
 |
 |
 |
 |
 |
Educação
| One-pot diastereoselective synthesis of chiral tricyclic L-cysteine and D-penicillamine derivatives: a laboratory experiment |
|
Maria I. L. Soares; Susana M. M. Lopes; Dina Murtinho; Teresa M. V. D. Pinho e Melo*
Departamento de Química, Centro de Química de Coimbra, Universidade de Coimbra, 3004-535 Coimbra, Portugal Recebido em 10/07/2019 *e-mail: tmelo@ci.uc.pt A one-pot diastereoselective synthesis of thiazolidine-ring fused systems derived from enantiomerically pure amino acids, L-cysteine or D-penicillamine, and achiral succindialdehyde is described as an experiment to be carried out by upper-division undergraduate students in a laboratory classroom. Reactions were performed under mild conditions, the products were isolated through simple experimental procedures and fully characterized. This study combines organic synthesis, determination of the purity of compounds (TLC analysis and melting point measurements), optical activity measurements as well as structural analysis (interpretation of 1D NMR and 2D NMR spectra). It offers a platform for the discussion of important organic chemistry concepts such as diastereoselectivity, kinetic control vs thermodynamic control and cyclization reactions via nucleophilic addition to imines/iminium cations. INTRODUCTION The total synthesis of relevant natural products, such as penicillin1 and biotin,2 involves as a first step the construction of the 1,3-thiazolidine ring via condensation of sulfur-containing amino acids, namely D-penicillamine or L-cysteine, with aldehydes. Thiazolidines (1,3-thiazolidines) are five membered heterocyclic compounds containing one sulfur and one nitrogen atom (e.g. 1). In this straightforward strategy to thiazolidines derived from natural enantiomerically pure amino acids (chiral pool), a new asymmetric centre is created while the chiral centre derived from the amino acid is retained. Condensation of D-penicillamine or L-cysteine with achiral aldehydes is not diastereoselective, affording mixtures of cis and trans diastereoisomers (Scheme 1).3-5
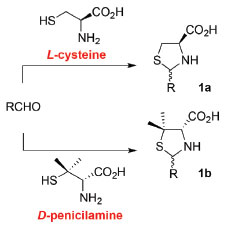 Scheme 1. Synthesis of 2-substituted-1,3-thiazolidine-4-carboxylic acids 1 derived from L-cysteine and D-penicillamine
1,3-Thiazolidine-4-carboxylic acids are useful building blocks in organic synthesis. The most interesting feature of these heterocycles is the possibility of carrying out diastereoselective reactions via a ring opening-ring closure mechanism. This chemical behavior has been explored for the synthesis of chiral thiazolidine-ring fused systems, as illustrated by the selective synthesis of thiazolo[2,3-b]isoindole derivative 2,6,7 in which the creation of a new asymmetric centre is involved (Scheme 2).
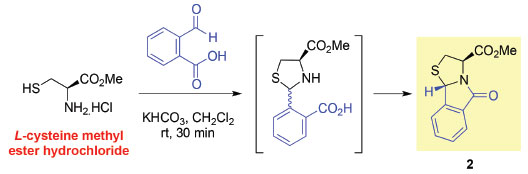 Scheme 2. Stereoselective synthesis of chiral tetrahydro-thiazolo[2,3-b]isoindole 2 derived from l-cysteine methyl ester
Using dialdehyde 4, it is possible to obtain tricyclic compounds in which two new asymmetric centres are created. Hexahydro-pyrrolo[1',2',5':3,4,5]thiazolo[3,4-c]oxazol-1-one 5 was obtained as a single stereoisomer via the condensation of L-cysteine with succindialdehyde 4 (generated in situ by treatment of dimethoxytetrahydrofuran 3 with HCl aqueous solution) (Figure 1a).8 The structure and stereochemistry of the chiral centres C2a, C4a and C6a of tricyclic L-cysteine derivative 5 was confirmed by X-ray crystallography studies (Figure 1b).8
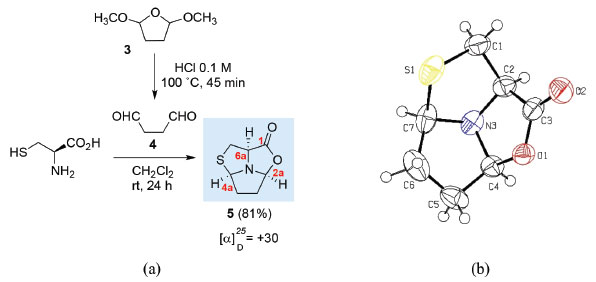 Figure 1. a) Synthesis of (2aS,4aR,6aR)-hexahydro-pyrrolo[1',2',5':3,4,5]thiazolo[3,4-c]oxazol-1-one 5 from l-cysteine; b) X-Ray crystal structure of compound 5
Györgydeák et al. have demonstrated that the acylation of the mixture of diastereoisomers (2R,4R)- and (2S,4R)-2-aryl-1,3-thiazolidine-4-carboxylic acid derivatives with acetic anhydride can lead to the selective synthesis of N-acylthiazolidines as pure stereoisomers (Scheme 3). In fact, 2-substituted-1,3-thiazolidine-4-carboxylic acid derivatives can undergo selective inversion of configuration at C-2 through a ring opening-ring closure mechanism. However, the N-protection with an acyl group prevents the epimerization process and allows the isolation of pure diastereoisomers.3,9
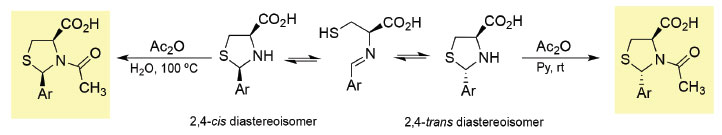 Scheme 3. Diastereoselective N-acylation of 1,3-thiazolidine-4-carboxylic acids
This is an example of a reaction where thermodynamic control and kinetic control lead to different outcomes. In fact, the stereochemistry outcome of the acylation reaction depends on the reaction conditions used. Carrying out the reaction using milder conditions (acetic anhydride/pyridine at 25 ºC), the 2,4-trans diastereoisomers are obtained whereas using acetic anhydride/water at 100 ºC the 2,4-cis isomers are isolated. These results indicate that the latter are the products of the thermodynamic control (the most stable products) and the 2,4-trans diastereoisomers are products of the kinetic control. The mechanism of the tandem ring closure reactions leading to tricyclic L-cysteine derivative 5 can be rationalized considering the initial synthesis of thiazolidine 7 as a mixture of (2R,4R) or (2S,4R) diastereoisomers followed by stereoselective ring closure to generate the iminium cation 8 (Scheme 4). The observed stereoselectivity can also be rationalized considering the ring-chain tautomeric equilibria of 1,3-thiazolidines and the corresponding Schiff base (imine 6), followed by selective intramolecular iminium cation formation. Subsequent diastereoselective 5-endo-trig cyclization of intermediate 8 gives the chiral tricyclic L-cysteine derivative 5.
 Scheme 4. Proposed mechanism for the synthesis of tricyclic compound 5
The same one-pot protocol can be applied to carry out the reaction of D-penicillamine with succindialdehyde 4, allowing the stereoselective synthesis of tricyclic compound 9 as single stereoisomer with (2aR,4aS,6aS) stereochemistry (Scheme 5).8 It is worth noticing that the stereochemistry of the newly created chiral centres (C-2a and C-4a) and the chiral centre C-6a derived from D-penicillamine is opposite to that observed for L-cysteine derived compound 5.
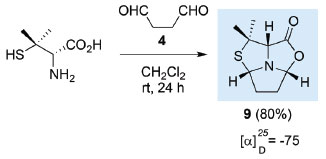 Scheme 5. Synthesis of (2aR,4aS,6aS)-6,6-dimethyl-hexahydro-pyrrolo[ 1',2',5':3,4,5]thiazolo[3,4-c]oxazol-1-one 9 from D-penicillamine
The purpose of this work is to carry out the diastereoselective synthesis of two tricyclic thiazolidine-ring fused systems, starting from L-cysteine or D-penicillamine and a succindialdehyde precursor, through a one-pot procedure.
EXPERIMENT OVERVIEW In the classroom environment, upper-division undergraduate students may work either individually or in groups of two during two lab periods of 3 hours. Each group runs one experiment, either starting with L-cysteine or D-penicillamine to obtain tricyclic compound 5 or 9, respectively (Figure 1 and Scheme 5). The detailed procedure is provided in the Supplementary Material. First Session The first session involves the in situ generation of succindialdehyde 4 and subsequent reaction with L-cysteine or D-penicillamine.8 Treatment of commercially available dimethoxytetrahydrofuran with 0.1 mol L-1 HCl aqueous solution (prepared by the students) at 100 ºC over a period of 45 min affords the target succindialdehyde 4. After cooling to room temperature, the reaction mixture is diluted with dichloromethane and the appropriate amino acid (L-cysteine or D-penicillamine) is added. The reaction is stirred 24 h at room temperature. Stirring should be vigorous since the reaction of the unprotected succindialdehyde with the amino acids is carried out using a biphasic solvent system. Second Session This session involves the isolation, determination of the purity and characterization of the tricyclic compounds. The biphasic mixture is diluted with water and the organic phase is separated. The aqueous phase is extracted with dichloromethane and the combined organic extracts washed with brine and dried. After removal of the solvent the tricyclic compound 5 is obtained as a white solid. However, compound 9 is initially obtained as an oil but becomes a white solid upon addition of diethyl ether. The yields are calculated and the purity of the products evaluated by thin layer chromatography (TLC) and by optical activity measurements. Samples are prepared to record IR and NMR spectra and to measure melting points (optional).
RESULTS AND DISCUSSION The synthesis of tricyclic L-cysteine and D-penicillamine derivatives8 was reproduced individually by twelve BSc students and three second-year masters students during two laboratory sessions (3 h/period). The students obtained the tricyclic compound 5 with yields ranging from 47 to 66% and compound 9 with yields ranging from 61 to 72%. The purity of the products was assessed by TLC analysis, by determining the optical rotation of both compounds and by 1H NMR analysis. The values of the optical rotation obtained by the students ranged from +22.1 to +23.7 for compound 5 (+30 is the optical rotation value of the pure compound)8 and from -66.7 to -71.4 for compound 9 (-75 is the optical rotation value of the pure compound).8 The characterization should be completed with the IR spectra and melting points measurement of both compounds. Additional 13C NMR and bidimensional COSY spectra, may also be recorded. The detailed experimental procedure and characterization data for tricyclic compounds 5 and 9 are available in Supplementary Material. Compound 5 was obtained as a white solid in pure form after final work-up. Additionally, it can be recrystallized from ethyl acetate/hexane (it is worth noting that this procedure may require a third laboratory session). Compound 9 was obtained as an oil in pure form after final work-up and its purity was determined by students in this form. Initially, the purity of the products was assessed by TLC developed in ethyl acetate/hexane (1:1) and staining with iodine. The TLCs performed by the students did not show any visible impurity. The 1H NMR spectra confirmed the purity and allowed identification of the products by comparison with the previously reported data.8 Analysis of the 1H and 13C NMR spectra allows the consolidation of concepts such as chemical shift and multiplicity. For example, in the 1H spectrum of compound 5 it is possible to observe an ABX system for the thiazolidine protons H-6 (3.43 ppm, dd, J = 12.2 and 6.1 Hz and 3.53 ppm, pseudo-d, J = 12.2 Hz) and proton H-6a (4.10 ppm, pseudo-d, J = 6.1 Hz) (Figure 2). In the case of compound 9, proton H-6a appears as a singlet (3.59 ppm) due to the presence of two methyl groups at C-6 (see Supplementary Material, Figure 8S). Analysis of the COSY spectrum of compound 5 allows the assignment of protons H-3 and H-4 (Figure 3). Cross-peaks were observed between protons H-4 and H-4a, as well as between protons H-4 and H-3 protons. On the other hand, correlation between H-3 and the more shifted signal, assigned to H-2a, could also be observed.
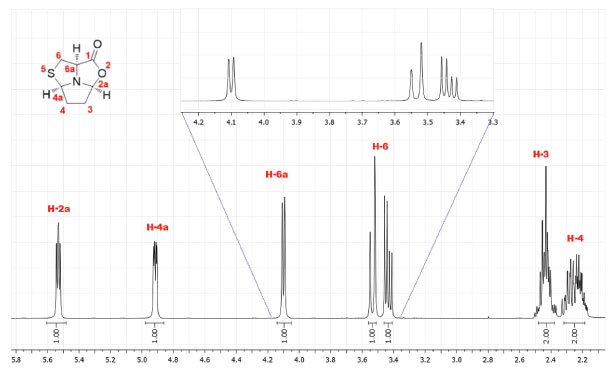 Figure 2. 1H NMR spectrum of compound 5 (400 MHz, CDCl3)
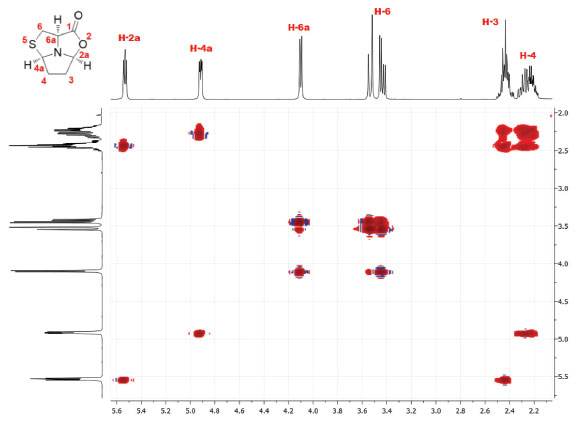 Figure 3. COSY spectrum of compound 5 (400 MHz, CDCl3)
Both tricyclic compounds 5 and 9 are optically active, having three chiral carbons. Thus, the optical rotation of compounds 5 and 9 was measured using solutions of 20 mg in 2 ml of dichloromethane (c 1). The calculated specific rotation for L-cysteine tricyclic derivative 5 was +30 and for D-penicillamine tricyclic derivative 9 was -75. CONCLUSIONS A synthetic methodology for the direct and stereoselective synthesis of chiral tricyclic compounds derived from amino acids (L-cysteine and D-penicillamine) and an achiral dialdehyde was implemented for educational purposes. This straightforward experiment, carried out under very mild conditions and using chiral natural compounds as starting materials (chiral pool), provides some student learning objectives, including the concept of one-pot and stereoselective heterocyclic synthesis. Additionally, this experiment gives a good background in concepts such as imine/iminium cation formation, kinetic control vs thermodynamic control and cyclization via nucleophilic addition to imines/iminium cations.
SUPPLEMENTARY MATERIAL Detailed experimental procedures, detailed spectroscopic data, optical activity measurements, IR and 1H and 13C NMR spectra and hints for students' discussion are available free of charge at http://quimicanova.sbq.org.br, in PDF format.
ACKNOWLEDGEMENTS The Coimbra Chemistry Centre (CQC) is supported by the Portuguese Agency for Scientific Research, "Fundação para a Ciência e a Tecnologia" (FCT), through Project UID/QUI/00313/2019. We acknowledge the UC-NMR facility for obtaining the NMR data (www.nmrccc.uc.pt).
REFERENCES 1. Sheehan, J. C.; Henerylogan, K. R.; J. Am. Chem. Soc. 1957, 79, 1262; Sheehan, J. C.; Henerylogan, K. R.; J. Am. Chem. Soc. 1959, 81, 3089. 2. Confalone, P. N.; Pizzolato, G.; Baggiolini, E. G.; Lollar, D.; Uskokovic, M. R.; J. Am. Chem. Soc. 1977, 99, 7020. 3. Szilágyi, L.; Györgydeák, Z.; J. Am. Chem. Soc. 1979, 101, 427. 4. Gilchrist, T. L.; Rocha Gonsalves, A. M. D.; Pinho e Melo, T. M. V. D.; Tetrahedron 1994, 50, 13709. 5. Santos, K.; Laranjo, M.; Abrantes, A. M.; Brito, A. F.; Gonsalves, C.; Ribeiro, A. B. S.; Botelho, M. F.; Soares, M. I. L.; Oliveira, A. S. R.; Pinho e Melo, T. M. V. D.; Eur. J. Med. Chem. 2014, 79, 273. 6. Pinho e Melo, T. M. V. D.; Santos, C. I. A.; Rocha Gonsalves, A. M. D.; Paixão, J. A.; Beja, A. M.; Tetrahedron 2004, 60, 3949. 7. Soares, M. I. L.; Cardoso, A. L.; Lopes, S. M. M.; Pinho e Melo, T. M. V. D. In Comprehensive Organic Chemistry Experiments for the Laboratory Classroom; Afonso, C. A. M., Candeias, N. R., Simão, D. P., Trindade, A. F., Coelho, J. A. S., Tan, B., Franzén, R., eds.; The Royal Society of Chemistry: London, 2017, Chap. 3.1.18, pp. 194-197. 8. Pinho e Melo, T. M. V. D.; Lopes, S. M. M.; Rocha Gonsalves, A. M. D.; Kaczor, A.; Fausto, R.; Paixão, J. A.; Beja, A. M.; Silva, M. R.; Lett. Org. Chem. 2006, 3, 820. 9. Györgydeák, Z.; Kajtár-Peredy, M.; Kajtár, J.; Kajtár, M.; Liebigs Ann. Chem. 1987, 756, 927; Benedini, F.; Ferrario, F.; Sala, A.; Sala, L.; Soresinetti, P. A.; J. Heterocycl. Chem. 1994, 31, 1343. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access