
 |
 |
 |
 |
 |
Artigo
|
|
| Uma releitura dos artigos científicos do Prof. Hans Stammreich Rereading the works of Prof. Hans Stammreich |
|
Mauro C. C. Ribeiro
Departamento de Química Fundamental, Instituto de Química, Universidade de São Paulo, 05508-000 São Paulo - SP, Brasil Recebido em em 03/05/2019 *e-mail: mccribei@iq.usp.br In this work we provide a revision of the studies by Raman spectroscopy carried out by Prof. Hans Stammreich in the São Paulo University. The works of Prof. Stammreich are discussed focusing on the most important conclusion of each paper and the scientific issue at the time of publication. Approximately twenty-five papers are discussed, which were separated into four groups according to the investigated systems: metal carbonyls, halogens and inter-halogens, halogen compounds, and oxoanions. INTRODUÇÃO Aspectos da vida do Prof. Stammreich e do seu trabalho na Universidade de São Paulo têm sido apresentados em textos anteriores.1-3 O leitor prontamente assimila que o Prof. Stammreich, além de introduzir a espectroscopia Raman no Brasil, teve contribuições fundamentais para ampliação do escopo de aplicação da técnica, em particular ao construir lâmpadas de hélio para excitação dos espectros. Amostras que absorviam a mais usual radiação de arcos de mercúrio ou eram fotossensíveis passaram a ser estudadas por espectroscopia Raman com excitação no amarelo ou vermelho. O Prof. Stammreich resume alguns dos avanços técnicos (espectrógrafos com redes de difração, fontes de luz para excitação, métodos fotográficos para detecção) em um artigo de 1956.4 A presente revisão está direcionada para os resultados efetivamente alcançados pelo grupo do Prof. Stammreich. Neste artigo pretendemos transmitir ao leitor o contexto científico e as conclusões alcançadas pelo grupo do Prof. Stammreich com o uso da espectroscopia Raman. Os aspectos experimentais de preparação de amostras e obtenção dos espectros Raman não serão comentados nesta revisão. Concentraremos a discussão no ponto central de cada artigo de tal forma a destacar a questão científica pendente no momento e, portanto, a principal conclusão do artigo. Cerca de vinte e cinco artigos publicados pelo Prof. Stammreich serão revistos, ou seja, aproximadamente metade do número obtido numa consulta a um serviço de indexação de publicações científicas. Os artigos a serem revistos compreendem o período 1955–1969, mas a discussão não seguirá ordem cronológica. Os artigos foram alocados em quatro grupos em função do tipo de composto: carbonil-metais; halogênios e inter-halogênios; compostos de halogênio; oxoânions.
CARBONIL-METAIS A determinação da estrutura do pentacarbonilferro(0), Fe(CO)5, por espectroscopia de absorção no infravermelho (IR) era ambígua ao final da década de 1950. Atribuição do espectro IR levou às proposições de pirâmide tetragonal5 (C4v) ou bipirâmide trigonal6 (D3h). A Figura 1 mostra estruturas esquemáticas para cada um desses dois grupos de ponto e o número de modos normais de cada espécie de simetria. A partir das atividades no IR e Raman indicadas na Figura 1, fica claro que espectroscopia Raman complementaria a atribuição do espectro vibracional e discriminaria a estrutura correta entre C4v ou D3h. Nesse sentido, a possibilidade do grupo do Prof. Stammreich de excitar o espectro Raman com radiações de He (667,8 nm e 706,5 nm) foi fundamental porque Fe(CO)5 é colorido e fotosensível.7 A atribuição do espectro Raman indicou três modos atribuídos como estiramentos C–O (2 A1' e 1 E'), três dobramentos de ângulo C–Fe–C (2 E' e 1 E") e sete modos de estiramento Fe–C ou dobramento de ângulo Fe–C–O (2 A1', 3 E' e 2 E"). Portanto, ao comparar o número de modos normais de cada espécie de simetria e as correspondents atividades Raman, concluiu-se que a estrutura de Fe(CO)5 é a bipirâmide trigonal (D3h).7 Uma ambiguidade análoga foi a motivação de um artigo póstumo de 19708 sobre a estrutura do íon pentacianidoniquelato(II) em solução aquosa. Stammreich e colaboradores8 propuseram que a estrutura de [Ni(CN)5]3- é bipirâmide trigonal (D3h) a partir da atribuição do espectro Raman. Devemos notar, no entanto, que Griffith e Lane9 propuseram poucos anos depois que a estrutura de [Ni(CN)5]3- é pirâmide tetragonal (C4v) a partir do espectro Raman da solução aquosa.
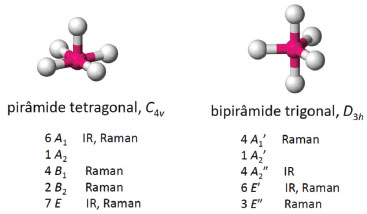 Figura 1. Ambiguidade das estruturas de Fe(CO)5 propostas a partir da atribuição de espectros IR: pirâmide tetragonal5 (C4v) ou bipirâmide trigonal6 (D3h). As estruturas esquemáticas mostram cada ligante carbonil como uma esfera branca. O número de modos normais de cada espécie de simetria e atividades IR e Raman estão indicados para cada estrutura
A esperada estrutura tetraédrica dos ânions [Co(CO)]4- e [Fe(CO)]42- foi confirmada pelos espectros Raman obtidos no grupo do Prof. Stammreich.10 A questão não trivial abordada nesse trabalho foi a estrutura do derivado [HFe(CO)]4-. Espectro IR de HCo(CO)4 sugeria que a simetria Td era mantida,11 i.e. posição do hidrogênio desconhecida, mas dublete de bandas do modo de estiramento do ligante carbonil indicava que o hidrogênio estava ligado a um carbonil, ou seja, simetria C3v. No entanto, o espectro IR não indicava a presença de um modo de estiramento OH.11 Edgell et al.12,13 propuseram um modelo de ponte para HCo(CO)4 a partir do espectro IR e de cálculo de orbitais moleculares. Nesse modelo (ver Figura 2) o hidrogênio está ligado a três carbonilas e ao metal numa estrutura de simetria C3v.
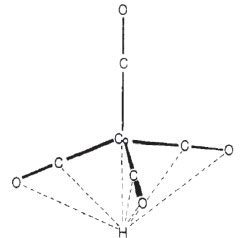 Figura 2. O modelo com simetria C3v proposto por Edgell et al.12,13 para HCo(CO)4. Reproduzido com permissão de Edgell, W. F.; Magee, C.; Gallup, G.; J. Am. Chem. Soc. 1956, 78, 4185. Copyright 1956 American Chemical Society
A redução da simetria implica em desdobramento das vibrações triplamente degeneradas do grupo de ponto Td em vibrações das espécies de simetria A1 e E do grupo C3v, mas este efeito não foi observado por Edgell et al.12 no espectro IR de HCo(CO)4. Ao comparar os espectros Raman de [Fe(CO)]42- e [HFe(CO)]4-, Stammreich et al.10 comprovaram que os modos F2 do tetraedro regular em [Fe(CO)]42- eram, de fato, desdobrados em [HFe(CO)]4-. A Tabela 1 mostra as frequências vibracionais dos modos relevantes reportadas no artigo de Stammreich et al.10
A indicação de estrutura C3v para [HFe(CO)]4- está citada na terceira edição do clássico livro de Linus Pauling (página 334).14 É curioso notar que Pauling cita o trabalho do Stammreich como sendo espectro infravermelho em vez de espectro Raman. O erro decorre do fato que no livro, o qual é do mesmo ano que o artigo do Stammreich (1960),10 Pauling faz referência ao estudo de HCo(CO)4 publicado por Cotton15 em que o autor menciona uma comunicação particular do Prof. Stammreich (sem explicitar que se tratava de espectroscopia Raman) sobre o desdobramento dos modos ao passar de [Fe(CO)]42- para [HFe(CO)]4-. Finalmente, mencionamos que Stammreich et al.10 consideraram que não estava clara a qual banda Raman atribuir a vibração esperada envolvendo hidrogênio e metal em [HFe(CO)]4-. No ano seguinte, Edgell e Summit16 analisaram o espectro IR de HCo(CO)4 em fase gasosa e atribuíram o estiramento ν(Co–H) à banda em 1934 cm-1. Difração de elétron em fase gasosa confirmou a simetria C3v de HCo(CO)4, sendo o comprimento da ligação rCo–H = 1.556 Å.17 Os estudos de carbonil-metais por espectroscopia Raman inserem-se no contexto científico mais amplo da natureza da ligação química com caráter parcial de ligação dupla. Pauling, no capítulo dedicado ao tema (ver capítulo 9 da ref. 14), considera como exemplo, entre outros sistemas, a distância relativamente pequena rNi–C = 1,82 Å do complexo tetraédrico Ni(CO)4 como manifestação do caráter duplo parcial da ligação Ni–C e das estruturas de ressonância. Comprimento de ligação é inversamente proporcional à ordem de ligação (por exemplo, ver Figura 7.3 da ref. 14) e à constante de força do modelo do oscilador harmônico (a chamada regra de Badger).18 Na relação direta entre ordem de ligação e constante de força reside a motivação da análise de modos normais realizada pelo grupo do Prof. Stammreich para a sequência Ni(CO)4, [Co(CO)4]- e [Fe(CO)4]2-. Stammreich et al.19 consideram a distribuição eletrônica desses complexos de acordo com os conceitos comumente encontrados em livros-textos atuais:20 ligação σ envolvendo doação de elétron do ligante para o metal e retrodoação de elétron d do metal para orbital π anti-ligante do carbonil. Assim, Stammreich et al.19 argumentam que na sequência de compostos isoeletrônicos Ni(CO)4, [Co(CO)4]- e [Fe(CO)4]2-, as ordens das ligações M–C e C–O, estimadas a partir das respectivas constantes de força, devem variar de forma complementar, i.e. a ordem da ligação M–C aumenta a expensas da C–O. A partir de um campo de força de valência, Stammreich et al.19 determinaram as constantes de força, algumas delas aqui reproduzidas na Tabela 2. Note a ordem decrescente de kC–O, enquanto que crescente de kM–C, ao longo da sequência Ni(CO)4, [Co(CO)4]- e [Fe(CO)4]2-. As tendências opostas de kC–O e kM–C indicam a conjugação na estrutura eletrônica. As ordens de ligação mostradas na Tabela 2,19 considerando monóxido de carbono (kCO = 18,53 mdina/ Å; nCO = 3) como referência, indicam o caráter parcial de ligações múltiplas e a tendência inversa de diminuição de nC–O concomitante ao aumento de nM–C. Note que a soma de nC–O e nM–C é aproximadamente quatro conforme a ordem de ligação total.
Espectroscopia IR indicou21 que as estruturas de carbonil-metais polinucleares, tais como as estudadas por Stammreich et al.,22 Cd[Co(CO)4]2 e Hg[Co(CO)4]2, envolviam ligação entre os metais. A evidência nesse sentido é a frequência do modo de estiramento ν(CO) ser observada acima de 2000 cm-1,21 i.e. sem o CO em ponte entre dois centros metálicos. A Figura 3 mostra esquemas de estruturas em duas conformações, alternada (D3d) e eclipsada (D3h), sendo que o espectro IR de Hg[Co(CO)4]2 em solução de n-heptano indicou a estrutura alternada.21 No entanto, a evidência mais direta da ligação metal–metal, ou seja, uma banda de baixa frequência correspondente ao modo de estiramento, não é facilmente acessível por espectroscopia IR. Os espectros Raman obtidos por Stammreich et al.22 confirmaram a ausência de CO em ponte e a estrutura alternada (D3d). O resultado mais importante do estudo foi a identificação do modo de estiramento metal–metal como uma banda Raman intensa, fina e polarizada. Os estiramentos ν(Cd–Co) e ν(Hg–Co) de Cd[Co(CO)4]2 e Hg[Co(CO)4]2 em solventes orgânicos foram observados em 152 e 161 cm-1, respectivamente.22
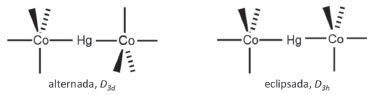 Figura 3. Esquema das estruturas de Hg[Co(CO)4]2 nas conformações alternada (grupo de ponto D3d) e eclipsada (grupo de ponto D3h)
O espectro IR de [(C5H5)Fe(CO)2]2 apresenta bandas de absorção em ∼800 cm-1 e em 1950–2060 cm-1 correspondentes aos estiramentos das carbonilas em ponte e terminais, respectivamente.23 A estrutura mostrada na Figura 4 corresponde à fase cristalina determinada por difração de raios-X. Uma vez que essa estrutura possui um centro de inversão, então, é válida a chamada regra de exclusão, ou seja, não há vibração ativa em comum, entre ambas as espectroscopias IR e Raman. No entanto, no caso de [(C5H5)Fe(CO)2]2 em solução, Cotton, Stammreich e Wilkinson23 observaram várias coincidências de bandas nos espectros IR e Raman na região dos estiramentos das carbonilas. A interpretação dada na ref. 23, embora de forma não categórica, foi que ocorre uma mudança estrutural na passagem do sólido para a solução e que existe mais de uma espécie em solução. De fato, estudo posterior de espectroscopias RMN e IR em função da temperatura caracterizaram a termodinâmica e cinética da conversão entre diferentes isômeros (ver a Figura 3 da ref. 24).
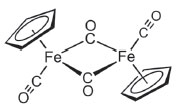 Figura 4. Estrutura de di-μ-carbonil-bis[carbonil(ciclopentadienil)ferro], [(C5H5)Fe(CO)2]2
HALOGÊNIOS E INTER-HALOGÊNIOS A técnica de excitação na região do vermelho é particularmente apropriada para obtenção do espectro Raman do vapor marrom de Br2. Stammreich25 obteve o espectro Raman de Br2(g) a 85 ºC e 900 mmHg usando a radiação 587,56 nm de uma lâmpada de He e tempo de exposição de 15 horas. O espectro Raman de Br2(g) reproduzido na Figura 5 mostra, além da transição fundamental em 316,8 cm-1, também a primeira e segunda harmônicas em 631,1 e 944,5 cm-1. Esse resultado foi "inesperado", nas palavras do próprio Stammreich, pois "harmônicas nunca haviam sido observadas em espectros Raman de moléculas diatômicas".25
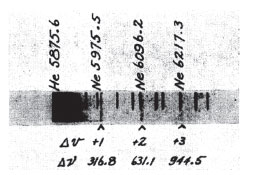 Figura 5. Espectro Raman de Br2(g) obtido por Stammreich.25 Reproduzido de Stammreich, H.; Phys. Rev. 1950, 78, 79, com permissão da APS
Observação de harmônicas relativamente intensas seria hoje explicada simplesmente como resultado do efeito Raman ressonante. No entanto, espectroscopia Raman ressonante não era uma área de pesquisa estabelecida em 1950, embora sabido teoricamente que o espectro Raman seria intensificado se a energia de excitação coincidisse com uma transição eletrônica da molécula. Por outro lado, Barrow26 mostrou que as linhas observadas por Stammreich25 no "espectro Raman" do Br2(g) correspondiam, em verdade, ao espectro de fluorescência devido à transição eletrônica B(3Π0u+) ← X(1Σg+) envolvendo os estados vibracionais v' = 13 e v" = 2, 3, 4 e 5. Assim, o artigo do Prof. Stammreich desencadeia uma discussão ainda mais fundamental, a saber, a distinção entre Raman e fluorescência quando o espectro é excitado em condição de ressonância. Kiefer e Schrötter27 mostraram que espectros de fluorescência e Raman ressonante de Br2(g) são observados simultaneamente se a linha de excitação é 694,3 nm, mas apenas espectro Raman ressonante se excitado com 488,0 nm. Holzer et al.28 resumiram a questão ao comparar os espectros de diferentes gases (Cl2, Br2, I2, BrCl, ICl, IBr) excitados com as várias linhas de um laser de íon argônio: espectro Raman ressonante é obtido quando a energia de excitação é maior do que o limite de convergência da transição B(3Π0u+) ← X(1Σg+), enquanto que fluorescência ressonante se a energia de excitação está dentro da estrutura rovibracional da banda de absorção. Várias propostas têm sido feitas para separar os termos atribuídos à Raman ressonante e fluorescência ressonante nas equações obtidas por teoria de perturbação da mecânica quântica para a interação radiação–molécula. A abordagem mais adequada da questão é pelo formalismo do tempo da espectroscopia, o qual mostra, no entanto, que a separação entre fluorescência e Raman ressonante não é formalmente válida sob um ponto de vista teórico matematicamente rigoroso.29 Por outro lado, é mais fácil distinguir entre fluorescência e Raman ressonante sob o ponto de vista experimental, por exemplo, diferenças em largura de banda, razão de despolarização e dependência com a pressão.28,30 A característica distintiva ainda mais evidente é que, se forem usadas diferentes radiações de excitação, a linha Raman manterá a mesma diferença de energia em relação à excitação. Nesse sentido, Stammreich e Forneris31 registraram o espectro Raman do bromo líquido usando simultaneamente sete radiações de uma lâmpada de argônio. A Figura 6 reproduz os espectros Raman de Br2(l), a partir dos quais a frequência da transição fundamental é 306,1 ± 0,6 cm-1. O espectro Raman de Br2(l) não exibe harmônicas, assim Stammreich e Forneris31 reconhecem que no caso de Br2(g) do artigo anterior25 não se tratava de espectro Raman. Recentemente, um estudo detalhado do espectro Raman de bromo líquido identificou até trinta harmônicas quando excitado com a radiação 405 nm de um laser de diodo.32
 Figura 6. Espectro Raman de bromo líquido obtido por Stammreich e Forneris.31 Reproduzido de Stammreich, H.; Forneris, R.; J. Chem. Phys. 1954, 22, 1624, com permissão da AIP
Os espectros Raman de moléculas diatômicas de halogênios e inter-halogênios permitiriam obter as constantes vibracionais de uma forma mais direta do que as reportadas a partir da análise da estrutura da banda do espectro eletrônico. Por exemplo, o valor tabelado por Herzberg33 para BrCl é ω = 430 cm-1. Stammreich e Forneris34 obtiveram ω = 428 ± 2 cm-1 a partir do espectro Raman de BrCl em solução de CCl4. No caso de cloro líquido, Stammreich e Forneris35 obtiveram o espectro Raman com alta resolução, sendo que linhas Raman foram atribuídas aos diferentes isótopos e transições fundamental e harmônica. A frequência harmônica e a constante de anarmonicidade resultantes, por exemplo, para 35Cl2, são ωe = 562,3 cm-1 e exe = 2,7 cm-1.35 As constantes de anarmonicidade obtidas a partir do espectro Raman são consideravelmente menores do que aquelas obtidas a partir do espectro eletrônico: Herzberg36 reporta para 35Cl2 os valores ωe = 564,9 cm-1 e exe = 4,0 cm-1. Stammreich e Forneris35 argumentam que a análise usual da progressão vibracional, bem conhecida em livros-texto de Físico-Química (gráfico de Birge-Sponer),37 depende da expressão para os termos vibracionais, G(v) = (v+½)ωe – (v+½)2ωexe, ser estritamente válida mesmo para números quânticos elevados. Por isso que Stammreich e Forneris35 consideram que as constantes de anarmonicidade corretas são as resultantes dos espectros Raman. De fato, uma consulta a um banco de dados atual (NIST, National Institute of Standards and Technology)38 indica os seguintes valores para 35Cl2: ωe = 559,7 cm-1 e exe = 2,67 cm-1. Stammreich e Forneris31 já salientam o efeito de fase condensada na frequência vibracional de Br2(l), pois a mesma é cerca de 10 cm-1 menor do que Br2(g). Na segunda parte do artigo sobre moléculas diatômicas de halogênios e inter-halogênios, Stammreich et al.39 compararam as frequências vibracionais de várias diatômicas (Cl2, F2, Br2, I2, ClF, BrCl, ICl, BrF, IF, IBr) em gás, líquido e solução em diferentes solventes. A comparação entre diatômicas homo- e heteronucleares teve a intenção de discriminar os efeitos das forças intermoleculares de dispersão de London e a presença adicional de momento de dipolo no caso das heteronucleares. A partir do conjunto de dados de frequências vibracionais, Stammreich et al.39 concluíram que o principal efeito das forças intermoleculares sobre a frequência vibracional nesse grupo de moléculas é devido à polarizabilidade das mesmas, sendo menos importante o efeito da polaridade da ligação nos inter-halogênios. Stammreich e Forneris40 analisaram os espectros Raman de moléculas AB4 que possuem uma estrutura de quadrado planar (D4h) em vez da simetria tetraédrica mais usual. No capítulo dedicado às ligações químicas envolvendo orbitais d, Pauling apresenta os quatro orbitais híbridos sp2d que apontam ao longo dos vértices de um quadrado (ver pag. 153 da Ref. [14]). Stammreich e Forneris40 partem dessa consideração para estudar os espectros Raman de (ICl4)-, (AuCl4)-, (AuBr4)- e (PtCl4)2-. Esses complexos são quadrados planares de acordo com dados de difração de raios-x, mas não havia um estudo por espectroscopia Raman, além de ser difícil estudá-los por espectroscopia IR por causa das frequências vibracionais baixas. Os espectros Raman dos quatro complexos apresentaram, de fato, as três frequências ativas no Raman esperadas pela simetria D4h: estiramento simétrico (A1g), estiramento anti-simétrico (B2g) e dobramento de ângulo no plano (B1g).40 No caso do íon poli-halogênio (ICl4)-, Stammreich e Forneris40 fazem notar que a simetria D4h decorre da hibridização sp3d2, a qual resulta em seis orbitais ao longo de um octaedro, sendo que dois pares de elétron não compartilhados ocupam as direções dos ápices do octaedro. Isso estava de acordo com considerações de Linus Pauling na segunda edição do seu livro (1944), em que íons poli-halogênios são incluídos na lista de exemplos (ver pág. 111 da ref. 41). No entanto, medidas de frequências de ressonância de quadrupolo nuclear realizadas por Cornwell e Yamasaki42,43 em sais de (ICl2)- e (ICl4)- indicaram que a participação de orbitais d na ligação química é menor do que antecipado. Em vez de ligações essencialmente covalentes entre orbitais σ do cloro e híbridos sp3d2 (octaedro) ou sp3d (bipirâmide trigonal) do iodo, foi proposto que a representação mais adequada da ligação química envolveria híbridos de ressonância entre estruturas do tipo -[Cl ... I – Cl] ↔ [Cl – I ... Cl]-.42,43 Isto motiva a análise de coordenadas normais realizada por Stammreich et al.:44 obter indícios sobre a natureza da ligação química a partir das constantes de força dos íons poli-halogênios. As frequências vibracionais dos modos de estiramento simétrico ν1 e antissimétrico ν3 observadas, respectivamente, nos espectros Raman e infravermelho dos íons triatômicos (ICl2)-, (BrCl2)- e (Br3)- prontamente permitem obter as constantes de força do estiramento X–Y, fr, e a constante de interação entre as ligações adjacentes, frr, de um campo de força de valência.45 Stammreich et al.44 encontraram que fr é significativamente menor nos íons triatômicos do que na molécula diatômica correspondente. Por exemplo, fr em (ICl2)- é 1.00 mdina/Å, a ser comparado com 2.38 mdina/Å em ICl. Além disso, as constantes de interação frr são relativamente grandes nos íons poli-halogênios: frr = 0.36 mdina/Å em (ICl2)-, enquanto que o usual, e.g. CO2, é frr apenas ca. 10% de fr. Os valores pequenos de fr, concomitante com valores grandes de frr, levaram Stammreich et al.44 a concordarem com a descrição proposta para a ligação química em termos de orbitais p e ressonância do tipo -[Cl ... I – Cl] ↔ [Cl – I ... Cl]-. Bell e Longuet-Higgins46 apresentaram um estudo muito detalhado sobre a análise de coordenadas normais de moléculas X2Y6 com simetria correspondente ao grupo de ponto D2h. A estrutura em questão é mostrada no painel esquerdo da Figura 7, sendo que os sistemas representativos abordados por Bell e Longuet-Higgins46 foram diborano, B2H6, e haletos de alumínio, e.g. Al2Cl6. Stammreich e Kawano47 estudaram o espectro Raman de I2Cl6, o qual é também de simetria D2h, mas com estrutura planar conforme ilustrado no painel direito da Figura 7. A estrutura cristalina de I2Cl6 já tinha sido determinada por difração de raios-X,48 mas não havia um estudo do espectro vibracional de uma molécula do tipo X2Y6 planar. Stammreich e Kawano47 atribuíram todas as nove vibrações ativas Raman esperadas para I2Cl6 de acordo com o grupo de ponto D2h: 4 Ag, 1 B1g, 1 B2g e 3 B3g. Destaque para as vibrações com contribuição de estiramentos I–Cl: quando envolve as ligações dos átomos de cloro em ponte entre os átomos de iodo é de frequência menor (ν8 (B3g) 106,2 cm-1) do que as vibrações envolvendo ligações I–Cl das extremidades (ν1 (Ag) 312,1 cm-1 e ν7 (B3g) 342,9 cm-1).47 Essa diferença de frequências vibracionais entre ligações I–Cl em ponte e na extremidade é consistente com as distâncias de ligação obtidas por difração de raios-X,48 respectivamente, 2,68 e 2,39 Å.
 Figura 7. Estruturas de moléculas X2Y6 com simetria D2h
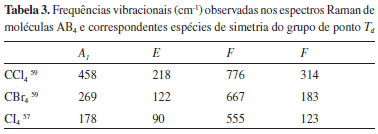
COMPOSTOS DE HALOGÊNIOS Stammreich obteve espectros Raman de diversos compostos de halogênios, sendo que análise de coordenadas normais foi realizada para vários sistemas e em alguns havia ambiguidade entre diferentes estruturas possíveis. A estrutura de dicloreto de enxôfre, SCl2, tinha sido determinada por difração de elétrons: dS–Cl = 1.99 Å, = 101º.49 Stammreich et al.50 registraram o espectro Raman de SCl2 líquido e, a partir das frequências observadas (208 cm-1 (A1), 514 cm-1 (A1) e 535 cm-1 (B2)), calcularam as constantes de força considerando um campo de força de valência. A constante de força de estiramento da ligação, kS-Cl = 2,58 × 105 dina cm-1, foi usada para estimar a distância da ligação, rS-Cl, a partir da conhecida regra de Badger que relaciona k e r.18 O valor rS-Cl = 2,02 Å estimado a partir do espectro Raman concorda com o valor determinado por difração de elétrons.50 A estrutura de cloreto de cromilo, CrO2Cl2, também foi determinada no estudo de difração de elétrons,49 o qual indicou que CrO2Cl2 pertence ao grupo de ponto C2v. Durante o andamento do estudo de CrO2Cl2 por espectroscopia Raman no grupo do Prof. Stammreich, o espectro infravermelho desse composto foi publicado por Hobbs.51 Quatro frequências fundamentais foram observadas no espectro infravermelho de CrO2Cl2, sendo que oito modos são ativos infravermelho. No caso do espectro Raman, Stammreich et al.52 atribuíram as noves frequências fundamentais esperadas para CrO2Cl2 de acordo com o grupo de ponto C2v: 4 A1 + 1 A2 + 2 B1 + 2 B2. A análise de coordenadas normais realizada com um campo de força quadrático resultou em frequências calculadas em excelente concordância com as experimentais. Assim como em vários sistemas discutidos nas seções anteriores, o escopo mais amplo da análise de coordenadas normais era obter inferência sobre a natureza da ligação química ao comparar com moléculas análogas. Por exemplo, a frequência vibracional 984 cm-1 atribuída ao estiramento Cr–O em CrO2Cl2 é maior do que em sistemas com estruturas de ressonância, tais como [CrO4]2- (859 cm-1) e [Cr2O7]2- (904 cm-1) que foram objetos de estudos anteriores no próprio grupo do Prof. Stammreich e que serão comentados na próxima seção.53,54 A ordem de ligação maior de Cr–O em CrO2Cl2 resulta na frequência de estiramento maior em comparação com a mesma ligação nos oxoânions.52 Análise de coordenadas normais para moléculas AB3, grupo de ponto C3v, foi realizada para vários trihaletos:55 PF3, PCl3, PBr3, AsF3, AsCl3, SbCl3 e BiCl3. O campo de força usado na análise incluiu quatro constantes de força para o estiramento da ligação, dobramento de ângulo e para as correlações entre ligações e entre ângulos. Stammreich et al.56 consideraram os derivados iodeto: PI3 e AsI3. A obtenção dos espectros Raman desses derivados é difícil pela cor vermelha dos compostos, sendo usada, então, a radiação 667,82 nm do He. A análise por espectroscopia de absorção no infravermelho também é difícil devido às frequências vibracionais baixas. Por exemplo, as frequências vibracionais observadas no espectro Raman de AsI3 diluído em solventes orgânicos são 216 cm-1 (A1), 94 cm-1 (A1), 221 cm-1 (E) e 70 cm-1 (E).56 As frequências vibracionais calculadas concordam com as frequências experimentais mesmo se as constantes de força de interação são ignoradas no campo de força proposto para moléculas AB3.56 Stammreich et al. trabalharam com outros derivados de iodo cuja análise vibracional era conhecida para moléculas análogas: tetraiodeto de carbono (CI4)57 e iodofórmio (CHI3).58 Simanouti59 fez a análise de coordenadas normais considerando o campo de força Urey-Bradley para moléculas tetraédricas AB4, inclusive CCl4 e CBr4. A Tabela 3 compara as frequências vibracionais dessas moléculas com as observadas por Stammreich et al.57 no espectro Raman de CI4. Como esperado, a constante de força de estiramento da ligação C–X diminui na sequência CCl4, CBr4 e CI4, respectivamente, 1,76, 1,43 e 1,12 × 105 dina/cm. No entanto, a constante de força do dobramento de ângulo em CI4 (0,094 × 105 dina/cm) é maior que em CCl4 e CBr4, respectivamente, 0,080 e 0,045 × 105 dina/cm. Stammreich et al.57 atribuíram esse fato ao volume atômico grande dos átomos de iodo em CI4. No caso do iodofórmio, a análise detalhada do espectro infravermelho foi publicada por Hexter e Cheung60 no mesmo ano do artigo de Stammreich e Forneris58 sobre o espectro Raman de CHI3. De fato, os autores compartilharam seus resultados antes da publicação no Oxford Meeting 1955 do European Molecular Spectroscopy Group.58 O estudo por espectroscopia infravermelho considerou monocristal de CHI3,60 enquanto que o estudo por espectroscopia Raman considerou CHI3 em solução em diversos solventes orgânicos.58 Excelente concordância foi obtida entre as frequências vibracionais dos espectros IR e Raman de CHI3.
Outros estudos de compostos de halogênios tiveram a geometria da molécula como questão principal. Espectros Raman61 e infravermelho62 de cloreto de tionilo, SOCl2, reportados na primeira metade da década de 1950 tinham confirmado que a estrutura não é planar C2v, mas sim piramidal Cs. No caso C2v são esperadas 3 A1 + 2 B1 + B2 vibrações, enquanto que no caso Cs são esperadas 4 A' + 2 A" vibrações. Todos esses modos são ativos no Raman e IR, mas medidas de polarização das bandas Raman permitem discriminar entre as geometrias planar e piramidal, pois são polarizadas as bandas correspondentes aos três modos A1 ou quatro modos A'. Stammreich et al.63 registraram o espectro Raman do correspondente derivado brometo, SOBr2. As medidas de polarização das bandas Raman confirmaram que a estrutura de SOBr2 também é piramidal, Cs.63 A determinação da estrutura da molécula P2I4 é mais complicada devido às várias conformações possíveis. A Figura 8 mostra possíveis estruturas de P2I4 com os respectivos grupos de ponto e as atividades dos modos no IR e Raman. Em um estudo por espectroscopia infravermelho, Cowley e Cohen propuseram que a estrutura de P2I4 é trans (C2h) no estado sólido e "provavelmente" gauche (C2) em solução em CS2.64 O uso combinado das espectroscopias IR e Raman por Stammreich et al.65 indicou, no entanto, a estrutura trans (C2h) para P2I4 em ambos os estados sólido e solução. Todas as vibrações esperadas para o grupo de ponto C2h foram identificadas, Ag: 316, 303, 114 e 78; Au: 327, 90 e 51; Bg: 330 e 95; Bu: 313, 109 e 65 cm-1 (valores em solução).65
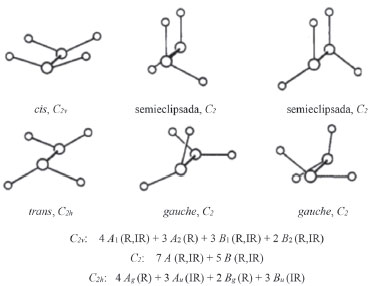 Figura 8. Estruturas possíveis para a molécula P2I4, respectivos grupos de ponto e atividades das vibrações no infravermelho e no Raman64
Diferentes estruturas também poderiam ser propostas para os complexos de iodeto de bismuto.66 No caso de (BiI4)-, se a estrutura fosse tetraedro (Td) ou quadrado planar (D4h), o espectro Raman seria relativamente simples com apenas quatro ou três bandas, respectivamente. A complexidade do espectro Raman em solução de acetona (bandas em 141, 129, 107, 80 e 53 cm-1) indicou que a geometria de (BiI4)- corresponde ao grupo de ponto C2v numa estrutura de bipirâmide trigonal, ou seja, análoga à estrutura mostrada na Figura 1, mas com uma posição equatorial ocupada pelo par isolado de elétrons. No caso de (BiI6)3-, as frequências observadas (130, 110 e 55 cm-1) são consistentes com uma estrutura de octaedro.66
OXOÂNIONS As frequências vibracionais do ânion cromato, [CrO4]2-, estavam estabelecidas pelos espectros Raman reportados por Venkateswaran.67 No entanto, Stammreich et al.53 notaram que as frequências atribuídas aos modos de dobramento de ângulo eram muito altas e, por consequência, a respectiva constante de força. Nesse sentido, Stammreich et al.53 obtiveram o espectro Raman da solução aquosa de K2CrO4 usando a radiação 587,56 nm do He para excitação do espectro. As frequências vibracionais obtidas por Venkateswaran67 e Stammreich et al.53 são comparadas na Tabela 4. Stammreich et al.53 interpretaram que o valor elevado das frequências dos modos ν2 e ν4 no artigo de Venkateswaran67 era devido ao fato de linhas de referência do espectro de comparação para as medidas de comprimento de onda terem sido identificadas como sendo linhas Raman da amostra. A partir das frequências corrigidas, Stammreich et al.53 determinaram a constante de força para o dobramento de ângulo consideravelmente menor que o valor encontrado por Venkateswaran,67 respectivamente, 0,61 e 1,00 mdina/Å. É interessante notar que Venkateswaran e Sundaram,68 em um artigo posterior, calcularam constantes de força para várias moléculas tetraédricas, inclusive [CrO4]2-, para o qual consideraram as novas frequências vibracionais corrigidas. Vários artigos do Prof. Stammreich são usados como referências de frequências vibracionais no conhecido livro do Nakamoto.69,70 Os valores citados por Nakamoto (ver tabela 2.6e, pag. 202 da Ref. [69]) para [CrO4]2- também estão na Tabela 4, mas referem-se ao artigo de Malchus e Jansen.71 As diferenças em frequências vibracionais deve-se ao fato de Malchus e Jansen71 considerarem cromato de tetrametilamônio no estado sólido. Em solução aquosa, Kiefer e Bernstein72 reportam, de fato, frequências vibracionais de [CrO4]2- consistentes com os valores obtidos por Stammreich et al.53

A atribuição dos espectros Raman e infravermelho do ânion dicromato, [Cr2O7]2-, realizada por Stammreich et al.54 considerou um modelo separando as vibrações em dois tipos. Ao visualizar a estrutura molecular como O3Cr–O–CrO3, o oxigênio central e os átomos de cromo (Cr–O–Cr) definem uma simetria local C2v, enquanto que cada extremidade (O3Cr–O') possui simetria local C3v. As frequências observadas em 220, 558 e 772 cm-1 no espectro Raman da solução aquosa foram atribuídas às vibrações da parte central, respectivamente, δ(CrOCr), νs(CrOCr) e νas(CrOCr). As frequências dos grupos das extremidades são 365, 904 e 946 cm-1, atribuídas às vibrações δ(CrO3), νs(CrO3) e νas(CrO3), respectivamente. Nesse modelo, a constante de força da ligação central, fCr–O' = 3,27 mdina/Å, é consideravelmente menor que da ligação da extremidade, fC–O = 6,07 mdina/Å.54 Por outro lado, Abbas e Davidson73 reavaliaram a frequência do modo de dobramento de ângulo δ(CrOCr), o qual foi atribuído à frequência 89 cm-1 observada no espectro Raman da solução aquosa. Cálculo recente das frequências vibracionais por método de química quântica computacional resultou em 117 cm-1 com 90% de distribuição de energia potencial no modo δ(CrOCr).74 O cálculo de química quântica confirma que a magnitude de fCr–O' é aproximadamente metade de fCr–O. Finalmente mencionamos os trabalhos sobre os derivados de halogênio de oxoânions: [CrO3Cl]- 75 e [CrO3F]-.76 As frequências observadas nos espectros Raman e infravermelho, assim como a polarização das bandas Raman, indicam que a simetria de [CrO3Cl]- corresponde ao grupo de ponto C3v. As constantes de força foram obtidas por análise de coordenadas normais considerando um campo de força de valência para o íon [CrO3Cl]-. Isso permitiu comparar as constantes de força de [CrO3Cl]- com as correspondentes determinadas nos trabalhos anteriores sobre [CrO4]2-,53 [Cr2O7]2- 54 e CrO2Cl2.52 A constante de força da ligação Cr–O aumenta na sequência de compostos [CrO4]2- < [Cr2O7]2- < [CrO3Cl]- < CrO2Cl2 e, consequentemente, aumenta também a ordem da ligação Cr–O. Os cálculos de química quântica realizados por Bell e Dines74 para diferentes oxoânions de cromo e seus derivados halogênios confirmam essa tendência, inclusive mostrando uma correlação linear entre a constante de força e o comprimento da ligação Cr–O de forma análoga à regra de Badger. No caso de [CrO3F]-,76 um ponto interessante é a identificação de uma atribuição errada feita por Dupuis77 no espectro infravermelho desse íon. Stammreich et al.76 notaram que bandas em 481 e 740 cm-1 apareciam no espectro infravermelho devido à impureza [SiF6]2- quando a amostra de KCrO3F em emulsão de nujol era preparada em contato com materiais de vidro ou cerâmica. O destaque na análise de coordenadas normais é que a distribuição de energia potencial dos modos ν2 e ν3, atribuídos ao estiramento Cr–X e ao dobramento dos ângulos O–Cr–O e O–Cr–X, respectivamente, indica que a mistura entre os modos é maior em [CrO3Cl]- do que em [CrO3F]-. Os resultados de Stammreich et al. para [CrO3Cl]- 75 e [CrO3F]- 76 foram comparados com os cálculos de frequências vibracionais e constantes de força por métodos modernos de química quântica computacional.74
AGRADECIMENTOS O autor agradece à FAPESP (2016/21070-5) e ao CNPq pelo suporte financeiro.
REFERÊNCIAS 1. Sala, O.; Kawano, Y.; Santos, P. S.; Temperini, M. L. A.; Quim. Nova 1983, 7, 320. 2. Sala, O.; Fundamentos da espectroscopia Raman e no infravermelho, 2ª ed., Editora UNESP: São Paulo, 2008, pag. 16-17. 3. Laboratório de Espectroscopia Molecular: http://lem.iq.usp.br, acessada em setembro 2019. 4. Stammreich, H.; Spectrochim. Acta 1956, 8, 41. 5. Odwyer, M. F.; J. Mol. Spec. 1958, 2, 144. 6. Pistorius, C.; Haarhoff, P. C.; J. Chem. Phys. 1959, 31, 1439. 7. Stammreich, H.; Sala, O.; Tavares, Y.; J. Chem. Phys. 1959, 30, 856. 8. Spragg, R. A.; Stammreich, H.; Kawano, Y.; J. Mol. Struct. 1970, 5, 359. 9. Griffith, W. P.; Lane, J. R.; J. Chem. Soc. - Dalton Trans. 1972, 158. 10. Stammreich, H.; Kawai, K.; Tavares, Y.; Krumholz, P.; Behmoiras, J.; Bril, S.; J. Chem. Phys. 1960, 32, 1482. 11. Friedel, R. A.; Wender, I.; Shufler, S. L.; Sternberg, H. W.; J. Am. Chem. Soc. 1955, 77, 3951. 12. Edgell, W. F.; Magee, C.; Gallup, G.; J. Am. Chem. Soc. 1956, 78, 4185. 13. Edgell, W. F.; Gallup, G.; J. Am. Chem. Soc. 1956, 78, 4188. 14. Pauling, L.; The Nature of the Chemical Bond, 3rd ed., Cornell University Press: Ithaca, 1960. 15. Cotton, F. A.; J. Am. Chem. Soc. 1958, 80, 4425. 16. Edgell, W. F.; Summitt, R.; J. Am. Chem. Soc. 1961, 83, 1772. 17. McNeill, E. A.; Scholer, F. R.; J. Am. Chem. Soc. 1977, 99, 6243. 18. Badger, R. M.; J. Chem. Phys. 1934, 2. 19. Stammreich, H.; Kawai, K.; Sala, O.; J. Chem. Phys. 1961, 35, 2168. 20. Atkins, P. W.; Overton, T. L.; Rourke, J. P.; Weller, M. T.; Armstrong, F. A.; Shriver and Atkins' Inorganic Chemistry, 5th ed., W. H. Freeman and Company: New York, 2010, p. 541. 21. Bor, G.; Marko, L.; Spectrochim. Acta 1960, 16, 1105. 22. Stammreich, H.; Kawai, K.; Krumholz, P.; Sala, O.; J. Chem. Phys. 1961, 35, 2175. 23. Cotton, F. A.; Stammreich, H.; Wilkinson, G.; J. Inorg. Nucl. Chem. 1959, 9, 3. 24. Bullitt, J. G.; Cotton, F. A.; Marks, T. J.; Inorg. Chem. 1972, 11, 671. 25. Stammreich, H.; Phys. Rev. 1950, 78, 79. 26. Barrow, R. F.; J. Chem. Phys. 1954, 22, 1775. 27. Kiefer, W.; Schrotter, H. W.; J. Chem. Phys. 1970, 53, 1612. 28. Holzer, W.; Murphy, W. F.; Bernstein, H. J.; J. Chem. Phys. 1970, 52, 399. 29. Lee, D.; Albrecht, A. C.; Adv. Infrared Raman Spectrosc. 1985, 12, 179. 30. Ziegler, L. D.; Acc. Chem. Res. 1994, 27, 1. 31. Stammreich, H.; Forneris, R.; J. Chem. Phys. 1954, 22, 1624. 32. Branigan, E. T.; van Staveren, M. N.; Apkarian, V. A.; J. Chem. Phys. 2010, 132, 044503. 33. Herzberg, G.; Molecular Spectra and Molecular Structure. I. Spectra of diatomic molecules, 2nd ed., D. van Nostrand Company: Princeton, 1950, p. 512. 34. Stammreich, H.; Forneris, R.; J. Chem. Phys. 1953, 21, 944. 35. Stammreich, H.; Forneris, R.; Spectrochim. Acta 1961, 17, 775. 36. Herzberg, G.; Molecular Spectra and Molecular Structure. I. Spectra of diatomic molecules, 2nd ed., D. van Nostrand Company: Princeton, 1950, p. 519. 37. McQuarrie, D. A.; Simon, J. D.; Physical Chemistry - A Molecular Approach, University Science Books: Sausalito, 1997, p. 539. 38. http://webbook.nist.gov/chemistry/, acessada em setembro 2019. 39. Stammreich, H.; Forneris, R.; Tavares, Y.; Spectrochim. Acta 1961, 17, 1173. 40. Stammreich, H.; Forneris, R.; Spectrochim. Acta 1960, 16, 363. 41. Pauling, L.; The Nature of the Chemical Bond, 2a ed., Cornell University Press: Ithaca, 1944. 42. Yamasaki, R. S.; Cornwell, C. D.; J. Chem. Phys. 1959, 30, 1265. 43. Cornwell, C. D.; Yamasaki, R. S.; J. Chem. Phys. 1957, 27, 1060. 44. Person, W. B.; Fordemwalt, J. N.; Stammreich, H.; Forneris, R.; Anderson, G. R.; J. Chem. Phys. 1961, 35, 908. 45. Herzberg, G.; Infrared and Raman spectra of poliatomic molecules, D. van Nostrand Company: Princeton, 1945, p. 187. 46. Bell, R. P.; Longuet-Higgins, H. C.; Proc. R. Soc. London, Ser. A 1945, 183, 357. 47. Stammreich, H.; Kawano, Y.; Spectrochim. Acta, Part A 1968, A 24, 899. 48. Boswijk, K. H.; Wiebenga, E. H.; Acta Crystallogr. 1954, 7, 417. 49. Palmer, K. J.; J. Am. Chem. Soc. 1938, 60, 2360. 50. Stammreich, H.; Forneris, R.; Sone, K.; J. Chem. Phys. 1955, 23, 972. 51. Hobbs, W. E.; J. Chem. Phys. 1958, 28, 1220. 52. Stammreich, H.; Kawai, K.; Tavares, Y.; Spectrochim. Acta 1959, 15, 438. 53. Stammreich, H.; Bassi, D.; Sala, O.; Spectrochim. Acta 1958, 12, 403. 54. Stammreich, H.; Bassi, D.; Sala, O.; Siebert, H.; Spectrochim. Acta 1958, 13, 192. 55. Howard, J. B.; Wilson, E. B.; J. Chem. Phys. 1934, 2, 630. 56. Stammreich, H.; Forneris, R.; Tavares, Y.; J. Chem. Phys. 1956, 25, 580. 57. Stammreich, H.; Tavares, Y.; Bassi, D.; Spectrochim. Acta 1961, 17, 661. 58. Stammreich, H.; Forneris, R.; Spectrochim. Acta 1956, 8, 52. 59. Simanouti, T.; J. Chem. Phys. 1949, 17, 245. 60. Hexter, R. M.; Cheung, H.; J. Chem. Phys. 1956, 24, 1186. 61. McDowell, C. A.; Trans. Faraday Soc. 1953, 49, 371. 62. Martz, D. E.; Lagemann, R. T.; J. Chem. Phys. 1954, 22, 1193. 63. Stammreich, H.; Forneris, R.; Tavares, Y.; J. Chem. Phys. 1956, 25, 1277. 64. Cowley, A. H.; Cohen, S. T.; Inorg. Chem. 1965, 4, 1200. 65. Frankiss, S. G.; Miller, F. A.; Stammreich, H.; Sans, T. T.; Spectrochim. Acta, Part A 1967, A 23, 543. 66. Spragg, R. A.; Stammreich, H.; Kawano, Y.; J. Mol. Struct. 1969, 3, 305. 67. Venkateswaran, C. S.; Proc. Indian Acad. Sci. 1938, 7, 144. 68. Venkateswarlu, K.; Sundaram, S.; J. Chem. Phys. 1955, 23, 2365. 69. Nakamoto, K.; Infrared and Raman Spectra of Inorganic and Coordination Compounds, Part A: Theory and Applications in Inorg. Chem., 6th ed., John Wiley & Sons: Hoboken, 2009. 70. Nakamoto, K.; Infrared and Raman Spectra of Inorganic and Coordination Compounds, Part B: Applications in Coordination, Organometallic, and BioInorg. Chem., 6th ed., John Wiley & Sons: Hoboken, 2009. 71. Malchus, M.; Jansen, M.; Z. Naturforsch., B 1998, 53, 704. 72. Kiefer, W.; Bernstein, H. J.; Mol. Phys. 1972, 23, 835. 73. Abbas, M. H.; Davidson, G.; Spectrochim. Acta, Part A 1994, 50, 1153. 74. Bell, S.; Dines, T. J.; J. Phys. Chem. A 2000, 104, 11403. 75. Stammreich, H.; Sala, O.; Kawai, K.; Spectrochim. Acta 1961, 17, 226. 76. Stammreich, H.; Sala, O.; Bassi, D.; Spectrochim. Acta 1963, 19, 593. 77. Dupuis, T.; C. R. Acad. Sci. 1958, 246, 3332.
FAPESP contribuiu para custear a publicação deste artigo |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access