
 |
 |
 |
 |
 |
Artigo
| Spectroscopic characterization of pedot:pss conducting polymer by resonance raman and serrs spectroscopies |
|
Pedro V. Almeida; Celly M. S. Izumi; Hélio F. Dos Santos; Antonio C. Sant'Ana*
Departamento de Química, Instituto de Ciências Exatas, Universidade Federal de Juiz de Fora, 36036-900 Juiz de Fora – MG, Brasil Recebido em 27/05/2019 *e-mail: antono.sant@ufjf.edu.br Poly(3,4-ethylenedioxythiophene)-poly(styrenesulfonate) (PEDOT:PSS) synthesis was monitored by resonance Raman (RR) and surface-enhanced resonance Raman scattering (SERRS) spectroscopies and both the oxidation of the polymer and the effects of chain length were assessed by vibrational analysis. It was supported by theoretical models of oligomers, with charge ranging from 0 to +6 and size varying from 2 to 16 3,4-ethylenedioxythiophene (EDOT) repeat units, described through density functional theory. The symmetric CαCβ stretching band observed at ca. 1426 cm-1 shifts to higher wavenumbers for reaction times varying from 2 to 9 hours and stabilizes at intermediate wavenumbers for longer times. The former behavior was ascribed to the increase of concentration of oxidized species, which was corroborated by calculated wavenumbers for oligomer models when their charges changed from neutral to +6. The behavior observed for longer reaction times was associated with the growth of chains. It was also observed through SERRS spectroscopy that small chains of the polymer, obtained in the early time of the synthesis, adsorbs on gold nanoparticle surfaces through interactions via oxidized residues. In this way, RR and SERRS spectroscopies were valuable tools to follow the synthetic procedures of PEDOT:PSS, allowing the determination of the structural evolution of the polymer. INTRODUCTION Poly[3,4-ethylenedioxythiophene] (PEDOT) is a semiconducting polymer widely used in the field of electronic organic devices to gather and generate light, such as bulk heterojunction solar cells and diodes.1 Such a kind of material has become very popular, as the world faces the necessity to improve its power generation capacity provided by more ambiently responsible sources.2,3 The greatest challenge related to the use of PEDOT as the main component of electronic organic devices resides on its very poor processability, which reduces the availability of methods to produce thin films that are needed to build these devices.4 Nevertheless, the oxidative polymerization also produces oligomers of very low solubility, turning PEDOT into a hard-to-characterize material.5 To overcome this, PEDOT was synthesized together with Sodium Polystyrene Sulfonate (PSS) to produce an aqueous dispersion known as PEDOT:PSS, with PSS working also as dopant.6 The use of polymers dispersed in low boiling point solvents allowed the application of low-cost deposition techniques, such as spin coating, to produce homogeneous thin films with high reproducibility, resulting in both the lowering of the costs of production and improvements in the power conversion efficiency.7–9 Many efforts have been done to parameterize the polymeric dispersion syntheses in order to find the most suitable applications. Due to the electronic structures of conducting polymers, the use of resonance Raman (RR)10–12 and surface-enhanced resonance Raman scattering (SERRS)13,14 spectroscopies can provide valuable information for the characterization of their dispersions and films. These techniques could lead to infer the structural parameters along with the molecular systems chemical environment, which interferes in the properties of interest. Nevertheless, the correct vibrational assignment tends to become very hard, as the occurrence of occasional degeneracy is frequent and the definitive association of normal modes with specific functional groups is unclear. In spite of this, some of PEDOT spectral features are well known to provide information about the oxidation, doping and organization of thin PEDOT:PSS films.15 In addition, computational methods of analysis provide a set of powerful tools to understand experimental vibrational spectra and the behavior of such materials. However, high-level ab initio calculations are restricted due to the size of the systems, which may range from hundreds to thousand atoms in polymeric systems. The modeling of oligomers, in this case, can provide very reliable information about the polymeric system.16 Many chemical properties have been determined with the support of computational methods of analysis, such as the elasticity of the chains by density functional theory (DFT),8 and the interaction forces and distribution of states between PEDOT and tosylates from PSS by using Perdew–Burke-Ernzerhof (PBE) exchange energy.16 The determination of the HOMO/LUMO gaps is of particular interest for the production of organic photovoltaic devices. The control of the changes in the energy gap through the addition of extra energy states during the doping process, that often also occurs in the oxidative polymerization, lead to significant variations in the sheet resistance and conductivity of the thin films.17 In this work PEDOT:PSS was synthesized by the chemical oxidative method.18 The oxidation and doping of the obtained dispersions were evaluated by RR and SERRS spectroscopy. SERRS spectra of the polymer were recorded on non-capped gold nanoparticles. DFT calculations of structures of EDOTn oligomers ranging from 2 to 16 units were used to evaluate the experimental parameters obtained by the vibrational spectroscopy techniques. The oxidation of the PEDOT chains was also studied from vibrational assignment supported by the frequency calculations of 16 units EDOT oligomer, with charge varying from neutral to +6 atomic units. These data were correlated to the experimental ones obtained by the variation of the reaction time, leading to the conclusion that the oxidative polymerization reaction evolves in two different regimes, where the early oxidation stage is followed by the effective polymerization reaction that leads to the growth of the polymer. In addition, SERRS spectra of PEDOT:PSS were obtained to understand the chemical interactions of the polymeric chain with the surfaces of gold nanoparticles (AuNPs) since such plasmonic systems have been used for enhancing the efficiency of photovoltaic devices.19,20
EXPERIMENTAL SECTION Materials and instrumentation Ethylenedioxythiophene (EDOT; 97%), sodium persulfate (Na2S2O8; ≥ 98.0%), poly(4-styrenesulfonate) sodium salt (PSS; ~70.000 g mol-1), tetrachloroauric(III) acid trihydrate (HAuCl4.3H2O; ≥ 99.9%), and trisodium citrate (C6H5Na3O7.2H2O; ≥ 99.0%) were purchased from Sigma-Aldrich and used without further purifications. All solutions were prepared by using deionized water (18.2 MΩ cm at 25 ºC). The glassware used in the polymerization reaction were rinsed with a 50% by weight nitrous acid (HNO3) bath, followed by 'aqua regia' solution (HCl:HNO3, 3:1) and copiously washed with deionized water. RR and SERRS spectra were recorded in a Bruker FT-Raman RFS 100 equipped with a Ge detector cooled with liquid nitrogen and Nd:YAG laser with wavelength at 1064 nm. The measurements were gathered using Duran glass tubes as support for the suspensions, and the power was set at 100 mW for 500 scans with a spectral resolution at 4 cm-1. RR and SERRS spectra were also acquired by using exciting radiation with wavelength at 785 nm in a Bruker Senterra dispersive Raman spectrometer coupled with an optical microscope and a CCD type detector with a thermoelectric cooling system. The power was set to 25 mW, with 2 scans, integration time of 100 seconds and resolution of 3-5 cm-1. UV-VIS-NIR spectra were recorded in an Ocean Optics USB2000+XR1-ES spectrophotometer equipped with an Ocean Optics DH-2000-BAL power supply. All UV-VIS-NIR spectra of PEDOT:PSS aqueous suspension were acquired in a quartz cuvette with 0.1 cm optical path and when diluted with water or AuNP aqueous suspension 1:9 in volume, the optical path was 5 mm. For obtaining the RR, SERRS and UV-VIS-NIR electronic spectra, PEDOT:PSS suspensions were dispersed in water or AuNPs aqueous suspension, respectively, in the 1:10 proportion under the simple mixture process. It was done with aliquots took from the synthesis and analyzed in the sequence, without further purification. Synthesis of PEDOT:PSS PEDOT:PSS was synthesized by the chemical method as described elsewhere.18 Briefly, 144 µL of EDOT and 384 mg Na2S2O8 were added to 6% by weight aqueous solution of PSS followed by the addition of 22 mL of deionized water. The synthesis was kept under controlled temperature (23.0 or 30.0 ºC) and vigorous stirring for 12 hours and samples were taken hour by hour for spectroscopic analyses. Synthesis of AuNPs Colloidal gold suspension was obtained by adapted method Frens21 as follow, 50 mL of 0.01% by weight HAuCl4.3H2O aqueous solution was heated to the boiling point under reflux conditions. Following, 500 µL of 1% by weight trisodium citrate aqueous solution were added dropwise. After the appearance of a red-wine color, the heating was interrupted and the suspension was allowed to cool to room temperature and then stored under refrigeration. Density Functional Theory Calculations PEDOT:PSS properties were obtained from computational methods of analysis using EDOT oligomers hanging from 2 to 16 residues. The static molecular properties were calculated by DFT with Becke's three parameters exchange functional22 along with Lee-Yang-Parr correlation functional (B3LYP).23 The split-valence double-zeta polarized basis set 6-31G(2d)24 were used to describe the system, and the singlet spin state was chosen to represent the charged species. Geometry optimizations and harmonic frequencies were calculated for free molecules in vacuum using the Linux version of Gaussian 09, revision B-01.25 Harmonic frequencies were determined by calculations in the static DFT method, avoiding increasing computational cost26 associated to determinations by time-dependent DFT of excited states of interest, which could show resonance effects with exciting radiations used in this work.
RESULTS AND DISCUSSIONS Figure 1a presents the RR spectra of PEDOT:PSS recorded by using 1064 nm exciting radiation, along with the chemical structure with usual nomenclature for carbon atoms of the conjugated system. The chosen structure to calculate PEDOT:PSS properties was 16 EDOT repeat units oligomer (EDOT16). For each residue of this oligomer, the dihedral angle CβCαCα'Cβ' was set to 180º to best represent the most stable structure of PEDOT. Figure 1b shows the Wiberg's bond order indexes obtained for the 32 CαCβ bonds of the oligomeric structure in terms of natural atomic orbitals (NAO), defined in terms of the off-diagonal contribution for each pairs of atoms in the density matrix.27 The bond order using such approach was first defined for semiempirical restricted closed-shell calculations, which use the first approximation of the orthonormality of basis sets.27 Orthogonalization procedure allows one to obtain bond order data based on NAOs for systems accounting the effects of the overlap matrix when non-orthogonalized basis sets are used.28 The literature shows that these methods are not very sensible to determine the size of the basis set, allowing one the search of tendencies for further evaluation when using the same method.29 Each subsequent pair of indexes represents two bonds located in the same residue. It is possible to note a strong effect in the CαCβ bonds located at the border of the oligomer. However, the index tends to converge to a value around 1.4156 for the bonds localized in between residue number 3 to 29, indicating the region of influence of border effect over the main chain of this oligomer. In the borders CαCβ double bond characters are greater and have higher order indexes, indicating that the π conjugation is less present.16,30 This parameter indicates that the EDOT16 is a suitable model to represent the PEDOT polymeric chains. EDOT16 oligomer model was used for calculation of vibrational modes of PEDOT polymer, but the absence of resonance effects precludes the use of its theoretical Raman spectra for analysing relative intensities of the experimental bands.
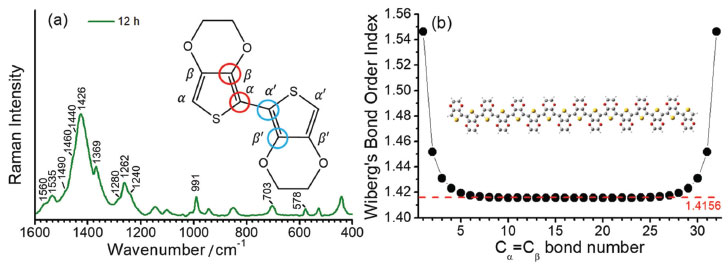 Figure 1. (a) Typical experimental RR spectrum of PEDOT:PSS as synthesized with 12 hours, when using exciting radiation with wavelength at 1064 nm. Inset: bisEDOT representing the nomenclature based on the position of the carbon atoms relative to the sulfur atom. The dihedral angle represented by the two pairs of bonds marked with red and blue circles were set to 180 ºC to represent the most stable conformation of the EDOTn oligomers; (b) variation of the bond order index, theoretically obtained for the CαCβ bond along the subsequent EDOT units, for the EDOT16 oligomer obtained from the B3LYP/6-31G(2d) level of theory calculations. Inset: structure of the EDOT16 oligomer
As the RR spectra of PEDOT:PSS is dominated by the strong bands in the region of the C=C stretching, the main features used to characterize PEDOT:PSS were at 1426 cm-1, assigned to the symmetric stretching of the CαCβ bond of reduced species; 1440 and 1460 cm-1, assigned to the symmetric stretching of the CαCβ bond of oxidized species; 1490 cm-1, assigned to the antisymmetric stretching of the CαCβ bond of reduced species and two bands at 1535 and 1560 cm-1, assigned to the antisymmetric CαCβ stretching of both oxidized and reduced EDOT units in polymer chains, respectively. These data are summarized in table 1, along with the other vibrational features observed for PEDOT:PSS.
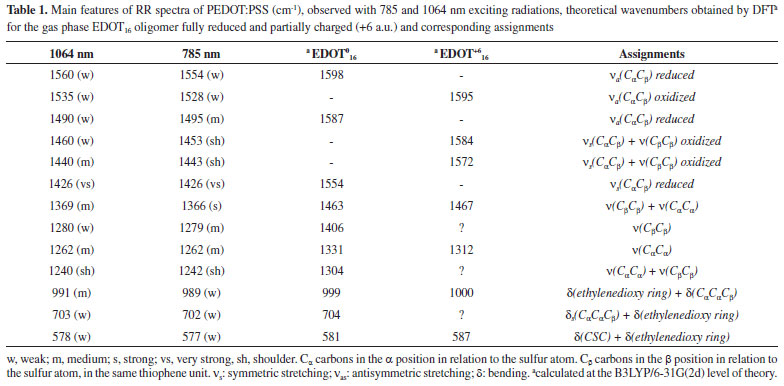
Figure 2 shows the UV-VIS-NIR absorption spectra of PEDOT:PSS (Figure 2a) and its RR spectra in the high-frequency region as a function of the reaction time (Figure 2b). For better visualization, Figure 2c shows the RR spectrum of PEDOT:PSS after 12 hour of synthesis, as the final reaction product. The wide overlapping of electronic states, observed in conducting polymers associated with the existence of different chemical structures along the chains, results in a wide electronic absorption band, which ranges from the purple to the near infrared region. The RR spectrum of PEDOT completely overtakes the Raman signal of PSS, and presents several differences in function of the exciting radiation since electronic transition resonances comprise different chromophore groups capable of being excited by distinct laser lines.
 Figure 2. (a) UV-VIS-NIR spectra of PEDOT:PSS as a function of reaction time; (b) RR spectra of PEDOT:PSS in the wavenumber range of double bond stretching, as a function of reaction time, and (c) in the same spectral interval after 12 hours of reaction. All spectra were obtained at 23.0 ºC and using exciting radiation with wavelength at 1064 nm
In all RR spectra of PEDOT:PSS it can be observed an intense band in the region of 1426 cm-1, relative to the symmetric CαCβ stretching, which is the normal mode with the highest Raman activity. For polymers with a higher degree of oxidation, this band loses intensity, shifts to regions of greater wavenumber and splits into other bands also characteristics of the symmetric CαCβ stretching, relative to species with greater localized charge in the polymeric structure. The variability of the maximum intensity and line profile of such a band at ca. 1426 cm-1 was observed in the RR spectra of all dispersions obtained with the increasing time of synthesis, as shown in Figure 2b. Such variations in the wavenumbers from 6 to 12 hours were plotted in Figure 3a.
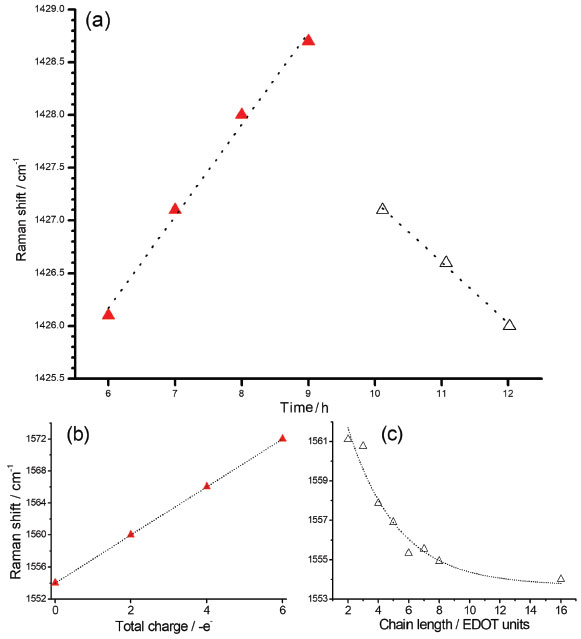 Figure 3. (a) Reaction time dependence of the maximum of the band observed at 1426 cm-1 region in the experimental RR spectra of PEDOT:PSS; (b) theoretical wavenumbers obtained for the band at 1554 cm-1 of EDOT16 oligomer (νs(CαCβ)oxidized) with charge density varying from 0 to +6 a.u.; (c) simulation data for the band at 1561 cm-1 (νs(CαCβ) reduced) as function of the number of the residues for a series of EDOTn oligomers with n varying from 2 to 16 residues. Two different regions marked in (a) are correlated with (b) as red triangles and (c) as white triangles. All spectra were obtained at 23.0 ºC and using 1064 nm laser line. Theoretical data obtained with the DFT methodology in the B3LYP/6-31G(2d) level of theory
The slow early stages of the strongly oxidative process in the synthesis of PEDOT:PSS, represented by the time interval between 6 to 9 hours, is responsible for the generation of small oligomers that further condense into high order oligomers and even larger species. Before 6 hours of reaction, the presence of radical monomers, dimers and very small oligomers with different oxidative stages precludes the observation of a reproducible behavior of the spectral pattern as observed in Figure 3. The latter stages, however, follow with considerably higher speeds.9 The increase in the proportion of oxidized species in the medium is reflected in the displacement of the band around 1426 cm-1 (Figure 3a) to the region of greater energy. Doping is also a slow and dynamic process that is dependent on the growth of polymeric chains.31 For times greater than 10 hours there is a combination of the spatial and charge reorganization effects over the units, with the insertion of electrons of PSS in the PEDOT moiety in the gaps created by the absence of electrons in the polaronic species.32 The harmonic frequency corresponding to the vibrational mode ascribed to the band observed experimentally in 1426 cm-1 are theoretically located at 1554 cm-1 for the reduced EDOT16 species. However, when the positive density of charge changes from 0 to +6 atomic units of charge, this band shifts linearly to 1572 cm-1 (Figure 3b), thus evidencing changes in the force constants associated to the CαCβ bond with the variation of charge, which can be related to the ongoing oxidative process observed in the band shift to the samples obtained with reaction time varying from 6 to 9 hours (Figure 3a). From theoretical results, it is possible to note that the harmonic frequency at 1561 cm-1 for the EDOT2 dimer decreases to 1554 cm-1 for EDOT16, which are assigned to the symmetric stretching of CαCβ bonds for reduced species. Such features present exponentially behavior when chains grow (Figure 3-c). This behavior evidences that the size of the chain affects this vibrational modes at this spectral region as a function of the stabilization energy achieved in the growth stage of the polymerization reaction. Such a shift to low wavenumber values can be correlated with the behavior of the band at 1426 cm-1 of the experimental RR spectra when reaction times were higher than 10 hours (Figure 3a). It is noteworthy that the growth of the chains occurs since early reaction times but apparently the prevalence of oxidized species initially dominates the observed spectral pattern and the fast growth of the chains is prevalent in the latter stages. Figures 4a and 4b show the RR spectra obtained with 785 and 1064 nm exciting radiations, respectively, in the high-frequency region in function of the reaction time, at 30.0 ºC. When 785 nm laser line was used, it can be observed three resolved bands at 1426, 1443 and 1453 cm-1, which are assigned to symmetric stretching modes of CαCβ for reduced and oxidized species (Table 1). Such features allow specifying the degree of oxidation of the polymer by the evaluation of their intensity ratios11 (Figure 4c). For shorter reaction times, it is observed that the band around 1426 cm-1, attributed to the reduced regions of the polymer, is the most intense band, while for longer times this band loses intensity and the band in 1443 cm-1, attributed to the oxidized regions, gains intensity. The presence of a band of low intensity near 1490 cm-1, relative to the CαCβ, antisymmetric stretching of the fully reduced species can also be observed. For 1064 nm excitation the higher intense band, around 1426 cm-1 is broadened, with the presence of shoulders at 1440 and 1460 cm-1. The variation in the intensity ratios, observed with 785 nm excitation cannot be evaluated with 1064 nm laser line, since oxidized species are in resonance with the first excitation. Throughout the synthesis, even though variations in the relative intensities of such three features cannot be separated with 1064 nm excitation, since it is embedded in a broad envelope, the global effect is the shift of the maximum of this feature to higher wavenumbers, indicating the increasing in the oxidized species concentration with reaction time (Figure 4d). In this way, the formation of regions with extended conjugation in the conducting polymer chains, ascribed to oxidized species, has higher resonance with 785 nm exciting radiation and can be monitored with this laser line with better precision.
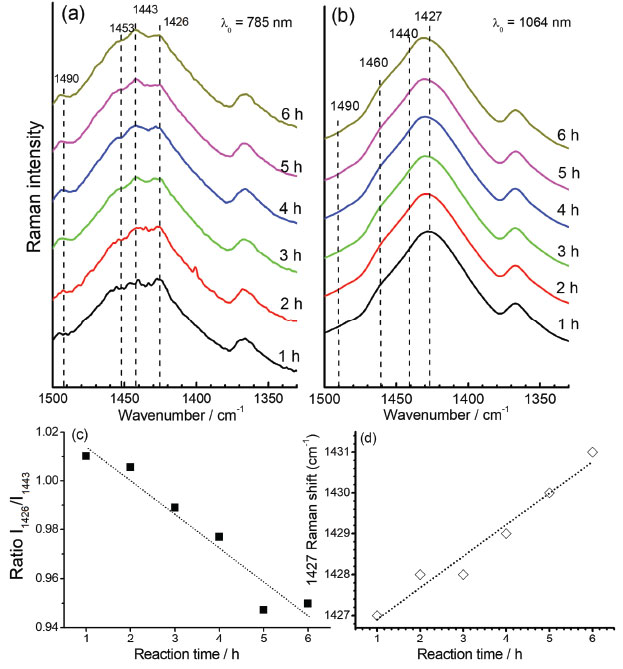 Figure 4. Time dependence of the RR spectra of PEDOT:PSS observed at 1350-1500 cm-1 region when using exciting radiation with wavelength at (a) 785 nm and (b) 1064 nm; (c) intensity ratio of 1426 cm-1 and 1443 cm-1 bands, as function of reaction times, taken from the spectra obtained with 785 nm excitation and (d) maximum of the band at 1427 cm-1 as function of the reaction time, taken from the spectra obtained with 1064 nm excitation. All syntheses were carried out with temperature fixed at 30.0 ºC
To understand the chemical interactions of polymeric chains and the metallic surfaces of AuNPs the SERRS spectra of oligomers with incident radiation at 785 nm were obtained by simple mixing aliquots of the synthesis of PEDOT:PSS with AuNPs aqueous suspension in 1:9 proportion, and compared to the RR spectra of the polymeric dispersions diluted in the same proportion in water (Figure 5). The UV-VIS-NIR spectra obtained of AuNPs and PEDOT:PSS aqueous suspension, before and after their mixture (Figure 5c) did not show any variation in the electronic transitions profile for the polymer as well as for the localized surface plasmon resonance transition, indicating both structures were preserved. However, SERRS spectra showed significant changes in the spectral pattern which can be ascribed to the chemical interactions of the oligomers with the metallic surface.
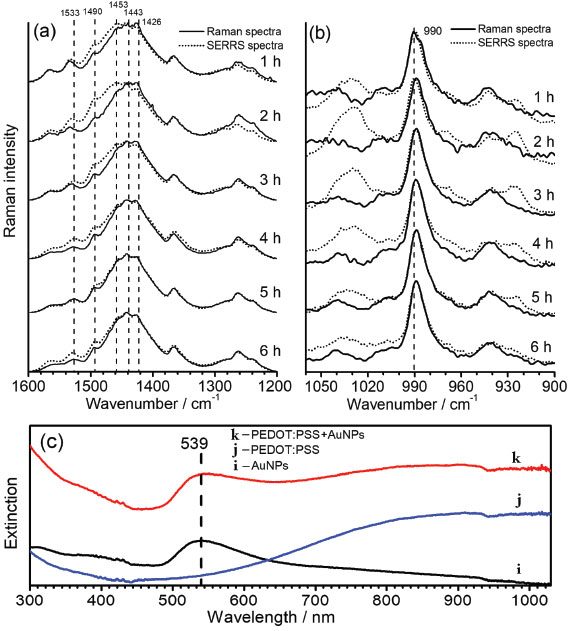 Figure 5. Time dependence of the SERRS spectral patterns of PEDOT:PSS, in comparison with its RR spectra, observed (a) from 1600 to 1200 cm-1, and (b) from 900 to 1050 cm-1; (c) UV-VIS-NIR spectra of (i) AuNPs aqueous suspension, (j) PEDOT:PSS diluted in water and (k) the mixture of AuNPs with PEDOT:PSS. All syntheses were obtained with temperature fixed at 30.0 ºC, and all RR and SERRS spectra were recorded by using λ0 = 785 nm. The bands at 1443 and 990 cm-1 were used as internal pattern to normalize each spectrum
Figure 5a and 5b shows the SERRS spectra for the high and low wavenumbers regions, which were separated in two plots since the intensities of the bands in the first region are stronger enough to preclude the evaluation of changes in only one plot. The variations in the relative intensities of the SERRS bands are definitive indication of the interactions of PEDOT oligomers with the metallic surfaces. In the higher wavenumber region (Figure 5a) for reaction times lower than 5 hours, the band at 1426 cm-1, attributed to the CαCβ, symmetric stretching of the fully reduced species, is the most intense band, while for the SERRS spectra it can be observed the enhancement of the bands at 1443 cm-1 and 1453 cm-1, assigned to the CαCβ symmetric stretching of the oxidized species of the polymer. Such results allow inferring that the small chains formed in early of synthesis have oxidized moieties interacting more freely with the metallic surface. This can be due to both the lower steric hindrance of small oligomers and higher affinity of conjugated moieties by the metallic surface. Such an assumption can be reinforced by the enhancement of the band at 1533 cm-1, which may be ascribed to the antisymmetric CαCβ stretching of the most oxidized species, which presents a maximum SERS intensity with two hours of synthesis. For reaction times of 5 and 6 hours, the SERS spectra presented little or none change in comparison with RR spectra. In this way, it is possible that greater oligomers, which take helical form around PSS chains, have steric hindrance for interactions with the surfaces of AuNPs, leading to the prevalence of RR spectral pattern over SERRS signal. It is noteworthy the complex overlapping of both RR and SERRS signals, which is function of the changes of the size distributions of oligomer chains with reaction times, precludes the calculations of SERRS enhancements. Figure 5b presents the RR and SERS spectra in the region between 900 and 1050 cm-1. The enhanced SERRS bands at 991 and 1030 cm-1, both assigned to the deformation of the ethylenedioxy ring, indicates that such a moiety is involved in the interaction with the metallic surface. Such a proposal is in agreement with the surface selection rules,33,34 which state that the molecular moiety in interaction with the metal will have normal vibrational modes ascribed to enhanced features in the spectrum. It is important to note that the thiol moiety is prevented from interacting with metallic surface since it is the doped site, which is in strong interaction with PSS structure. The SERS spectra of PEDOT:PSS were recorded by Stavytska-Barba and Kelley35 by using 633 nm exciting radiation on silver or gold thin film. They observed silver nanostructures can lead to the reoxidation of reduced moieties while gold surfaces do not lead to significant interactions with the polymers, resulting in small changes in the spectral pattern in comparison with Raman spectra in absence of nanostructures. It is in agreement with the results presented here, since it were observed changes in SERRS spectral pattern only when small oligomers were presented, reinforcing the assumption that the interactions of the molecules of PEDOT with gold surfaces are precluded by the growth of the chains.
CONCLUSIONS The use of RR and SERRS spectroscopies, supported by DFT calculations of oligomeric EDOTn models, with charge ranging from 0 to +6 atomic units and size varying from 2 to 16 EDOT units, allowed to follow the synthetic procedure of PEDOT:PSS and to characterize its structural evolution. It was evidenced by the diverse nature of the suspensions obtained with different reaction times when vibrational analysis was done. The symmetrical CαCβ stretching band located at ca. 1426 cm-1 for dispersions synthesized with small reaction times, evolved to 1428 cm-1 band for times ranging from 6 to 9 hours of synthesis, showed that more oxidized species are formed predominantly in this stage, which is a strong characteristic of the oxidative polymerization reaction. This fact was also corroborated by the evaluation of the ratio between the symmetrical CαCβ stretching band located at ca. 1426 cm-1 and the band located at ca. 1440 cm-1, which showed a reduction of the values with the reaction time. While the former is associated with the vibration of reduced EDOT units in the polymeric chain, the latter are related to the same vibration of more oxidized units. The application of AuNPs as SERRS substrate showed diverse spectral patterns from that observed in the RR analysis, with the bands observed at ca. 1443 cm-1 and 1453 cm-1, assigned to the symmetrical CαCβ stretching vibration of more oxidized species, being the most enhanced for shorter reaction times. This difference, however, becomes null for higher reaction times, with the predominance of the RR pattern of PEDOT:PSS, which indicated that, for early reaction times, the small and more oxidized oligomers interact preferentially with the surface of the nanoparticles. Nonetheless, the SERRS spectral pattern tends to become very similar to one from RR when the PEDOT chains are longer, probably due to steric hindrance involving gold surfaces and PEDOT molecules that are interacting strongly with long chains of PSS. This work showed that the application of vibrational spectroscopies along with the support of computational methods of analysis could provide an outstanding tool to analyze the synthesis of PEDOT:PSS, allowing one to obtain different materials with different chemical properties, which could be more suitable for distinct applications of PEDOT:PSS in the field of electronic organics.
ACKNOWLEDGEMENTS This work was financially supported by the CNPq and FAPEMIG (CEX-APQ-02392-15; PRONEM APQ-01283-14) Brazilian Funding Agencies. This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - Brasil (CAPES) - Finance Code 001. H. F. dos Santos also would like to thank CNPq for his grant.
REFERENCES 1. Zheng, Y.; Zeng, H.; Zhu, Q.; Xu, J.; J. Mater. Chem. C 2018, 6, 8858. 2. de Castro Camioto, F.; Moralles, H. F.; Mariano, E. B.; Rebelatto, D. A. do N.; J. Clean. Prod. 2016, 122, 67. 3. Zaman, K.; Abdullah, A. Bin; Khan, A.; Nasir, M. R. B. M.; Hamzah, T. A. A. T.; Hussain, S.; Renewable Sustainable Energy Rev. 2016, 56, 1263. 4. Bharti, M.; Singh, A.; Samanta, S.; Aswal, D. K.; Prog. Mater. Sci. 2018, 93, 270. 5. Hui, Y.; Bian, C.; Xia, S.; Tong, J.; Wang, J.; Anal. Chim. Acta 2018, 1022, 1. 6. Du, Y.; Xu, J.; Paul, B.; Eklund, P.; Appl. Mater. Today 2018, 12, 366. 7. Gan, Q.; Bartoli, F. J.; Kafafi, Z. H.; Adv. Mater. 2013, 25, 2385. 8. Agbaoye, R. O.; Adebambo, P. O.; Akinlami, J. O.; Afolabi, T. A.; Karazhanov, S. Z.; Ceresoli, D.; Adebayo, G. A.; Comput. Mater. Sci. 2017, 139, 234. 9. Kirchmeyer, S.; Reuter, K.; J. Mater. Chem. 2005, 15, 2077. 10. Ouyang, J.; Xu, Q.; Chu, C. W.; Yang, Y.; Li, G.; Shinar, J.; Polymer (Guildf). 2004, 45, 8443. 11. Garreau, S.; Louarn, G.; Buisson, J. P.; Froyer, G.; Lefrant, S.; Macromolecules 1999, 32, 6807. 12. Duvail, J. L.; Rétho, P.; Garreau, S.; Louarn, G.; Godon, C.; Demoustier-Champagne, S.; Synth. Met. 2002, 131, 123. 13. Tsai, M. H.; Lin, Y. K.; Luo, S. C.; ACS Appl. Mater. Interfaces 2019, 11, 1402. 14. Moraes, B. R.; Campos, N. S.; Izumi, C. M. S.; Vib. Spectrosc. 2018, 96, 137. 15. Chiu, W. W.; Travaš-Sejdic, J.; Cooney, R. P.; Bowmaker, G. A.; Synth. Met. 2005, 155, 80. 16. Alemán, C.; Armelin, E.; Iribarren, J. I.; Liesa, F.; Laso, M.; Casanovas, J.; Synth. Met. 2005, 149, 151. 17. Dai, Q.; Li, Y.; Zhai, L.; Sun, W.; J. Photochem. Photobiol., A 2009, 206, 164. 18. Diah, A. W. M.; Quirino, J. P.; Belcher, W.; Holdsworth, C. I.; Macromol. Chem. Phys. 2016, 217, 1907. 19. Ranganathan, K.; Wamwangi, D.; Coville, N. J.; Sol. Energy 2015, 118, 256. 20. Coville, N. J.; Matsoso, B. J.; Wamwangi, D.; Ranganathan, K.; Mutuma, B. K.; J. Nanosci. Nanotechnol. 2018, 19, 2747. 21. Frens, G.; Nature 1973, 242, 117. 22. Becke, A. D.; J. Chem. Phys. 1993, 98, 5648. 23. Lee, C.; Yang, W.; Parr, R. G.; Phys. Rev. B 1988, 37, 785. 24. Ditchfield, R.; Hehre, W. J.; Pople, J. A.; J. Chem. Phys. 2004, 54, 724. 25. Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H. P.; Izmaylov, A. F.; Bloino, J.; Zheng, G.; Sonnenberg, J. L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgomery, J. A.; Peralta, J. E.; Ogliaro, F.; Bearpark, M.; Heyd, J. J.; Brothers, E.; Kudin, K. N.; Staroverov, V. N.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, J. M.; Klene, M.; Knox, J. E.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Zakrzewski, V. G.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Dapprich, S.; Daniels, A. D.; Farkas; Foresman, J. B.; Ortiz, J. V; Cioslowski, J.; Fox, D. J.; Gaussian 09, Rev. B.01, Gaussian, Inc., Wallingford CT, 2009. 26. Foerster, D.; Phys. Rev. B: Condens. Matter Mater. Phys. 2005, 72, 7. 27. Wiberg, K. B.; Tetrahedron 1968, 24, 1083. 28. Mayer, I.; Salvador, P.; Chem. Phys. Lett. 2004, 383, 368. 29. Sizova, O. V.; Skripnikov, L. V.; Sokolov, A. Y.; J. Mol. Struct. THEOCHEM 2008, 870, 1. 30. Casanovas, J.; Zanuy, D.; Alemán, C.; Phys. Chem. Chem. Phys. 2017, 19, 9889. 31. Shi, H.; Liu, C.; Jiang, Q.; Xu, J.; Adv. Electron. Mater. 2015, 1, 1. 32. Kalagi, S. S.; Patil, P. S.; Synth. Met. 2016, 220, 661. 33. Moskovits M.; Suh, J. S.; J. Phys. Chem. 1984, 88, 5526. 34. Moskovits, M.; J. Chem. Phys. 1982, 77, 4408. 35. Stavytska-Barba, M; Kelley, A. M.; J. Phys. Chem. C 2010, 114, 6822. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access