
 |
 |
 |
 |
 |
Artigo
|
|
| Desenvolvimento de metodologias analíticas para determinação de clorpirifós em amostras aquosas empregando a microextração líquido-líquido dispersiva seguida por HPLC-UV Development of analytical methodologies for the determination of chlorpyryphes in aqueous samples employing the dispersive liquid microextraction followed by HPLC-UV |
|
Adão S. Oliveira; Leandro Honório; Mayara L. de Matos; João Batista G. de Souza*
Instituto de Química, Universidade Federal de Mato Grosso do Sul, 79074-460, Campo Grande - MS, Brasil Recebido em 15/05/2019 *e-mail: jb.souza@hotmail.com The miniaturized extraction techniques use less aggressive methods to the environment, since they generate less waste and spend a minimum of organic solvents, besides having a good selectivity and specificity in the extractions. In this context, liquid-liquid dispersive microextraction (DLLME) has been highlighting. Thus, the present article describes an optimization of the DLLME using low environmental impact extractor solvent to determine the chlorpyrifos insecticide in water. The analytical curves presented values for the correlation coefficient (r) higher than 0.999. The limits of detection and quantification were 0.24 and 0.80 µg L-1, respectively. There were recoveries between 93 and 97 %, with relative standard deviation (RSD) varying between 1.00 and 3.61%. The developed methods presented accurate and reliable results, are simple and safe, characteristics important for the implementation of a methodology as a routine analysis, and were applied in the determination of chlorpyrifos in water samples from five points along the Rio Dourados, near the city of Fátima do Sul. INTRODUÇÃO Os agrotóxicos (inseticidas, herbicidas, fungicidas, etc.) apresentam uma grande variedade química de princípios ativos utilizados em sua composição, sendo esses classificados como carbamatos, piretróides, organoclorados, organofosforados, dinitroanilinas, anilinas, entre outros.1 Desses, os inseticidas se mostram como sendo o grupo mais representativo dos poluentes ambientais, pois possuem uso intensificado na agricultura devido ao número crescente de pragas.2 Assim, a exposição humana aos inseticidas decorrem da ingestão, inalação ou absorção cutânea, sendo essas últimas vias de intoxicação mais comuns entre os trabalhadores responsáveis por sua aplicação no campo, manufatura ou formulação desses produtos na indústria química.3 Em relação aos inseticidas organofosforados, grupo ao qual pertence o clorpirifós, esses são ésteres do ácido fosfórico ou de ácidos derivados desse e pertencem à classe dos inibidores das enzimas colinesterases.4 O mecanismo de ação dessas substâncias ocorre por meio da fosforilação do sítio ativo dessas enzimas.5 Os efeitos na saúde humana devido à exposição ao clorpirifós em níveis baixos (miligramas) pode gerar tonturas, fadigas, náuseas e alterações na frequência cardíaca; em níveis altos (gramas) pode ocasionar paralisia, convulsões, perda de consciência e morte; em exposições a longo prazo (exposição crônica) pode ocorrer alteração comportamental, efeitos no sistema nervoso e desenvolvimento da Doença de Parkinson.6-8 O clorpirifós é um dos princípios ativos mais comercializados no Brasil, tendo registro de venda superior a 6.000 toneladas, no ano de 2017.9 É aplicado principalmente nas culturas do milho, da soja e do algodão, no combate a diversas infestações de insetos, tais como a broca e pulgão do algodoeiro, lagarta de cartucho e lagarta da soja. Posteriormente à aplicação dos agrotóxicos nas lavouras, de modo geral, uma parcela considerável atinge rios, lagos, aquíferos e oceanos, nos quais o processo de lixiviação e o escoamento superficial são tidos como os responsáveis pela contaminação das águas subterrâneas e superficiais, respectivamente.10 No processo de lixiviação, os agrotóxicos são carreados em solução, simultaneamente com a água que abastece os mananciais subterrâneos. Já a contaminação superficial ocorre em função da adsorção dos agrotóxicos com partículas do solo ou em solução, nesse processo ocorre a exposição dos organismos aquáticos em níveis que podem ser tóxicos para várias espécies.11 Alguns artigos publicados recentemente vêm apontando para a presença de clorpirifós em águas fluviais com lavouras próximas, em concentrações variadas, tais como 38,94 µg L-1; 1,01 µg L-1 e 2,86 ng L-1.12-14 Assim, o risco da contaminação de mananciais destinados ao abastecimento de cidades é preocupante, pois, assim como diversas outras substâncias tóxicas, os agrotóxicos não são removidos adequadamente em um sistema de tratamento de água convencional, exigindo processos adicionais mais complexos para que ocorra sua remoção.15 Dentre as técnicas utilizadas para determinar clorpirifós, assim como outras classes de agrotóxicos, nas mais diversas matrizes, as mais utilizadas são a cromatografia a gás e a líquido. Em relação aos métodos de extração, tem se destacado a microextração em fase líquida (LPME),16 uma vez que apresentam alta seletividade e especificidade na extração, geram baixos volumes de resíduos orgânicos e possuem a possibilidade do uso de solventes de baixo impacto ambiental.17 Uma das técnicas de LPME amplamente utilizada é a Microextração Líquido-Líquido Dispersiva (DLLME), desenvolvida por Rezaee et al.18 A técnica consiste no equilíbrio de distribuição do analito entre as fases doadora (amostra) e receptora (solvente orgânico), sendo indicada para extração de compostos com propriedades lipofílicas moderadas a altas, podendo ser estendida para compostos que possam ter seu coeficiente de distribuição alterado pelo controle de pH.19 Uma transferência parcial de analitos caracteriza a DLLME como uma técnica de equilíbrio,18 em que a quantidade de analito transferida para a fase aceptora ao final da extração é expressa em porcentagem. O tempo de extração é curto, pois o estado de equilíbrio é alcançado rapidamente, visto que as gotas finamente divididas do solvente extrator proporcionam uma grande área de contato superficial entre o solvente extrator e a fase aquosa.20 A técnica de DLLME pode ser executada em duas etapas: a) Injeção de uma mistura adequada de solventes dispersor e extrator ou somente o solvente extrator em uma amostra aquosa contendo os analitos; b) Centrifugação da solução turva para separação das fases, seguida da transferência da fase separada para um microvial, permitindo assim a determinação dos analitos. Na etapa (a) o solvente extrator é disperso na amostra aquosa na forma de microgotas (emulsão de gotículas), realizando a extração. A dispersão pode ser favorecida pelo uso de solvente dispersor que é solúvel na amostra aquosa e no solvente extrator, pelo aquecimento da amostra ou uso de agentes surfactantes.21 A Figura 1 ilustra as diferentes etapas da DLLME.
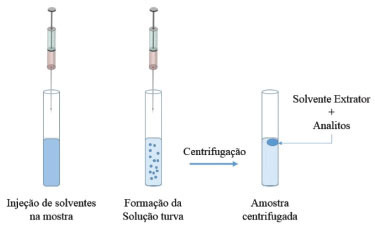 Figura 1. Diferentes etapas da DLLME
Diversos fatores, tais como os solventes de extração e dispersão e seus respectivos volumes, aquecimento da amostra, força iônica, pH e tempo de centrifugação, influenciam na eficiência de extração da DLLME, sendo necessário otimizá-los.22 Em relação ao desenvolvimento de metodologias de análise, pesquisadores têm se dedicado à elaboração de métodos de extração que façam uso de solventes como os líquidos iônicos e álcoois de cadeia longa, que além de menos prejudiciais ao meio ambiente, são altamente eficientes no processo de extração de diversos analitos orgânicos.12,22 No estado de Mato Grosso do Sul (Brasil), boa parte da produção agrícola dos municípios pertencentes à região da Grande Dourados (Caarapó, Douradina, Fátima do Sul, Jateí, Itaporã, Maracaju, Rio Brilhante e Vicentina) ocorre em regiões próximas a Bacia do Rio Dourados.23 A região produziu em 2017 mais de 2,5 milhões de toneladas de soja e mais de 3,7 milhões de tonelada de milho, sendo que o principal agrotóxico aplicado a essas culturas é o clorpirifós. O estado de Mato Grosso do Sul neste mesmo período consumiu 309 toneladas dessa substância.24 Esse elevado consumo desta substância gera motivos de preocupação, pois os resíduos dos agrotóxicos aplicados nas lavouras podem ser carreados para o leito do rio e assim desenvolver seus efeitos tóxicos tanto para as espécies aquáticas, quanto para seres humanos que utilizam tais águas para consumo, como por exemplo, a cidade de Dourados, onde 50% da população é abastecida com água coletada diretamente no Rio Dourados.25 Com base no exposto, o objetivo deste trabalho foi o desenvolvimento de metodologias analíticas simples, robustas e rápidas utilizando a microextração líquido-líquido dispersiva e a cromatografia líquida de alta eficiência com detector ultravioleta (UV) para detecção e quantificação de resíduos do inseticida clorpirifós em águas do Rio Dourados - MS.
PARTE EXPERIMENTAL Materiais, reagentes e condições cromatográficas Os reagentes utilizados neste estudo foram: 1-Dodecanol (98,00% de pureza), Sigma Aldrich; 1-Octanol (99% de pureza), Sigma Aldrich; Acetona (grau UV-HPLC-Espectroscópico), Vetec - Química Fina; Acetonitrila (grau HPLC), J. T. Baker; Ácido Clorídrico (37% PA), Vetec; Água Ultrapura; Cloreto de Sódio (99,00% de pureza), Impex; Hidróxido de Sódio (98% de pureza), Vetec; Metanol (grau HPLC), J.T. Baker; Padrão Clorpirifós (99,80% de pureza), Sigma Aldrich. As soluções padrão de clorpirifós foram preparadas por diluições, conforme a necessidade experimental, em acetonitrila. Brancos foram realizados regularmente para a verificação do analito nos reagentes utilizados. Para o processo de extração, os principais materiais utilizados foram: Agitador Magnético (Fisatom 751); Agitador Vortex (Fisatom 772) com velocidade fixa em 2800 rpm; Centrífuga (Sigma 4K15); Filtro para seringa (PVDF 0,45 µm); Medidor de pH de bancada (HANNA HI221); Purificador de água (GEHAKA OS 50LX TQ); Seringa Hipodérmica (50 mL). Utilizou-se para análise cromatográfica um Cromatógrafo a líquido Varian ProStar com detector UV modelo 320 - injetor manual (20 µL). Fase móvel de acetonitrila/água, utilizando o método isocrático 75/25 (v:v), com tempo total de análise de 10 minutos numa vazão de 1 mL min-1 e comprimento de onda ajustado a 230 nm. Coluna Zorbax ODS 4.6 x 150 mm x 5 µm (Agilent). Procedimentos de extração As metodologias utilizadas foram definidas nas seguintes condições: Método de extração 1: O método 1 de DLLME proposto baseou-se na adição de uma mistura de 300 µL acetonitrila/70 µL de 1-dodecanol em um volume de 5 mL de amostra aquosa com 1% de NaCl. Após, realizou-se a agitação no vortex, com velocidade fixa em 2800 rpm, durante 15 segundos seguido da etapa de centrifugação, 3000 rpm por 3 minutos. Ao final, o volume de solvente extrator suspenso na parte superior da amostra foi recolhido com auxílio de uma microsseringa, avolumado a 200 µL com metanol, filtrado com auxílio de um filtro seringa PVDF (0,45 µm) e injetado, em triplicata, no sistema cromatográfico. O método 2 de DLLME proposto baseou-se na adição de 70 µL de 1-dodecanol em um volume de 5 mL de amostra aquosa com 1% de NaCl, seguido do aquecimento da amostra a 30 °C, em Banho Maria, durante 5 minutos e submissão da amostra a uma agitação manual por 10 s. Posteriormente, realizou-se a etapa de centrifugação, 3000 rpm por 3 minutos. Ao final, o volume de solvente extrator suspenso na parte superior da amostra foi recolhido com auxílio de uma microsseringa, avolumado a 200 µL com metanol, filtrado com auxílio de um filtro seringa PVDF (0,45 µm) e injetado, em triplicata, no sistema cromatográfico. Ambos os métodos estão demonstrados na Figura 2.
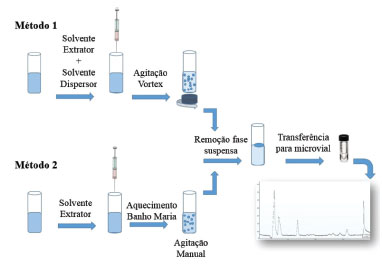 Figura 2. Procedimentos de DLLME empregados na extração do clorpirifós
Avaliação dos parâmetros otimizados na microextração líquido-líquido dispersiva A avaliação foi realizada para determinar e otimizar os parâmetros envolvidos na extração, sendo eles o tipo e volume de solvente extrator e dispersor, tempo e temperatura de aquecimento, agitação no vortex, velocidade e tempo de centrifugação, força iônica e pH. Validação dos métodos de extração Um método analítico deve gerar resultados confiáveis e adequados a finalidade no qual se destina,26 sendo, portanto, submetido a uma validação, cujos critérios são regidos por órgãos como a ANVISA (Agência Nacional de Vigilância Sanitária) e INMETRO (Instituto Nacional de Metrologia, Normalização e Qualidade Industrial). A validação dos métodos de DLLME, para extração de clorpirifós, utilizando o HPLC/UV, foi realizada seguindo os procedimentos otimizados. As soluções utilizadas nos procedimentos foram preparadas em acetonitrila/água ultrapura e as extrações realizadas em amostras de água coletadas no Rio Dourados. Os seguintes parâmetros foram avaliados: seletividade, curva analítica e faixa linear, efeito matriz, limites de detecção e quantificação, exatidão (ensaio de recuperação) e precisão (ensaio de repetitividade intra-dia e inter-dia).
RESULTADOS E DISCUSSÃO Otimização dos parâmetros de extração Escolha do solvente extrator e dispersor O clorpirifos possui uma solubilidade em água na ordem de 1,39 mg L-1 e uma constante de partição octanol/água (Log P) de 4,9, esses valores indicam que a solubilidade do clorpirifós é baixa em meio aquoso, tendo uma maior afinidade com o meio orgânico. Para escolha do solvente extrator, foram realizados testes com o 1-dodecanol e 1-octanol. Os testes com ambos os solventes apresentaram uma boa separação das fases, havendo a formação da gota de solvente extrator no limite superior da amostra ao final das etapas de extração. Entretanto, o 1-octanol não promoveu uma extração efetiva do clorpirifós, diferentemente do 1-dodecanol, que durante os testes iniciais propiciou uma extração efetiva do clorpirifós, livre de interferentes e, portanto, escolhido como solvente extrator. Esse resultado está de acordo com o valor de Log P: o 1-dodecanol possui uma cadeia carbônica maior que o 1-octanol, o que lhe proporciona uma característica apolar maior, sendo mais eficiente na extração do clorpirifós. Em seguida, avaliou-se sua dispersão perante os solventes acetona, acetonitrila e metanol. Um sistema de duas fases e boas recuperações ocorreram com todos os solventes estudados. Entretanto, a acetonitrila, por apresentar um menor valor de desvio padrão, bem como ser compatível com a fase móvel, foi escolhida como solvente dispersor para as análises subsequentes. Os resultados estão apresentados na Figura 3.
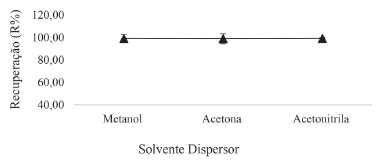 Figura 3. Efeito do tipo de solvente dispersor na extração do clorpirifós. Condição da extração: 5 mL de amostra; 100 µL de solvente extrator/300 µL de solvente dispersor; 1% NaCl; 1 min vortex; centrifugação: 3000 rpm/2 minutos
Escolha do volume de solvente dispersor e extrator O volume de solvente dispersor impacta na formação da solução turva constituída por microgotas de água/solvente orgânico, bem como relaciona-se com o grau de dispersão do solvente extrator no meio aquoso e consequentemente, influencia na eficiência da extração.27 Neste sentido, selecionaram-se, para testes, os volumes de 50; 100; 200; 300 e 400 µL de acetonitrila. Os resultados estão apresentados na Figura 4.
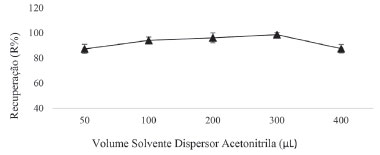 Figura 4. Otimização do volume de solvente dispersor. Condições da extração: 5 mL de amostra; 100 µL de solvente extrator; 1% de NaCl; 1 min vortex; centrifugação: 3000 rpm/2 minutos
O solvente extrator não foi disperso adequadamente em volumes mais baixos de solvente dispersor e isso se deve ao fato de ocorrer uma diminuição na emulsificação no meio o que pode levar a uma diminuição do fator de enriquecimento. Entretanto, a solubilidade do solvente extrator, na solução aquosa, pode ser aumentada para volumes maiores de solvente dispersor, diminuindo o volume final da gota, portanto, em ambos os casos, influenciando na eficiência da extração.28 O volume de solvente dispersor que apresentou melhor eficiência na extração foi de 300 µL de acetonitrila. O volume de solvente extrator também é determinante na quantidade de emulsão formada, afetando a razão do volume de solvente extrator/dispersor, influenciando diretamente na eficiência da extração.29 Desse modo, foram testados diferentes volumes de solvente extrator 1-dodecanol, sendo eles 30; 50; 70; 100 e 150 µL. Os resultados estão apresentados na Figura 5.
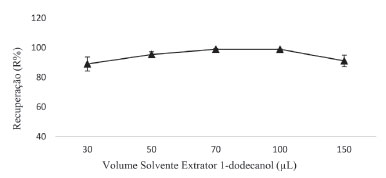 Figura 5. Otimização do volume de solvente extrator. Condições da extração: 5 mL de amostra; 300 µL de solvente dispersor; 1% de NaCl; 1 min vortex; centrifugação: 3000 rpm/2 minutos
Verificou-se que para volumes mais elevados de solvente extrator, houve redução na razão solvente extrator/dispersor, influenciando sua dispersão no meio aquoso. Em relação aos volumes menores de solvente extrator, observaram-se variações no volume final da gota formada, fato esse relacionado à solubilização do solvente extrator na amostra e consequentemente, em ambos os casos, limitando a eficiência da extração. Desse modo, o volume selecionado para posteriores análises foi de 70 µL. Temperatura e tempo de aquecimento da amostra Avaliou-se o emprego da microextração com 1-dodecanol sem o uso do solvente dispersor, substituindo-o por uma etapa de breve aquecimento controlado da amostra, pois esse processo favorece a dispersão do solvente extrator na amostra aquosa, visto que em temperaturas mais altas a solubilidade do solvente extrator é aumentada, em comparação com a amostra em temperatura ambiente ou inferior.30 O ponto de fusão do 1-dodecanol ocorre a 24 oC, assim, as temperaturas escolhidas para o teste iniciaram a partir da primeira temperatura em que o álcool se tornou líquido. A Figura 6 expõe os resultados encontrados para o estudo da temperatura de aquecimento da amostra, que foi avaliada em 25,0; 30, 0; 50,0; 70,0 e 90,0 °C.
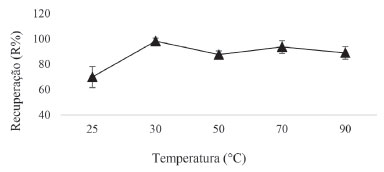 Figura 6. Otimização da temperatura de aquecimento da amostra. Condições das extrações: 70 µL de solvente extrator; aquecimento em Banho Maria: 5 minutos; 1% de NaCl; Agitação manual, centrifugação: 3000 rpm/2 minutos
O 1-dodecanol é considerado como sedo um álcool insolúvel em água, no entanto, ele pode ser solubilizado em outros solventes, como etanol, e em seguida ser diluído em água, nesse caso o etanol atuaria também como solvente dispersor. O uso de aquecimento favoreceu a dispersão do 1-dodecanol em meio aquoso sem a necessidade de utilizar um solvente para dispersar o álcool no meio. Na execução dos experimentos, observou-se um aumento na perda de solvente extrator para a amostra em temperaturas acima de 30 °C, gerando maiores erros nas análises, além da queda no percentual de recuperação. Assim, selecionou-se 30 °C, como a temperatura ideal de aquecimento para uso no método 2. Encontrada a temperatura, avaliaram-se as possíveis influências do tempo de aquecimento da amostra na eficiência da extração, variando-se o tempo em 1; 3; 5; 7 e 9 minutos. Embora possa ter ocorrido uma variação praticamente insignificativa na recuperação em função dos tempos de aquecimento avaliados, observou-se que para os tempos de 5 e 7 minutos foram obtidas melhores estabilidades analíticas, em relação ao desvio padrão. Assim, optou-se por escolher o menor tempo de aquecimento, 5 minutos. Efeito da agitação no vortex Nos estudos de agitação da amostra aquosa no vortex, manteve-se a velocidade fixa em 2800 rpm, variando em 0; 15; 30; 45 e 60 segundos, o tempo de agitação. Os resultados mostraram um aumento significativo na recuperação do clorpirifós no tempo de 15 segundos, sem grandes variações para os tempos seguintes de 30, 45 e 60 segundos, demostrando que o vortex ajuda na dispersão do solvente extrator, melhorando o equilíbrio de partição do clorpirifós na fase orgânica. Desse modo, para as próximas etapas avaliadas no método 1, fixou-se em 15 segundos o tempo de agitação no vortex. Estudo da força iônica da amostra Avaliou-se a adição de pequenas concentrações de cloreto de sódio (NaCl), na amostra aquosa, visto que ele possui capacidade de conferir melhores recuperações ao método.22 A adição de sal pode diminuir a solubilidade dos analitos e, dessa forma, aumentar a eficiência de extração em virtude do efeito salting-out.31 Assim, para avaliar a influência da força iônica na extração, realizaram-se ensaios sem a adição de sal e com adições de 0,5; 1,0; 2,0 e 3,0% de NaCl. Observou-se um aumento gradativo na recuperação até a concentração de 1% de NaCl, sem efeitos significativos na recuperação, para as concentrações de 2 e 3%. Estima-se que, para concentrações elevadas de sal, a interação das moléculas do analito com os íons adicionados reduz a transferência do analito para a fase extratora, ademais, concentrações elevadas de sal também aumentam a viscosidade da amostra, podendo reduzir a mobilidade do analito,32 assim, em ambos os casos, influenciando na eficiência da extração. Desse modo, para as análises seguintes, adicionou-se na amostra aquosa 1% de NaCl Efeito do pH da amostra Avaliou-se o comportamento da DLLME na extração do clorpirifós variando o pH da amostra de 2 a 8. Constatou-se que as melhores recuperações ocorreram do pH ácido ao neutro e, para o pH 8, verificou-se uma diminuição da recuperação. Esse fenômeno pode estar relacionado com a tendência do clorpirifós, de sofrer reações de hidrólise em pH básico,33 interferindo, assim, na sua partição no solvente orgânico. Visto que as amostras de água utilizadas nos experimentos, tanto na otimização, quanto na validação dos métodos, encontraram-se na faixa de pH 4-6, optou-se por não acidificar a amostra. Velocidade e tempo de centrifugação As velocidades de centrifugação avaliadas foram 500; 1000; 2000; 3000 e 4000 rpm; a temperatura da centrifuga foi mantida a 25 oC, evitando variações de temperatura durante a centrifugação. Observou-se um aumento na recuperação do clorpirifós, conforme aumentou-se a velocidade de centrifugação de 500 para 1000 rpm, sem variações significativas para velocidades acima de 1000 rpm. Entretanto, visto que uma maior estabilidade analítica foi alcançada mantendo a velocidade da centrífuga em 3000 rpm, comparando-se os valores do desvio padrão, optou-se por fixar a velocidade da centrífuga em 3000 rpm para as próximas análises. Os tempos de centrifugação estudados foram 0,50; 1,0; 2,0, 3,0 e 4,0 minutos. Verificou-se que um aumento no tempo de centrifugação promove uma ligeira melhoria na eficiência na extração, bem como uma melhor estabilidade analítica, visto que o tempo de permanência na centrífuga, sob uma velocidade ideal determinada, influencia na separação das fases. Desse modo, na etapa de centrifugação da amostra, selecionou-se uma velocidade de 3000 rpm com tempo de permanência na centrífuga por 3 minutos. Validação dos métodos de extração Seletividade No ensaio para determinar a seletividade dos métodos, submeteram-se ao processo de extração amostras de água fortificadas na concentração de 4,0 µg L-1 e amostras de água sem fortificação. A seletividade dos métodos 1 e 2, demonstrada na Figura 7, foi assegurada, pois não se constatou a presença de substâncias eluídas no tempo de retenção correspondente ao pico do clorpirifós. Desse modo, as amostras de águas coletada são matrizes isentas de interferentes e, portanto, ideais para a validação do método DLLME-UV.
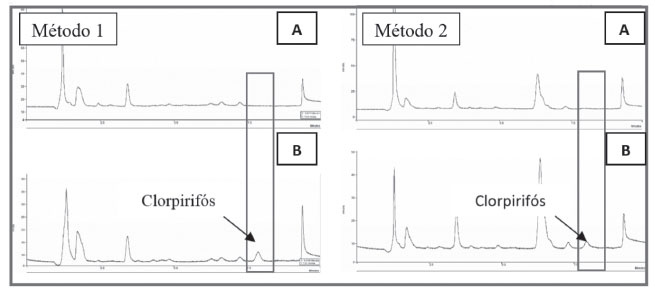 Figura 7. Cromatogramas para amostras de água empregando o método de extração 1 e 2. A) Sem fortificação; B) Com fortificação
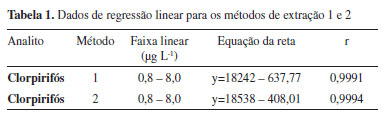
Efeito Matriz O efeito matriz é observado pelo aumento ou supressão do sinal analítico, para um determinado composto presente no extrato da matriz, quando comparado com o sinal analítico obtido para o mesmo analito em solventes orgânicos.26 O efeito matriz pode ser analisado a partir da comparação entre os coeficientes angulares das curvas de calibração construídas com o padrão diluído no solvente e com a amostra fortificada com o padrão e em seguida submetidas ao processo de extração.26 Assim, o efeito matriz é considerado baixo se os valores encontrados estiverem entre -20% e +20%; médio se estiverem entre -50% e -20% ou entre +20% e +50% e alto quando estiverem abaixo de -50% ou acima de +50%.34-36 Os cálculos efetuados indicaram um baixo efeito da matriz, com valores correspondentes a -8,23 e -6,75% para os métodos 1 e 2 respectivamente. Curvas analíticas e faixa linear Avaliou-se a linearidade dos métodos propostos, por meio da construção de curvas analíticas por fortificação das amostras de água (branco), em cada nível de concentração (0,8; 1,60; 2,40; 4,00; 5,60 e 8,00 µ L-1), com soluções padrão do analito, sendo então aplicada a extração otimizada, seguida por análise no HPLC-UV. A faixa linear de trabalho de todas as curvas analíticas empregando os métodos de extração foram construídas a partir do LQ dos métodos, em seis níveis de concentração: 1x, 2x, 3x, 5x, 7x e 10x e tiveram o desígnio de verificar a linearidade dos métodos analíticos. Em ambos os métodos se verificou uma boa linearidade para os compostos estudados, visto que os resultados obtidos (Tabela 1) demonstraram possuir coeficientes de correlação (r) maiores que 0,999, estando assim em conformação com o órgão regulamentador ANVISA, que sugere valores iguais ou superiores a 0,99.25
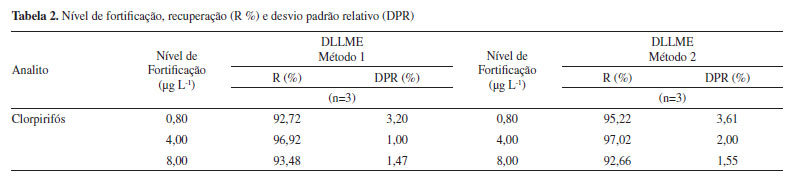
Limites de detecção e quantificação dos métodos Os valores dos limites observados de detecção (LOD) e de quantificação (LOQ) podem ser estimados por meio da relação sinal/ruído, baseado na determinação do branco ou em parâmetros baseados na curva analítica.26 Neste trabalho, os limites de detecção (LD) e quantificação (LQ) foram calculados a partir de parâmetros baseados na curva analítica, em função do desvio padrão residual da linha de regressão. Assim, a partir dos dados obtidos das curvas analíticas, pode-se estimar os valores correspondentes a 0,24 µg L-1 e 0,80 µg L-1 para os valores dos limites de detecção e quantificação, respectivamente. Exatidão A exatidão do método de extração foi avaliada em termos de recuperação, que corresponde a quantidade do analito que é transferida para a fase extratora ao final da extração, representado em porcentagem.29 Sendo assim, os valores encontrados para a recuperação, estão mostrados na Tabela 2. Verifica-se que os ensaios de recuperação, nos três níveis de concentração, para o clorpirifós, apresentaram valores entre 92,72 a 97,02%, tendo o DPR variando entre 1,00 e 3,61%, indicando que ambos os métodos propostos apresentam conformidade com a faixa estabelecida pela ANVISA (70 a 120%, com a precisão de até ± 20%).25
Precisão A precisão para os métodos propostos foi avaliada em termos de repetitividade, através da fortificação e extração de amostras de água em um nível de concentração intermediário da curva analítica (4,0 µg L-1) e injetados no sistema cromatográfico no mesmo dia (intra-dia) e em um dia diferente (inter-dia). As análises foram realizadas em triplicatas e o DPR calculado para as recuperações obtidas. Assim, a precisão dos métodos pode ser avaliada por meio dos valores de DPR, demostrado na Tabela 3.
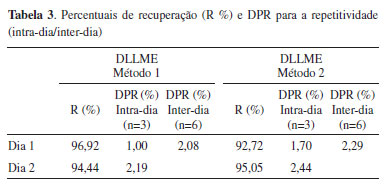
Os resultados obtidos para o estudo da repetitividade foram adequados para as determinações analíticas, visto que os valores encontrados são inferiores a 20%, estando assim, em conformidade com parâmetros estipulados para análise de concentrações traço.37 Comparação com outros métodos de extração de clorpirifós em amostras aquosas Para comparar os métodos propostos com outros métodos de extração, detecção e quantificação de clorpirifós em amostras aquosas publicados na literatura, alguns parâmetros como o volume de amostra, limite de detecção e quantificação, tipos de solventes utilizados e aquecimento da amostra foram considerados e demonstrados na Tabela 4.
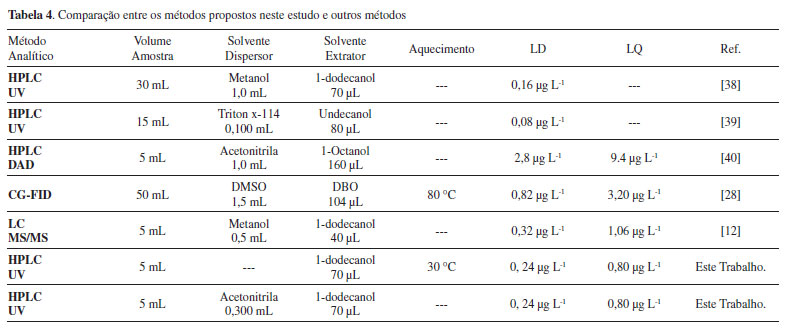
Os métodos propostos apresentam limites de detecção e quantificação comparáveis ou inferiores a outros métodos publicados na literatura, exceto pelos trabalhos envolvendo detectores com maior sensibilidade ou volumes de amostra maiores, que são fatores influenciáveis nos valores LD e LQ. Em ambos os métodos de extração priorizou-se o uso de menores volumes de amostra e de solvente orgânico possíveis. O método de extração 2 propôs a substituição do solvente dispersor por uma etapa de aquecimento, seguida da rápida agitação manual (10 s) da amostra e apresentou um alto valor de recuperação para o clorpirifós, com baixos valores de desvio padrão relativo. Ao final do processo, o solvente extrator foi removido com o auxílio de uma microsseringa, dispensando o congelamento da gota formada, que é o procedimento normalmente utilizado em métodos envolvendo o solvente 1-dodecanol. Desse modo, esses resultados mostram que os métodos desenvolvidos apresentam algumas vantagens sobre outros métodos relatados, como ser simples, sensível, confiável e eficiente. Ademais, usar o solvente de baixo impacto ambiental 1-dodecanol como extrator torna o método menos prejudicial à saúde e ao meio ambiente, quando comparado com solventes halogenados ou aromáticos, frequentemente usados em técnicas de microextração em fase líquida. Aplicação dos métodos desenvolvidos Para aplicabilidade dos métodos, foram coletadas cinco amostras de água oriundas do Rio Dourados, nas proximidades da cidade de Fátima do Sul. As amostras foram coletadas no centro do rio, em uma profundidade máxima de aproximadamente 50 cm, com o auxílio de um coletor construído a partir de um cano de PVC de 100 mm com tampa e armazenadas em frascos de polietileno. Posteriormente, as amostras coletadas foram acondicionadas e transportadas em uma caixa térmica de isopor com gelo. No processo de amostragem não houve a adição de conservantes e nem filtração das amostras. As extrações foram realizadas conforme os parâmetros de extração otimizados e não se encontrou resíduos deste inseticida nas amostras analisadas, pois sua concentração pode estar abaixo do limite de detecção dos métodos.
CONCLUSÃO Os métodos de DLLME propostos mostraram-se eficientes e com base na validação analítica, aptos para aplicações em amostras reais. Dentre os principais pontos da técnica encontram-se o manuseio simples, a necessidade de baixos volumes de amostras e solventes, o uso de materiais de baixo custo e fácil acesso, nos procedimentos de extração. A proposta do método envolvendo uma etapa de aquecimento da amostra apresentou-se vantajosa, uma vez que, além do uso de solvente com maiores características ecológicas, dispensou o uso de solvente dispersor acetonitrila e as recuperações foram elevadas mesmo sem o uso do agitador vortex. Ademais, as recuperações mostraram-se eficientes e estáveis, sendo, portanto, reprodutíveis e aplicáveis na determinação de clorpirifós em amostras de água. Desse modo, frente a precisão, exatidão e aos limites alcançados pelos métodos, esses podem ser empregados em análises traços de resíduos de clorpirifós, tendo em vista a conformidade com os parâmetros exigidos na validação de métodos cromatográficos.
MATERIAL SUPLEMENTAR Em http://quimicanova.sbq.org.br, podem ser encontradas, na forma de arquivo doc, algumas figuras adicionais, disponíveis com acesso livre.
AGRADECIMENTOS Agradecemos o apoio financeiro da Fundação de Apoio ao Desenvolvimento do Ensino, Ciência e Tecnologia do Estado de Mato Grosso do Sul (FUNDECT) e a Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPEs).
REFERÊNCIAS 1. Villa, R. D.; Oliveira, A. P.; Nogueira, R. F.; Quim. Nova 2011, 34, 1501. 2. http://sindiveg.org.br/balanco-2015-setor-de-agroquimicos-confirma-queda-de-vendas/. Acessada em Novembro 2019. 3. Lopes, R. M.; Filho, M. V. S.; Marsden, M.; Alves, N. G.; Quim. Nova 2011, 34, 1275. 4. Balali-mood, M.; Balali-mood, K.; Arch. Iran. Med. 2008, 11, 65. 5. Fukuto, R.; Environ. Health Perspect. 1990, 87, 245. 6. O'shea, S.; Rauh, V.; Alcalay, R.; Neurology 2018, 90, 15. 7. Freire, C.; Koifman, S.; NeuroToxicology 2012, 33, 947. 8. Narayan, S.; Liew, Z.; Paul, K.; Lee, P-C.; Sinsheimer, J. S.; Bronstein, J. M.; Ritz B.; International Journal of Epidemiology 2013, 42, 1476. 9. http://www.ibama.gov.br/agrotoxicos/relatorios-de-comercializacao-de-agrotoxicos#boletinsanuais. Acessada em Novembro 2019. 10. Spadotto, C. A.; Scorza Junior, R. P.; Dores, E. F. G. C.; Gebler, L.; Moraes, D. A. C.; Embrapa Monitoramento por satélite 2010. 46 p. (Documentos, 78). 11. Rebelo, R. M.; Caldas, E. D.; Quim. Nova 2014, 37, 1199. 12. Asati, A.; Satyanarayana, G. N. V.; Patel, D. K.; J. Chromatogr. A 2017, 1513, 157. 13. Ccanccapa, A.; Masiá, A.; Navarro-Ortega, A.; Picó, Y.; Barceló, D.; Environ. Pollut. 2016, 211, 414. 14. Tsaboula, A.; Emmanouil-Nikolaos, P.; Vryzas, Z.; Kotopoulou, A.; Kintzikoglou, K.; Papadopoulou-Mourkidou, E.; Environ. Int. 2016, 91, 78. 15. Pereira, A. R.; Pereira, R. O.; Bottrel, S. E. C.; Revista DAE. 2018, 66, 35. 16. Oliveira, A. R. M.; Magalhaes, I. R. S.; De Santana, F. J. M.; Bonato, P. S.; Quim. Nova 2008, 31, 637. 17. Moreira, B. J.; Yokoya, J. M. C.; Gaitani, C. M.; Sci. Chromatogr. 2014, 6, 186. 18. Rezaee M.; Assadi Y.; Milani Hosseini, M. R.; Aghaee E.; Ahmadi F.; Berijani S.; J. Chromatogr. A 2006, 1116, 1. 19. Zang X. H.; Wu, Q. H; Zhang, M. Y.; Xi, G. H.; Wang, S.; Chin. J. Anal. Chem. 2009, 37, 161. 20. Primel, E. G.; Caldas, S. S.; Marube, L. C.; Escarrone, A. L. V.; Trends Environ. Anal. Chem. 2017, 14, 1. 21. Ahmad, W.; Al-Sibaai, A. A.; Bashammakh, A. S.; Alwael, H.; El-Shahawi, M. S.; Trends Anal. Chem. 2015, 72, 181. 22. Martins, M. L.; Primel, E. G.; Caldas, S. S.; Prestes, O. D.; Adaime, M. B.; Zanella, R.; Sci. Chromatogr. 2012, 4, 35. 23. SEMADE-MS; Índice de Responsabilidade Social de Mato Grosso do Sul, 2a ed., Campo Grande - MS, 2011. 24. IBAMA; Relatórios de comercialização de agrotóxicos. Disponível em: https://www.ibama.gov.br/agrotoxicos/relatorios-de-comercializacao-de-agrotoxicos. Acessada em Novembro 2019. 25. http://www.sanesul.ms.gov.br/noticias/a-nossa-agua-de-cada-dia. Acessada em Novembro 2019. 26. ANVISA; Ministério da Saúde (MS) - Agência Nacional de Vigilância Sanitária. RDC No 166, de 24 de julho de 2017. Diário Oficial da República Federativa do Brasil. 2017. 27. Saraji, M.; Boroujeni, M. K.; Anal. Bioanal. Chem. 2014, 406, 2027. 28. Yang, X.; Wang, C.; Shao, H., Zheng, Q.; Sci Total Environ. 2019, 694, 133494. 29. Rezaee, M.; Yamini, Y.; Faraji, M.; J. Chromatogr. A 2010, 1217, 2342. 30. Farajzadeh, M. A.; Food Chem. 2016, 212, 198. 31. Merib, J.; Carasek, E.; Sci. Chromatogr. 2013, 5, 249. 32. Oliveira, A. R. M.; Magalhães, I. R. S.; Santana, F. J. M.; Bonato, P. S.; Quim. Nova 2008, 31, 3, 637. 33. Liu, B.; Mcconnell, L. L.; Torrents, A.; Chemosphere 2001, 44, 1315. 34. Gosetti, F.; Mazzucco, E.; Zampiere, M. C. G.; J. Chromatogr. A 2010, 1217, 3929. 35. Economou, A.; Botitsi, H.; Antoniou, S.; Tsipi, D.; J. Chromatogr. A 2009, 1216, 5856. 36. Muhammad, M.; Muhammad, R. S.; Shah, J.; Ara, B.; Akhtar, S.; Rahman, H. U.; J. Anal. Sci. Technol. 2017, 8, 12. 37. Ribani, M.; Bottoli, C. B. G.; Collins, C. H.; Jardim, I. C. S. F.; Melo, L. F. C.; Quim. Nova 2004, 27, 771. 38. Xiong, J.; Zhou, G.; Zhou, G.; Tang, X.; He, Q.; Wu, L.; J. Liq. Chromatogr. Relat. Technol. 2014, 37, 1499. 39. Bedassa, T.; Gure, A.; Megersa, N.; J. Anal. Chem. 2015, 70, 1199. 40. Peng, G.; Lu, Y.; He, Q.; Mmereki, D.; Zhou, G.; Chen, J.; Tang, X.; J. Sep. Sci. 2015, 38, 4204. |
On-line version ISSN 1678-7064 Printed version ISSN 0100-4042
Qu�mica Nova
Publica��es da Sociedade Brasileira de Qu�mica
Caixa Postal: 26037
05513-970 S�o Paulo - SP
Tel/Fax: +55.11.3032.2299/+55.11.3814.3602
Free access