken by centrifugation at 5000 rpm, and in this way the solids can be separated and isolated after washing many times with water, in order to remove the contaminant species present in solution. Electronic spectra The electronic spectra of the complexes were obtained using a Hewlett-Packard, model HP-8453-A diode array spectrophotometer in the 200 – 1100 nm range. For the solid samples, the spectra were obtained by suspending the particles in polyvinyl alcohol (PVA) gel and allowing to dry, in order to generate thin, transparent films, suitable for optical monitoring. Resonance Raman spectra The Raman spectra were recorded on a WITec alpha 300-R confocal Raman microscope equipped with 488, 532 and 633 nm lasers. The samples were prepared in aqueous solution, or as PVA films. The confocal Raman measurements were monitored with a laser spot area of 1 µm2, using 20x lens with 0.40 numerical aperture, 600 grating, and CCD detector of 1600 x 200 pixels. The laser wavelength, power and integration time were 488 nm, 0.06 W cm–2, 60 s; 532 nm, 0.1 mW cm–2, 20 s; and 633 nm, 0.08 W cm–2, 60 s, respectively. Theoretical calculations DFT calculations of the geometry and vibrational frequencies for the [Fe(phtpy)2]2+ complex were carried out using Gaussian-09, with the functional MH06-HF and cc-pvtz basis. Simple geometry optimization in this case demanded 9 days of computational work using the laboratory workstation, comprising an Octacore Intel Processor i7-3770 CPU @ 3.40GHz, DDR3 2666 MHz, 16 GB and SSD 240 GB. The molecular structure exhibited orthogonal terpyridine rings, with the phenyl rings adopting 40.3º orientation in relation to the central pyridine ring. Due to the molecular complexity of the complexes and their supramolecular species, DFT and TDTFD theoretical calculations were considered exceedingly time demanding for the purposes of this work. For convenience, the ZINDO-S semiempirical method from the Hyperchem 8.05 computational package was here employed.13 As demonstrated by Gorelski and Lever14 and according to our own experience, there is a reasonable convergence between the two methods. However, the last one requires only few seconds and can be carried out simultaneously with the spectroscopic measurements, thus allowing the planning and evaluation of the experiments in real time. In the theoretical calculations, the atomic dipoles were initially used to start the MM+ geometry optimization, and then replaced by the atomic charges obtained from the ZINDO-S method, with a convergence limit of about 10-5 kcal Å-1 mol-1. The electronic distribution was generated from single CI excitations in an active space involving 20 frontier molecular orbitals (10 highest occupied and 10 lowest unoccupied MOs). The semiempirical optimized geometry reproduced an orthogonal orientation of the terpyridine or terpyrazine ligands (Figure 1) very similar to that obtained by DFT; but yielding an angle of 9.0º for the phenyl ring with respect to the central pyridine ring, which is much closer to the crystallographic data for the free ligand, e.g. 10.9º.15 After the MM+/ZINDO-S geometry optimization, the final electronic spectra were obtained, and the molecular orbitals plotted as 2D energy contours. Such plots were expected to indicate the atomic bonds involved in the resonance Raman spectra, thus facilitating the interpretation of the observed peaks.
RESULTS AND DISCUSSION The [Fe(phtpy)2]2+ and [Fe(phtpz)2]2+ complexes exhibit a typical low spin configuration, with a peculiar symmetry, where the ligands adopt a local C2v point group, but yielding a global D2d symmetry because of their orthogonal configuration, as illustrated in Figure 1. It should be noted that the four pyrazine ligands are located at orthogonal positions, and are available for interacting with metal complexes, allowing the generation of polynuclear coordination compounds. Electronic Spectra of the [Fe(phtpy)2]2+ and [Fe(phtpz)2]2+ complexes The electronic spectra of the [Fe(phtpy)2]2+ and [Fe(phtpz)2]2+ complexes consist of two sets of bands: one set below 400 nm, associated with internal π→π* transitions in the organic ligand, and another set in the range of 400-700 nm, which is characteristic of the Fe(II)-terimine chromophore, being ascribed to iron(II)-phtpy or iron(II)-phtpz charge-transfer (CT) transitions (Figure 2).
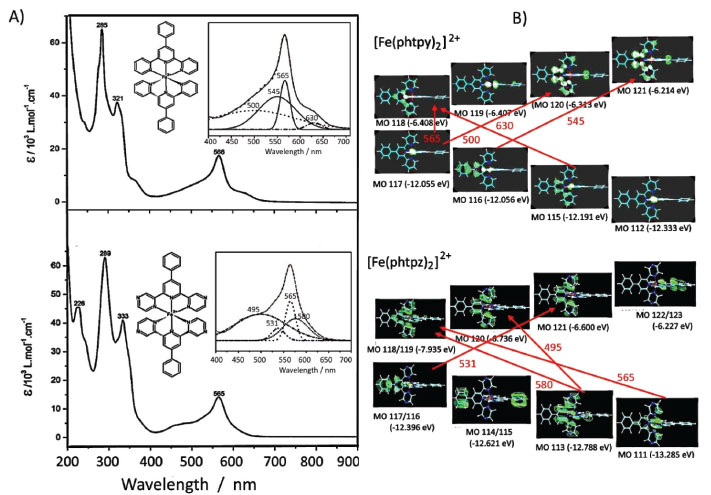 Figure 2. (A) Electronic spectra of the [Fe(phtpy)2]2+ and [Fe(phtpz)2]2+ complexes in the UV-visible region, showing the spectral deconvolution in the inset, and (B) the relevant HOMO and LUMO levels associated with the respective charge-transfer bands
DFT calculations of the structure and vibrational spectra of the iron(II)-bipyridine complexes have already been reported16 in the literature, but not for the terimine analogues. In general, such calculations do not include the resonance Raman effect17,18 which requires more specific considerations on the electronic states involved in the molecules, and depend on the Raman exciting wavelengths. As a matter of act, resonance Raman is chromophore selective and this is a very important feature which has been explored in a number of applications, especially in biological systems.19 In order to discriminate the electronic levels involved in the resonance Raman effect, the use of semiempirical methods, such as ZINDO/S can be very convenient, taking into account that the large molecular complexity would require very long computational times, using for instance, TDDFT or multiconfigurational methods. The observed visible bands can be deconvoluted into four bands as shown in the Figure 2. ZINDO/S theoretical calculations provided an approximate simulation of such bands, allowing to access the electronic distribution of the relevant HOMO and LUMO levels, and their corresponding energies, as shown in Figure 2 B and Table 1. According to the theoretical simulation, the HOMO levels of the [Fe(phtpy)2]2+ complex are essentially localized on the Fe(II) center, while for the [Fe(phtpz)2]2+ complex there is a substantial delocalization all over the molecule including the phenyl group (Figure 2 B). The LUMO levels for the [Fe(phtpy)2]2+ complex are also more localized on the central pyridine ring as compared with the delocalization over the terpyrazine moiety for the [Fe(phtpz)2]2+ complex.
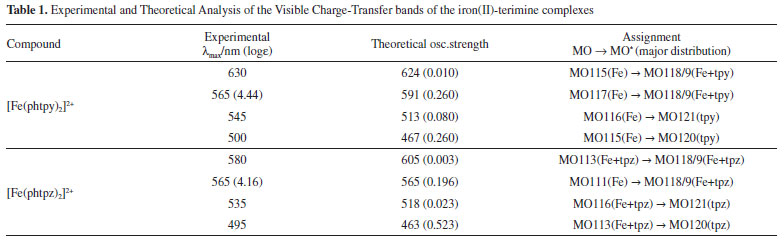
The lowest energy CT bands for the [Fe(phtpy)2]2+ and [Fe(phtpz)2]2+ complexes are quite distinct, and can be observed at 630 and 580 nm, respectively, in Figure 2 A. For the terpyridine complex, the electronic delocalization in the HOMO levels (MO 115, MO 113) is smaller than in the terpyrazine case (see Figure 2 B). The same reasoning applies to the remaining levels. In the [Fe(phtpy)2]2+ complex the electronic distribution seems more localized on the iron(II)-terimine chromophore, while in the[Fe(phtpz)2]2+ species, the delocalization extends to the pyrazine moieties, thus involving an iron(II)-terpyrazine chromophore. Raman spectra of the [Fe(phtpy)2]2+ and [Fe(phtpz)2]2+ complexes As observed for the iron(II) diimine complexes, the Raman spectra of the terimine analogues excited in the visible region, exhibited a selective enhancement, reflecting the the chromophore groups in the process. It should be noted that in the resonance Raman spectra, only the vibrational groups of the chromophores are enhanced, contrasting with the general vibrational spectra in the infrared region. The enhancement of the vibrational peaks can be demonstrated by using an internal standard, such as KNO3. As a matter of fact, in Figure 3, the intensity of the Raman signals for the complexes, obtained at a very low concentration (5 x10-5 mol L-1), are comparable to the normal Raman scattering of the NO3- internal standard (0.3 mol L-1), thus corroborating a large enhancement, typical of the resonance Raman effect.
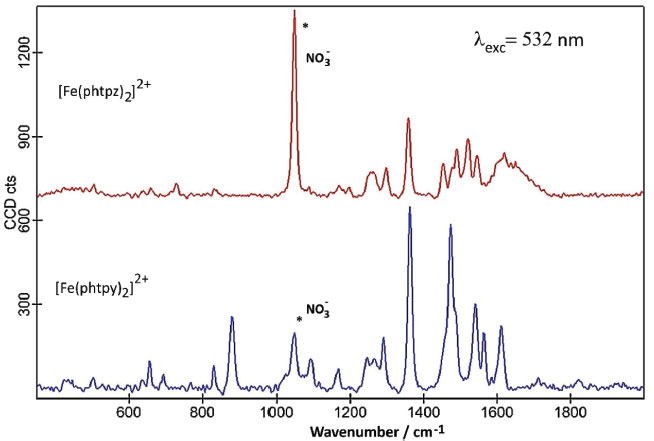 Figure 3. Raman spectra of the [Fe(phtpy)2]2+ and [Fe(phtpz)2]2+ complexes (5 x 10-5 mol L-1) in the presence of KNO3 (0.3 mol L-1), exc = 532 nm
DFT calculations of the Raman spectrum of the [Fe(phtpy)2]2+ complex led to more than 200 peaks, but the most strongly enhanced peaks were observed at 1711, 1687, 1681, 1671, 1558, 1546, 1403 and 1277 cm-1 involving a rather complex composition of the vibrational modes of the phenyl and pyridyl rings. Although the theoretical frequencies are close to the those experimentally observed for the complex (Figure 3), their intensities do not reproduce the observed spectral patterns because of the wavelength dependence introduced by the resonance Raman effect. The structural characteristics of the two complexes turn the assignment the phenyl, pyridine and pyrazine bands a very challenging task. The comparison of the two complexes may be helpful, since they have in common a phenyl group and a central pyridine moiety, so that the differences should arise from the four peripheral pyridyl groups in the [Fe(phtpy)2]2+ complex, and the four peripheral pyrazyl groups in the [Fe(phtpz)2]2+ case. The Raman spectra of the [Fe(phtpy)2]2+ complex is shown in Figure 4. In principle there are four spectral regions; a) from 1400 to 1700 cm-1 encompassing C-C stretching vibrations in the common phenyl and central pyridyl moieties, and in the distinct peripheral pyridyl groups; b) from 1000 to1400 cm-1 involving composite νCC, νCN, and δCH vibrational modes of the phtpy ligand; c) from 500 to 1000 cm-1 corresponding to δCH and δCCN modes and d) below 500 cm-1 involving νFe-N and low frequency skeleton modes.20,21
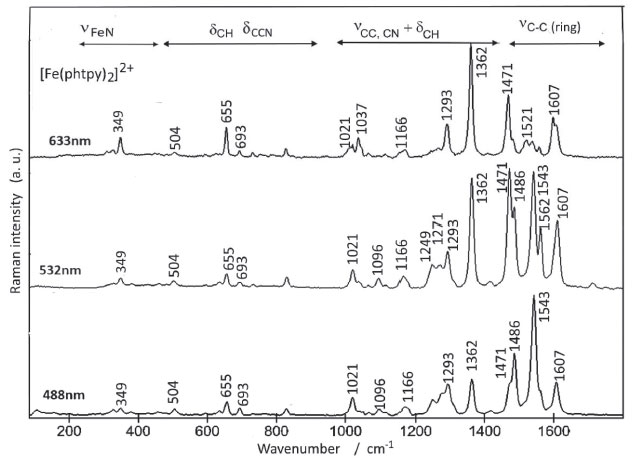 Figure 4. Raman spectra of [Fe(phtpy)2]2+ at several excitation wavelengths
As shown in Figure 2 B, at 633 nm the excitation of [Fe(phtpy)2]2+ complex involves the electronic band at 630 nm, corresponding to the excited MO 118/119 mainly localized on the central pyridyl ring with a small distribution on the neighboring diamine bonds. The phenyl group is not involved in this excitation. Therefore, it is plausible that the observed bands arise from the central pyridyl and the interconnected diimine bonds. The peaks at 504 and 349 cm-1 correspond to the νFe-N stretching vibrations. The excitation at 488 nm coincides with the absorption band at 500 nm, involving MO 120 localized on the Fe-tpy chromophore. Excitation at 532 nm was less conclusive, since it involves a strong overlap of the absorption bands at 565, 545 and 500 nm, and this is reflected in a complex vibrational profile encompassing more than a single excited state. Considering the characteristics of the molecular orbitals involved (118/119 and 120, in Figure 2 B) it is plausible that the peaks simultaneously enhanced at 488 and 630 nm belong to Fe-tpy vibrations; while those preferentially enhanced at 630 nm are associated with the Fe-py(central) core and those preferentially enhanced at 488 nm involve Fe-py(peripheral) vibrations. Therefore, the common peaks at 630 and 488 nm, e. g. 1607, 1471, 1362, 1293, 1166, 1021, 693, 655 cm-1 peaks should correspond to Fe-tpy vibrations. The peaks at 1521 and 1037 should correspond to Fe-py(central) vibrations, while the peaks at 1543, 1486, 1271, 1249, and 1096 cm-1 should correspond to Fe-py(peripheral) vibrations. The Raman spectra of the [Fe(phtpz)2]2+ complex are shown in Figure 5. In this case, the 633 nm excitation involves the MO 118/119, which is delocalized over the iron(II)-terpyrazine (Fe-tpz) chromophore. The phenyl group is not involved in this excitation. Therefore, it is plausible that the observed bands arise from Fe-tpz chromophore. The peaks at 506 and 350 cm-1 are compatible with the νFe-N stretching vibrations. On the other hand, the excitation at 488 nm involves preferentially MO 120 which is also localized on the Fe-tpz chromophore. The excitation at 532 nm involves a strong overlap of the absorption bands at 565, 535 and 495 nm, and this is reflected in the complex vibrational profile encompassing more than one single excited state.
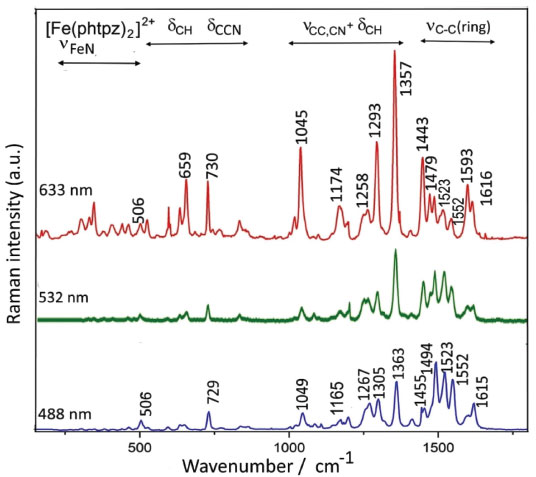 Figure 5. Resonance Raman spectra of [Fe(phtpz)2]2+ at several excitation wavelengths, showing the distribution of the vibrational modes
As one can see in Figure 5, although the Raman profiles observed at 633 and 488 nm are quite different, the corresponding molecular orbitals are spread over the Fe-tpz chromophore (Figure 2 B), and the contributions of the central pyridine and lateral pyrazine rings in the Raman spectra cannot be readily discriminated. In summary, the vibrational frequencies of the two complexes were collected in Table 2, with a tentative assignment as discussed before. For comparison purposes, a new column has also been added involving the corresponding pentacyanidoferrate (PC) species, commented in the following section.
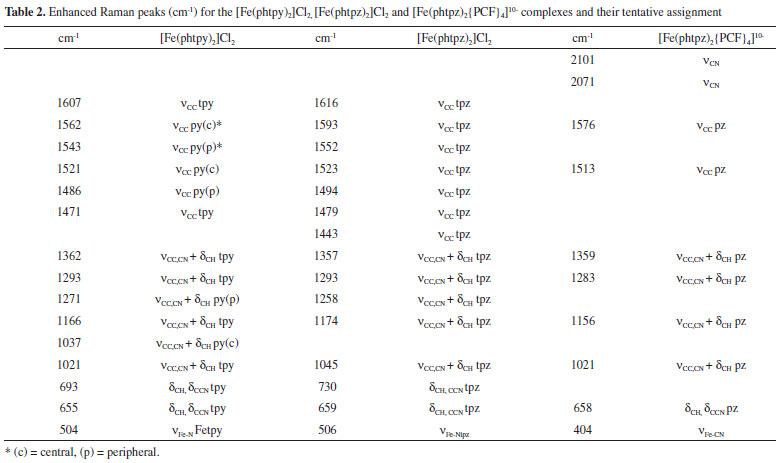
As shown in Table 2, the differentiation of the pyridine and pyrazine vibrational modes in the [Fe(phtpz)2]2+ complex has not been possible based on the molecular orbitals involved, in spite of the contrasting Raman profiles observed at distinct excitation wavelenghts. However, the presence of the pyrazine moieties allow a local perturbation by forming coordination bonds with pentacyanidoferrate(II) ions.10 Polynuclear [Fe(phtpz)2{Fe(CN)5}4]10- complex The [Fe(phtpz)2]2+ complex exhibits four pyrazyl groups which can bind additional metal ions, in contrast to the pyridyl group, and this strategy can be explored to differentiate the pyrazyl and pyridyl group vibrations. As a matter of fact, the binding of the [FeII(CN)5]3– groups to the pyrazyl moieties leads to the rise of two new MLCT bands, at 460 and 700 nm, as shown in Figure 5. No such change is observed when the [Fe(phtpy)2]2+ complex is treated with [Fe(CN)5]3- ions. As a matter of fact, the absorption band at 460 nm is typical of [Fe(CN)5L]3– complexes containing pyrazine ligands,10 and correspond to the peripheral complexes around the iron(II)-terpyrazine core. On the other hand, the absorption band at 700 nm can be ascribed to a new charge-transfer transition from the iron(II)-center to the bridging pyrazine-Fe(CN)5 complex. It is also observed when the pyrazine moieties are protonated, indicating the lowering of an excited π* orbital of the pyrazyl group induced by the peripheral metal ion or protons. The [Fe(phtpz)2]2+ complex reacts with [Fe(CN)5]3- ions in solution, increasing the absorption at 460 and 700 nm up to the 1:4 proportion, corresponding to the saturation point. The pentanuclear complex has a deep blue-violet color, and remains stable in aqueous solution, with no evidence of decomposition even after months. In spite of the apparent repulsion of the [Fe(CN)5]3- negative charges, one can see in the stereochemical model (Figure 7) that they are located quite far, practically in distant orthogonal positions.
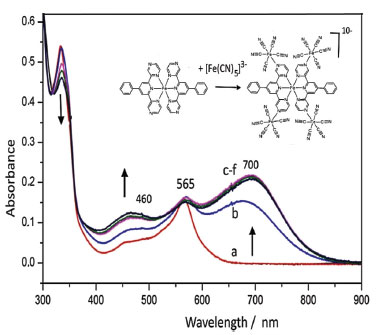 Figure 6. Changes in the electronic spectrum of [Fe(phtpz)2]2+ (a) in the presence of successive amounts of Na3[Fe(CN)5NH3], in aqueous solution, up to the saturation point (b-f)
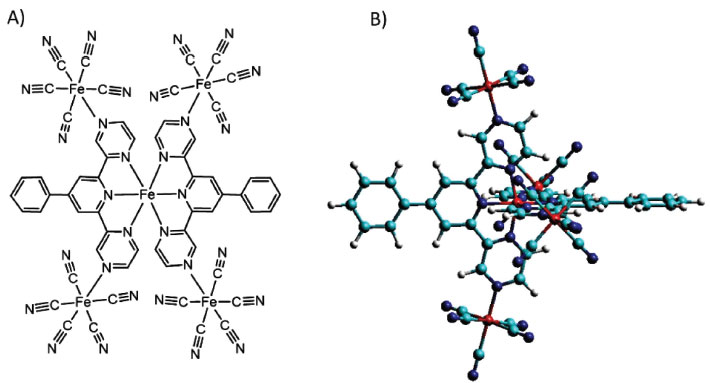 Figure 7. Structural representation of the pentanuclear complex [Fe(phtpz)2{Fe(CN)5}4]10- and its stereochemical view
The Raman spectrum of the [Fe(phtpz)2{Fe(CN)5}4]10- complex can be seen in Figure 8, in comparison with the spectrum of the [Fe(phtpz)2]2+ species at the same excitation wavelength (633 nm). The presence of the [Fe(CN)5]3- complexes is evidenced by the characteristic22 νCN stretching vibrations at 2070 and 2101 cm-1, and the νFe-C band at 404 cm-1 (Figure 8 B). Their selective binding to the pyrazine moieties should promote a selective enhancement of the new Fe-pz-Fe chromophore group absorbing at 700 nm (Figure 6), allowing to discriminate the characteristic frequencies of the pz ring. For this reason, the Raman spectra of the pentanuclear complex was actually much simpler than that for the [Fe(phtpz)2]2+ core. In this way, the νCN, νCC vibrations at 1593 and 1523 cm-1 can be ascribed to the pyrazine moieties (Figure 8 A) converting into the 1576 and 1513 cm-1 peaks in the pentanuclear complex, thus corroborating the proposed binding to the pentacyanidoferrate(II) ions (Figure 8 B). The CC peaks at 1616, 1552, 1494, 1479, 1443 cm-1 and the δCCN peak at 730 cm-1 are not enhanced in the pentanuclear complex and should belong to the central Fe-pyridyl group. Analogously, the νCC,CN peaks at 1357, 1293, 1174, the νring peak at 1045 cm-1 and the δCCN peak at 659 cm-1 are selectively enhanced in the pentanuclear complex and can be associated with the local pyrazine modes influenced by the [Fe(CN)5]3- coordination. In this way, the elucidation of the enhanced pyrazine modes could be performed, improving the assignment of the Raman spectra of the [Fe(phtpz)2]2+ complex, as shown in Table 2.
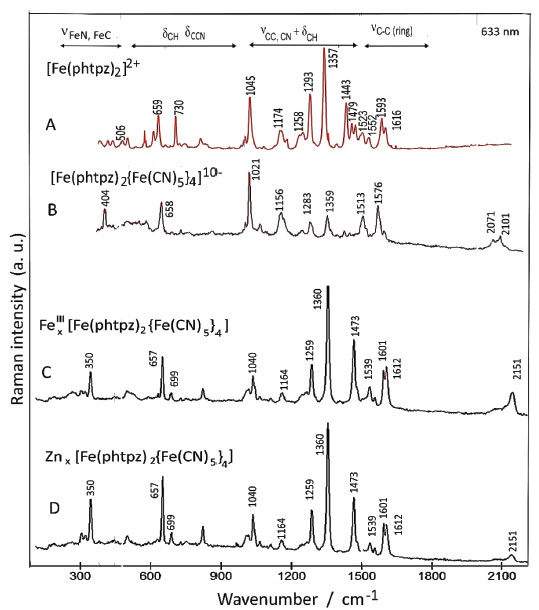 Figure 8. Comparative resonance Raman spectra (633 nm) of the [Fe(phtpz)2]2+ (A) , [Fe(phtpz)2{Fe(CN)5}4]10- (B) and Fex[Fe(phtpz)2{Fe(CN)5}4] and Znx[Fe(phtpz)2{Fe(CN)5}4] complexes in PVA films (C)
In the presence of transition metal ions, such as Fe(III) and Zn(II) the [Fe(phtpz)2{Fe(CN)5}4]10- complex forms insoluble extended polymers, analogous to the Prussian Blue compound, through the formation of FeII-CN-FeIII or FeII-CN-ZnII bonds.23 Their electronic species can be seen in Figure 9. The Fex[Fe(phtpz)2{Fe(CN)5}4] exhibit a broad intervalence transfer band around 750 nm, as in the Prussian Blue complex. The lowest energy charge transfer band is also apparent the polymeric complex, showing the perturbation of the pyrazine groups by the pentacyanidoferrate complexes.
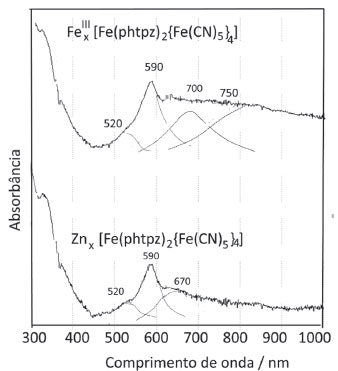 Figure 9. Absorption spectra of the Fex[Fe(phtpz)2{Fe(CN)5}4] and Znx[Fe(phtpz)2{Fe(CN)5}4] complexes in PVA films
The corresponding resonance Raman spectra are quite well defined as shown in Figure 8 C, D. The νCN frequency is shifted to 2151 cm-1 indicating the binding of the cyanide ligand to the Fe(III) and Zn(II) metal ions.21 Analogously, the peak at 350 cm-1 can be ascribed to νFe-CN22 The vibrational spectra of the [Fe(phtpz)2{Fe(CN)5}4]10– is nearly preserved in the extended polymer, showing a large enhancement of the νCC,CN vibration of the peripheral pz vibration at 1360 cm-1. The vibrational peaks of tpz at 1494, 1523 and 1552 cm-1 as well as at 730 cm-1 practically disappear, reflecting their strong perturbation by the attached pentacyanidoferrates.
CONCLUSION The iron(II) terpyrazine complex [Fe(phtpz)2]2+ exhibits a strong electronic delocalization on the Fe(II) terimine chromophore, encompassing the lateral pyrazyl groups and contrasting with the iron(II) terpyridine derivative. The assignment of the resonance Raman peaks has been tentatively proposed by comparing the spectra of these two systems. The pyrazine groups were explored to bind [Fe(CN)5]3- species, generating stable pentanuclear complexes displaying additional charge-transfer bands in the visible. The resonance Raman spectra reflected the presence of the pentacyanidoferrate(II) complexes, and the strong influence on the resonance Raman bands of the pyrazine groups allowed their discrimination from a rather complicated resonance Raman study. The pyrazine coordination features were also explored in the formation of polymeric complexes, similar to Prussian Blue, in the presence of transition metal ions.
ACKNOWLEDGEMENTS The financial support from Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP 2018/21489-1) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq 405301/2013-8) is gratefully acknowledged.
REFERENCES 1. Krumholz, P.; J. Am. Chem. Soc. 1953, 75, 2163. 2. Krumholz, P.; Serra, O. A.; DePaoli, M. A.; Inorg. Chim. Acta 1975, 15, 25. 3. Sone, K.; Krumholz, P.; Stammreich, H.; J. Am. Chem. Soc. 1955, 77, 777. 4. Li, L.; Niu, S.-Y.; Li, D.; Jin, J.; Chi, Y.-X.; Xing, Y.-H.; Inorg. Chem. Commun. 2011, 14, 993. 5. Pai, S.; Schott, M.; Niklaus, L.; Posset, U.; Kurth, D. G.; J. Mater. Chem. C 2018, 6, 3310. 6. Zedler, L.; Krieck, S.; Kupfer, S.; Dietzek, B.; Molecules 2019, 24, 245. 7. Assumpcao, A. M. C.; Bonacin, J. A.; Toma, S. H.; Zamarion, V. M.; Toma, H. E.; Can. J. Chem. 2014, 92, 918. 8. Wang, J. H.; Hanan, G. S.; Synlett 2005, 8, 1251. 9. Toma, H. E.; Malin, J. M.; Inorg. Chem. 1973, 12, 2080. 10. Toma, H. E.; Malin, J. M.; Inorg. Chem. 1973, 12, 1039. 11. Toma, H. E.; Lever, A. B. P.; Inorg. Chem. 1986, 25, 176. 12. Matsumoto, F. M.; Temperini, M. L. A.; Toma, H. E.; Electrochim. Acta 1994, 39, 385. 13. Zerner, M. C.; ZINDO PROGRAM V.98.1 - Quantum Project, University of Florida, Gainesville, Florida, 1998. 14. Gorelsky, S. I.; Lever, A. B. P.; J. Organomet. Chem. 2001, 635, 187. 15. Constable, E. C.; Lewis, J.; Liprot, M. C.; Raithby, P. R.; Inorg.Chim Acta 1990, 178, 47. 16. Alexander, B. D.; Dines, T. J.; Longhurst, R. W.; Chem. Phys. 2008, 352, 19. 17. Albrecht, A. C.; J. Chem. Phys. 1961, 34, 1476. 18. Clark, R. J. H.; Dines, T. J.; Angew. Chem., Int. Ed. 1986, 25, 131. 19. Efremov, E. V; Ariese, F.; Gooijer, C.; Anal. Chim. Acta 2008, 606, 119. 20. Burger, K.; Wagner, F. E.; Vertes, A.; Bencze, E.; Mink, J.; Labadi, I.; Nemes-Vetessy, Z.; J. Phys.Chem. Solids 2001, 62, 2059. 21. Mallick, P. K.; Danzer, G. D.; Strommen, D. P.; Kincaid, J. R.; J. Phys. Chem. 1988, 92, 5628. 22. Hipps, K. W.; Williams, S. D.; Mazur, U.; Inorg. Chem. 1984, 23, 3500. 23. Shinohara, J. S.; Grasseschi, D.; Almeida, S. N.; Toma, H. E.; An. Acad. Bras. Ciênc. 2019, 9, 2.
FAPESP helped in meeting the publication costs of the article |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access